

Abstract
Protein acetylation and phosphorylation can be key modifications that regulate both normal and pathological protein functions. Current gel systems used to analyze modified proteins require either expensive reagents or time–consuming second dimension electrophoresis. In this manuscript, we present a neutral pH gel system that allows the analysis of acetylated and phosphorylated proteins. This neutral pH urea Triton-polyacrylamide gel electrophoresis system, or NUT-PAGE, separates proteins based on their charge at pH 7 and generates discrete bands from each acetylated and phosphorylated species. In addition, the gel is composed of common and inexpensive laboratory reagents, and requires only a single dimension of electrophoresis. We are able to demonstrate the effectiveness of this system by analyzing phosphorylated species of an acidic protein, α-synuclein, and both acetylated and phosphorylated species of a basic protein, histone H3. NUT-PAGE thus provides a cost-effective alternative to resolving acetylated and phosphorylated proteins, and potentially proteins with other post-translational modifications that alter net charge.
Method Summary
Here we present a single-dimension neutral pH urea Triton-polyacrylamide gel electrophoresis (NUT-PAGE) system affording high-resolution separation of acetylated and phosphorylated proteins.
Keywords: acetylation, phosphorylation, α-synuclein, histone H3
Introduction
Protein lysine (Lys) acetylation and serine/threonine (Ser/Thr) phosphorylation are among the most pervasive posttranslational modifications that control the normal and even the pathological functions of numerous proteins. It has been estimated that 30% of human proteins are phosphorylated at any given moment, and that acetylation of histones and non-histones are increasing found to play critical roles in a variety of cellular and nuclear functions, including metabolism, nutrient sensing, and gene regulation (see, e.g., 1, 2). A gel system that can effectively resolve acetylated and phosphorylated proteins is of tremendous values in biomedical research. While SDS-PAGE (sodium dodecyl sulfate-polyacrylamide gel electrophoresis) (3) has been an essential laboratory technique, in most cases, it is unable to resolve protein isoforms resulting from such modifications as acetylation and phosphorylation. This is because acetylation and phosphorylation add only 42 and 80 daltons, respectively, to the protein. On the other hand, these two modifications reduce the net charge per modification site by one (acetylation) or by one to two (phosphorylation) at physiological pH, rendering acetylated and phosphorylated proteins amenable to electrophoresis systems based on separation by protein charge. Isoelectric focusing (IEF) is one such method. In IEF, proteins are introduced to a gel matrix with a stable pH gradient generated by ampholytes. Electric current pushes proteins to migrate until they reach the zone where the pH is equivalent to the pI of the protein (4). IEF can resolve proteins differing by a small charge variation. However, proteins with similar charges may differ significantly in their sizes. To further separate such proteins, IEF is paired with a second dimension SDS-PAGE (5), increasing the cost and labor of the assay. In addition to IEF, other charge-based gel systems are available as well. These systems typically use urea to denature proteins, and various chemicals and buffers to maintain a set pH in the gel and the running buffer, so that proteins are ionized and resolved by both the size and the charge. One of the most successful gel systems in this category is the Triton acetic acid urea gel (TAU). TAU-PAGE has been used widely to resolve acetylated histones (6,7,8). However, because of the acidity of the gel system, the phosphate group of certain phosphorylated proteins might become protonated and less charged, rendering acid urea gels less effective in separating phosphoproteins. In principle, at neutral pH, the charge differences caused by Lys acetylation and Ser/Thr phosphorylation are maintained. Thus, a gel system that runs at pH 7 should be a useful and versatile tool.
Protocols for urea-containing PAGE at or near neutral pH were described in several reports (9,10,11). However, these methods have been relatively underutilized, in particular in the realm of post-translational modifications. In this paper, we present a neutral urea Triton-polyacrylamide gel electrophoresis system (NUT-PAGE). NUT-PAGE maintains neutral pH via the use of imidazole and MOPS (3-(N-morpholino)propanesulfonic acid), two common and inexpensive chemicals in biochemistry and molecular biology laboratories. For a proof of principle, we used α-synuclein, an acidic protein, and histone H3, a basic protein, as the examples to demonstrate the feasibility of NUT-PAGE in resolving both acetylated and phosphorylated proteins. This method provides a versatile and affordable alternative to IEF and 2-D systems.
Materials and Methods
Cloning, Protein Expression, and Purification
Human α-synuclein (AAS83394.1, GI:46242542) and budding yeast histone H3 (GI: 855700) were cloned into the PIMAX system vectors developed in our lab (Sui et al, unpublished data) allowing the co-expression of a substrate with the cognate modifying enzyme, resulting in highly efficient modification of the substrate protein. α-synuclein was co-expressed with the human Aurora A kinase (NP_940835.1, GI:38327564), and histone H3 was co-expressed with either S. cerevisiae Gcn5 histone acetyltransferase (GI: 853167) or S. cerevisiae Ipl1 kinase (GI: 855892).
α-synuclein and histone H3 constructs were transformed into BL21–CodonPlus E. coli cells. All bacterial growth and induction were conducted in LB medium containing 100 µg/ml ampicillin. For induction, cells were seeded from an overnight culture at an OD600 of 0.1 and grown until OD600 was 0.3 at 37° C. α-synuclein and histone H3 were induced by 0.5 mM and 0.25 mM IPTG respectively at 37° C for two hours. Cells were then pelleted at 5,000 × g at 4° C and resuspended in Buffer A (100 mM NaCl, 20 mM Tris•HCl pH 7.4, 10% glycerol) and disrupted by sonication using a Misonix Sonicator 3000 (Farmingdale, NY) with ten 15-second bursts at 20% output.
α-synuclein Preparation
α-synuclein was purified by differential precipitation of unwanted protein with 0.5% perchloric acid (12). After ice incubation for 10 minutes, precipitated proteins were removed by centrifugation at 15000 × g for 20 minutes, and the soluble α-synuclein was collected, and the pH was adjusted to 7.4 by the addition of 2 M unbuffered Tris base. The supernatant was further purified by passing through a 40 µm ceramic hydroxyapatite column (Bio-Rad, Hercules, CA) and α-synuclein was eluted with a 75 mM sodium phosphate buffer (pH 6.8). Purified α-synuclein was concentrated using a Ultra-15 10K spin column (Millipore, Billerica, MA) with a molecular weight cutoff of 10,000 daltons, and diluted into Buffer A for storage at −80° C.
Recombinant Histone H3 Preparation
After sonication, bacterial cell debris was removed by centrifugation at 5,000 × g at 4° C for 5 minutes. The supernatant was then centrifuged again for 20 minutes at 10,000 × g at 4° C. The insoluble pellet, which contained H3 inclusion bodies, was collected and washed twice with Triton wash buffer (50 mM Tris•HCl pH 7.4, 100 mM NaCl, 1 mM EDTA, 1% Triton X-100), then washed twice with wash buffer (50 mM Tris•HCl pH 7.4, 100 mM NaCl, 1 mM EDTA). Proteins were then denatured in 2 mL of unfolding buffer (8 M Urea, 20 mM Tris•HCl pH 7.4) and gently rocked at room temperature for one hour. Denatured proteins were further clarified by centrifugation at 10,000 × g for 10 minutes. The supernatant was collected and dialyzed step-wise against 6, 4, 2 and 0 M urea in 20 mM Tris•HCl pH 7.4 using a 12–14 kD cutoff tube (Spectrum Laboratories, Rancho Dominguez, CA) at 4°C for 2 hours. 10% of glycerol was added to the final dialyzed product before −80° C storage.
HeLa Treatment and Histone Preparation
HeLa cells were grown in DMEM supplemented with 5% FBS and 1% Pen/Strep L-glut. For sodium butyrate and nocodazole treatments, HeLa cells were grown to 60% and 40% confluency, respectively. Sodium butyrate treatments were performed as described in Koprinarova et al. (13). Briefly, HeLa cells were treated with fresh DMEM containing 5 mM sodium butyrate for 24 hours. Cells were then washed twice with PBS pH 7.4, scraped, and pelleted. Nocodazole treatment was performed by first treating cells with DMEM containing 2 mM thymidine for 24 hours. Cells were then washed twice with PBS pH 7.4 and released into fresh medium for three hours. Cells were then treated with DMEM containing 100 ng/mL nocodazole for 12 hours, followed by two PBS pH 7.4 washes. Cells were pelleted and crude histones were prepared by acid extraction as described by Shechter et al. (6).
Reverse Phase HPLC Separation of Core Histones
Histone H3 was isolated from the crude histones by Reverse Phase High Performance Liquid Chromatography (RP-HPLC), using a Zorbax Rx-C8 column (4.6 mm inner diameter × 250 mm) (Agilent, Santa Clara, CA), with a multistep gradient method as previously described (6). RP-HPLC fractions containing histone H3 were pooled, proteins were vacuum dried, and dissolved in distilled water. The identity and purity of histone H3 was assessed by SDS-PAGE.
SDS-PAGE
Protein samples boiled in 1 × SDS-PAGE loading dye (60 mM Tris•HCl pH 6.8, 0.02% bromophenol blue, 2% SDS, 10% glycerol, 400 mM β-mercaptoethanol) for 5 minutes. Protein species were then resolved in a 15% SDS polyacrylamide gel at 180V for 70 minutes in a SE250 Mighty Small II Mini Vertical Electrophoresis Unit (Hoefer, Holliston, MA). Gels were then stained using the Coomassie blue protocol described by Wong et al. (14).
NUT-PAGE
To resolve proteins by NUT or TAU gels, it is imperative to remove any residual salt by precipitating proteins in 25% TCA (trichloroacetic acid) or 80% acetone. For acetone precipitation, protein samples were mixed with four volumes of −20° C acetone, incubated at −20° C for one hour, and pelleted by centrifugation at 24,000×g for 10 minutes at 4° C. Air dried protein samples were then dissolved in 2 µL distilled water, to which 6 µL of scavenger/loading dye [6 M urea, 60 mM MOPS pH 8.0, 5% glycerol, 12.5 mg/mL protamine sulfate and 0.15% methyl green (for histone H3 and other basic proteins) or bromophenol blue (for α-synuclein and other acidic proteins)] was added and mixed.
NUT gels were prepared by pipetting 5 mL of resolving gel (as described by Table 1) in 10 cm × 10.5 cm × 0.075 cm assembly, layered with 1 mL of water, and allowed to polymerize for 10 minutes at room temperature. 2 mL of stacking gel solution was prepared as described in Table 1 and layered on top of the resolving gel. The comb, 0.075 cm in thickness, was inserted immediately. The gel was allowed to polymerize for 30 minutes at room temperature. It is recommended to trace the bottom of each well with a marker. This aides subsequent sample loading.
Table 1.
Composition of NUT polyacrylamide gel.
| Resolving Gel | 6 mL (8.2 cm × 9.7 cm × 0.075 cm) | Final Concentration |
| Urea | 2.16 g | 6 M |
| Acrylamide, 60% (60:0.4) | 1.5 mL (15%)/1 mL (10%) | 15%/10% |
| MOPS, 0.5 M, pH 7.0 | 1.2 mL | 100 mM |
| 10% (v/v) Triton X-100 | 0.222 mL | 0.37% (v/v) |
| ddH2O | 1.34 mL (15%)/1.84 mL (10%) | N/A |
| APS, 10% (w/v) | 16 µL | 0.027% (w/v) |
| TEMED | 8 µL | 0.13% (v/v) |
| Stacking Gel | 2 mL (8.2 cm × 9.7 cm × 0.075 cm) | Final Concentration |
| Urea | 0.72 g | 6 M |
| Acrylamide, 60% (60:0.4) | 0.3 mL | 9% |
| MOPS, 0.5 M, pH 8.0 | 0.4 mL | 100 mM |
| 10% (v/v) Triton X-100 | 0.074 mL | 0.37% (v/v) |
| ddH2O | 0.646 mL | N/A |
| APS, 10% (w/v) | 12 µL | 0.06% (w/v) |
| TEMED | 6 µL | 0.3% (v/v) |
Gel assemblies were placed in a SE260 Mighty Small II Deluxe Mini Vertical Electrophoresis Unit (Hoefer, Holliston, MA) and the upper and lower reservoirs were filled with NUT-PAGE running buffer (22 mM MOPS pH 7.0, 100 mM imidazole). The comb was then removed carefully. Gel debris, if visible, were flushed out of the wells by injecting a stream of running buffer through the use of a 25G needle and a syringe. To load protein samples, a 10-µl Hamilton syringe was used. The negatively charged α-synuclein was run from cathode to anode at 125 V for 20 min, then at 100 V for 12 hours (1,200 V•hr). The positively charged histone H3 was run from anode toward cathode at 125 V for 20 min, then 100 V for 2,500 V•hr. The electrophoresis was typically run at room temperature, but could be done in a cold room (without pre-chilling). The lower temperature reduced protein migration by about 50%.
Triton Acetic Acid Urea Gel Electrophoresis
Histone samples were prepared by acetone precipitation as described above, and dissolved in no more than 6 uL of loading dye comprised of 6 M urea, 5% glycerol, 12.5 mg/mL protamine sulfate, 0.15% methyl green, and 5% glacial acetic acid. Triton acid-urea gels were prepared by pipetting 5 mL of resolving gel in 10 cm × 10.5 cm × 0.075 cm assembly with a gel mixture prepared as described in Table 2. Gels were layered with 1 mL water and allowed to polymerize for 20 minutes at room temperature. 2 mL of stacking gel solution were formulated as shown in Table 2 and layered on top of the resolving gel. A comb of 0.075 cm thickness was quickly inserted, and the gel was allowed to polymerize for 45 minutes at room temperature.
Table 2.
Composition of TAU polyacrylamide gel.
| Resolving Gel | 6 mL (8.2 cm × 9.7 cm × 0.075 cm) | Final Concentration |
| Urea | 2.16 g | 6 M |
| Acrylamide, 60% (60:0.4) | 1.5 mL | 15% |
| Glacial Acetic Acid | 0.3 mL | 5% (v/v) |
| 10% (v/v) Triton X-100 | 0.222 mL | 0.37% (v/v) |
| ddH2O | 2.24 mL | N/A |
| APS, 10% (w/v) | 80 µL | 0.13% (w/v) |
| TEMED | 40 µL | 0.67% (v/v) |
| Stacking Gel | 2 mL (8.2 cm × 9.7 cm × 0.075 cm) | Final Concentration |
| Urea | 0.72 g | 6 M |
| Acrylamide, 60% (60:0.4) | 0.3 mL | 9% |
| Glacial Acetic Acid | 0.1 mL | 5% (v/v) |
| 10% (v/v) Triton X-100 | 0.074 mL | 0.37% (v/v) |
| ddH2O | 0.946 mL | N/A |
| APS, 10% (w/v) | 60 µL | 0.3% (w/v) |
| TEMED | 30 µL | 1.5% (v/v) |
For electrophoresis, gels were placed in a SE260 Mighty Small II Deluxe Mini Vertical Electrophoresis Unit (Hoefer, Holliston, MA). Both reservoirs were filled with 5% glacial acetic acid. Histone H3 samples were then loaded and run from anode to cathode at 150 V for 20 min, followed by 100 V for 1,200 V•hr. Leaked urea was flushed out of each well immediately before sample loading.
Protein Staining and Visualization
Gels were stained using Coomassie blue dye (14) or silver nitrate stain (15) for general purposes. Phospho-specific stain was conducted with ProQ Diamond staining (Invitrogen, Carlsbad, CA) per the user manual. Briefly, gels were removed from the gel box and fixed at room temperature in 50% methanol/10% acetic acid for 30 minutes, followed by overnight fixation in fresh methanol/acetic acid solution with gentle agitation. Gels were then washed three times in ultrapure water for 10 minutes with gentle agitation, then soaked with ProQ Diamond stain for 90 minutes, followed by three 30-minute washes in de-stain solution (20% acetonitrile/50 mM sodium acetate pH 4.0). Finally, gels were washed twice in ultrapure water for five minutes and imaged on a Typhoon 9200 (GE Healthcare, Little Chalfont, UK) with excitation at 532 nm and emission at 560 nm.
NUT-PAGE Transfer and Western Blotting
For immunoblotting analysis following NUT-PAGE, gels were removed from the assembly after finishing the run, and proteins were transferred to a nitrocellulose membrane using a PhosPhor semi-dry blotter (Hoefer, Holliston, MA) under constant current of 1.0 mA/cm2 for 2 hours. Transfer was conducted in either NUT-PAGE running buffer supplemented with 20% methanol or Towbin’s transfer buffer (25 mM Tris•HCl pH 7.4, 192 mM glycine, and 20% methanol) (16). When using the NUT-PAGE running buffer for electroblotting, positively charged proteins such as histones moved from anode to cathode, whereas negatively charged proteins such as α-synuclein traveled in an opposite direction. If the Towbin’s buffer was used, all proteins moved from cathode to anode. The assembly of gel and membrane was adjusted accordingly. It is not necessary to equilibrate the NUT polyacrylamide gel in either transfer buffer before transferring to the membrane. After transfer, membranes were blocked with 5% milk in TBST (150 mM NaCl, 27 mM KCl, 250 mM Tris pH 7.4, and 0.05% Tween 20) at room temperature for 1 hour. Standard procedures for immunoblotting were followed using 1:2,000 dilution of α-synuclein antibody (ab27766) (Abcam, Cambridge, UK) or 1:1,000 dilution of phospho-S129 α-synuclein antibody (ab59264) (Abcam, Cambridge, UK) in TBST containing 0.4% gelatin at 4°C overnight. Blots were washed with TBST, probed with goat anti-mouse horseradish peroxidase conjugated secondary antibody (1:5,000 dilution) for 90 minutes, washed three times with TBST, incubated with Lumilight chemiluminescent substrate (Roche, Indianapolis, IN) for 5 minutes, and exposed to film.
Results
Overview of NUT-PAGE
The NUT-PAGE system is designed to separate proteins by exploiting the differences of net charge at neutral pH. This gel system uses urea to denature proteins while preserving their ionization status, unlike SDS that coats proteins with negative charges. Electrophoresis is conducted at pH 7 that is maintained by a combination of two common laboratory chemicals, MOPS and imidazole. Neutral pH allows both acidic and alkaline proteins to be separated. Acidic proteins (pI < 7) and basic proteins (pI > 7) are negatively and positively charged at pH 7.0, respectively, thus migrate toward different electrodes. Electric current direction is adjusted to match the pI of the protein. In addition, the NUT-PAGE system is compatable with common immunoblotting techniques and equipment. The following sections demonstrate the setup and applications of NUT-PAGE.
Gel Electrophoresis of α-synuclein
α-synuclein is an acidic protein with a close link to the development of Parkinson’s disease and several other neurodegenerative disorders collectively known as the synucleinopathies. These diseases are manifested by the intraneuronal Lewy bodies containing high abundance of phosphorylated α-synuclein (17, 18). Phosphorylation of α-synuclein has been reported to occur at several residues, including serines 87 and 129, and tyrosines 125, 133 and 136 (19). Oligomerization and neurotoxicity of α-synuclein can be regulated by phosphorylation(20). A simple gel system that enables the separation of α-synuclein based on its degree of phosphorylation will likely have a positive impact on the basic and translational research of Parkinson’s disease.
Unmodified and Aurora A kinase phosphorylated α-synuclein samples were first analyzed by SDS-PAGE (Figure 1A). Like many other phosphorylated proteins, reproducible but subtle migration retardation was seen in the phosphorylated isoform (right lane). This relatively minor alteration of mobility was not sufficient to reveal the relative abundance of the phosphorylated species, nor did it show the degree of α-synuclein phosphorylation. In contrast, the charge negation caused by each deprotonated phosphate group at the neutral pH is likely to cause a substantial alteration in the electrophoretic mobility of α-synuclein (pI = 4.7; net charge at pH 7.0 is −8.8 without phosphorylation). Indeed, when resolved by NUT-PAGE, phosphorylated α-synuclein ran as multiple faster-migrating species, suggesting different degrees of phosphorylation (Figure 1B). This notion was further supported by phosphatase treatment (Figure 1C) in that the “weight” of the bands shifted up toward the unphosphorylated α-synuclein band, coinciding with the increasing phosphatase doses.
Figure 1. NUT-PAGE resolves phosphorylated α-synuclein into multiple species.


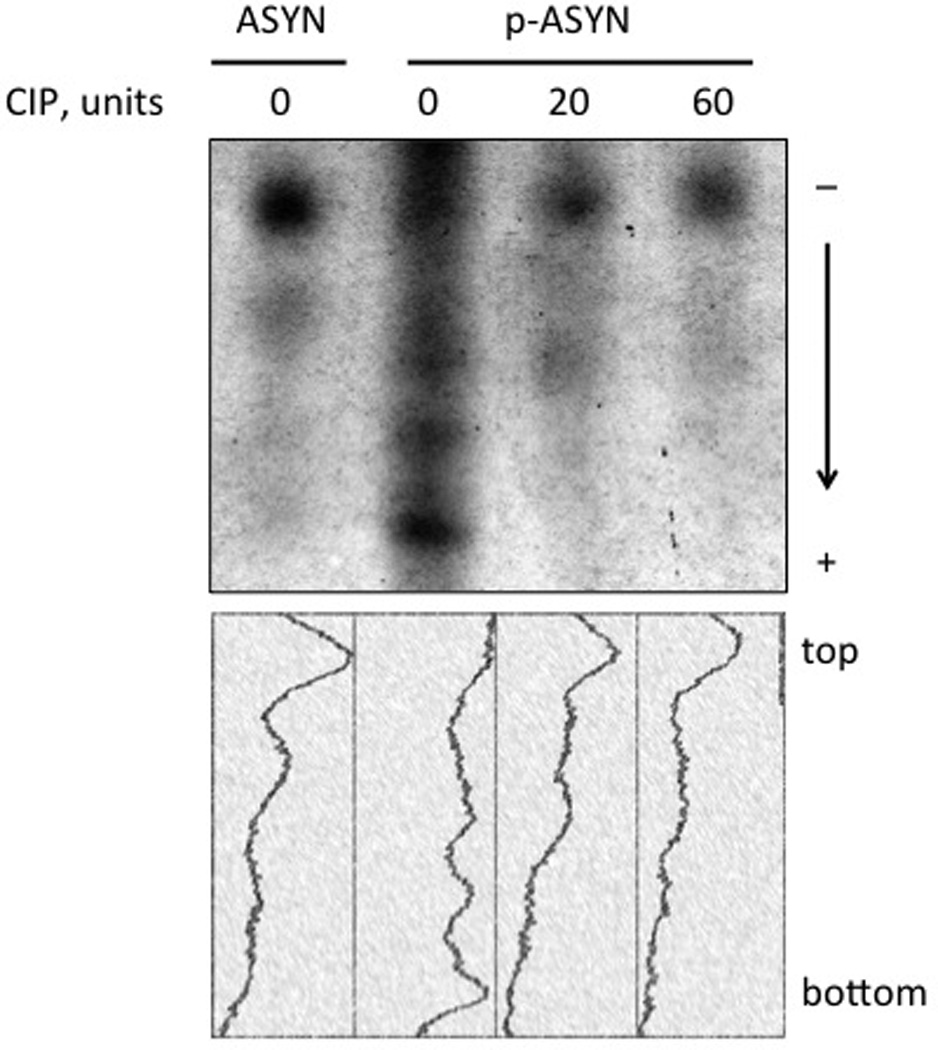
(A) Phosphorylated α-synuclein exhibits minimal mobility shift in SDS-PAGE. Unmodified and phosphorylated α-synuclein were resolved by 12% SDS-PAGE and detected by Coomassie blue staining. (B) Multiple isoforms of phosphorylated α-synuclein were resolved by NUT-PAGE. Purified α-synuclein proteins were analyzed by 10% NUT-PAGE and stained by Coomassie blue. Unmodified α-synuclein migrated as a single band, whereas phosphorylated α-synuclein were separated into multiple, higher-mobility bands. Note that these proteins migrated from cathode (−) to anode (+); more negatively charged phosphorylated species thus exhibited faster mobility under the electrophoresis condition. (C) Phosphatase treatment reduces mobility of phosphorylated α-synuclein. Phosphorylated α-synuclein was treated with increasing amounts of calf intestinal phosphatase (CIP) at 37°C for 60 minutes before NUT-PAGE and Commassie blue staining. The relative abundance of the faster-migrating species decreased upon CIP action. Densitometry traces below were generated by ImageJ (http://rsbweb.nih.gov/ij/). ASYN, α-synuclein, p-ASYN, phosphorylated α-synuclein.
Western blot analysis of α-synuclein of proteins resolved by NUT-PAGE
Western blotting following typical SDS-PAGE provides a convenient method for protein identification. To establish a protocol for immunoblotting for proteins resolved by NUT-PAGE, we used a standard semi-dry blotting approach (see Materials and Methods) to transfer α-synuclein isoforms from a NUT gel to nitrocellulose membrane. Using the NUT-PAGE running buffer supplemented with methanol or the Towbin buffer for SDS-PAGE blot transfer, we successfully transferred α-synuclein to nitrocellulose membrane, as evidenced by the subsequent immunoblotting with a general α-synuclein antibody and one recognizing phosphorylated Ser129 of α-synuclein (21) (Figure 2). As expected, the pSer129 antibody did not recognize unphosphorylated α-synuclein, but bound three faster migrating species of the phosphorylated isoform. As this antibody failed to bind to the second highest band (i.e., the likely monophosphorylated species), it seems likely that Ser129 is a secondary phosphorylation site for Aurora A kinase under the experimental condition. Indeed, preliminary mass spectrometry and mutation studies suggested that Ser84 was the primary phosphorylation site by Aurora A (data not shown), an observation that is yet to be examined for its biological relevance. Regardless of the sites of phosphorylation, the results of Figure 2 confirmed that the NUT-PAGE system can be readily integrated into the daily operation of many biochemistry and molecular biology laboratories.
Figure 2. Immunoblotting application following NUT-PAGE.

Following 10% NUT-PAGE resolution, unmodified and phosphorylated α-synuclein were transferred to a nitrocellulose membrane, and probed with a general anti-α-synuclein antibody (A) or anti-phosphorylated Ser129 antibody (B) using standard western blotting procedures. Note that panels A and B were from two independent protein preps and PAGE runs. A parallel anti-α-synuclein immunoblot of that shown in panel B allowed us to mark the bands not detectable by the phosphorylation-specific antibody (dotted circles), suggesting that Ser129 is phosphorylated only in the presence of other pre-existing phosphorylation events.
Histone H3 Acetylation and Phosphorylation
At neutral pH, many proteins are sufficiently charged, rendering them good subjects for NUT-PAGE analysis. However, the pI of the underlying protein determines its net charge at any given pH. For example, basic proteins have a pI that is higher than pH 7.0, and will be positively charged in the neutral environment of NUT-PAGE. These proteins, such as histones, shall be loaded at the positive end (anode) of the gel. Acidic proteins including α-synuclein will have to be loaded on the cathode (negative) end of the gel (Figure 1). By simply switching the two electrodes of the gel apparatus, these protein-specific running direction requirements can be met. To test this prediction, histone H3 was chosen for its well-documented acetylation and phosphorylation, which are linked to a variety of nuclear activities (22, 23, 24). Acetylation neutralizes the positive charge of lysine, whereas phosphorylation adds up to two negative charges to serine or threonine at pH 7. In either case, the net charge of histone H3 decreases. The mobility is thus predicted to be retarded by acetylation and phosphorylation when H3 migrates toward the cathode.
The resolution of acetylated and phosphorylated H3 by NUT-PAGE is shown in Figure 3. Unmodified H3 ran as a single band, whereas both acetylated and phosphorylated H3 showed multiple discrete bands with retarded mobility through the NUT gel (Figure 3A), consistent with the negation of overall positive charge on the protein. Interestingly, densitometry tracing shows that acetylation and phosphorylation caused discernible differences in mobility retardation. This distinction probably results from the charge difference between these two modifications, although the sizes of acetyl and phosphate groups (42 Da vs. 80 Da) may also contribute to this phenomenon.
Figure 3. Both acetylated and phosphorylated histone H3 isoforms can be resolved by NUT-PAGE, whereas TAU-PAGE can only resolve the acetylated H3 species.


(A) 15% NUT-PAGE was used to separate the positively charged histone H3. The densitometric traces (bottom) of the silver-stained histone H3 species were generated by ImageJ (http://rsbweb.nih.gov/ij/). Both acetylation and phosphorylation reduce the net positive charge on histone H3, resulting in multiple bands that display reduced mobility in the gel matrix. Note that histone H3 is a basic protein, which thus was run from annode to cathode. (B) Triton-acetic acid-urea (TAU) gel does not resolve phosphorylated histone H3. A conventional TAU gel (15%) was used to resolve different H3 isoforms. The left panel shows silver stained unmodified and acetylated histone H3 after electrophoresis. Acetylated species of histone H3 migrate at a slower rate, with comparable resolution to NUT-PAGE. The right panel shows unmodified and phosphorylated histone H3 stained by either silver stain or the phospho-specific ProQ Diamond stain following TAU-PAGE. Phosphorylated histone H3 displays only a marginal decrease in mobility. ProQ Diamond staining confirmed phosphorylation was present on the modified protein.
The same batch of acetylated and phosphorylated H3 were also analyzed using a traditional TAU gel system. Figure 3B shows the separation of acetylated H3 isoforms in the acidic matrix. Multiple bands were observed but the number of acetylated H3 isoforms was frequently smaller in TAU gels. Additionally, the phosphorylated forms of H3 co-migrated in a single band and displayed only a modest mobility shift. The phosphorylation status of histone H3 was confirmed by utilizing the ProQ Diamond stain that specifically detected phosphorylated proteins (right panel, Figure 3B). The inability of TAU gel to resolve phosphorylated H3 species was most likely because of the acidity of the gel (pH 2.4) that is close to pKa1 (2.15) and well below pKa2 (~7) (25, 26) of the phosphate group within a protein, leading to significant protonation and hence a minimal charge difference. Therefore, while the TAU gel has been used widely to resolve acetylated H3, it might not be as useful in resolving phosphorylated H3.
Analysis of Modified Histones in Cell Culture
Results of Figure 3 have displayed the utility and effectiveness of NUT-PAGE for investigating the modification status of recombinantly expressed proteins purified from E. coli. To test whether NUT-PAGE can also be utilized to probe the post-translational modifications of proteins isolated from cultured cells, we prepared HeLa histones after treating the cells with sodium butyrate and nocodazole that, respectively, triggered histone hyperacetylation and hyperphosphorylation (27). Total histones were obtained from the untreated and treated cultures for NUT-PAGE (Figure 4A). Purified chicken histones were used as the indicators for their HeLa counterparts. Hyperactylation can be observed in the sodium butyrate treated sample by noting the shift of “weight” in the histone H4 species to slower migrating species, indicating acetylation and the consequent loss of charge. However, the anticipated H3 phosphorylation resulting from mitotic arrest was difficult to detect, as histone H3 and histone H2B display similar mobility, and therefore overlap in NUT-PAGE. To solve this problem, we used reverse phase HPLC to fractionate H3 from H2B. Silver staining of H3 separated by NUT-PAGE revealed clear mobility retardation in the nocodazole-treated sample (Figure 4B). This change in mobility was associated with Ser10 phosphorylation, as confirmed by western analysis with a phospho-S10 H3 antibody (Figure 4B). We therefore conclude that NUT-PAGE also can be used for probing the post-translational modification status of histones from a natural source.
Figure 4. Post-translationally modified histones isolated from cell culture can be resolved by NUT-PAGE.


(A) HeLa cells were treated with sodium butyrate to induce hyperacetylation or nocodazole to block progression through mitosis and enrich for phosphorylated serine 10 of histone H3. Histone proteins were then isolated and separated by NUT-PAGE. Sodium butyrate treatment resulted in increased histone H4 acetylation, as can be seen by a decreased mobility of multiple bands resulting from the loss of charge. Histone H3 phosphorylation cannot be noted due to co-migration of histones H2B and H3. Purified chicken histones were used as mobility references for each core histone protein. (B) Histone H3 was isolated from crude histones by RP-HPLC and modified species of histone H3 were resolved by NUT-PAGE and visualized by silver staining. Histone H3 collected from untreated HeLa cells migrates as three distinct bands, while histone H3 from nocodazole treated HeLa cells displayed additional bands with overall lowered mobility in NUT-PAGE, consistent with the fact that metaphase H3 is phosphorylated at Ser10. Western analysis using an antibody against H3 phosphorylated at Ser10 confirmed that phosphorylation is associated with retarded gel mobility of NUT-PAGE.
Experimental results presented above show that the NUT-PAGE system can resolve both acetylated and phosphorylated species of acidic and basic proteins. Inexpensive and common laboratory chemicals are used. We also provided the protocols for blotting and western detection. Together, the NUT-PAGE system affords an attractive alternative to acid urea gels and isoelectrofocusing.
The neutral pH of NUT-PAGE ensures that each phosphate group is deprotonated (pKa1 = ~2; pKa2 = ~7) (26), whereas the acidic pH of the widely used TAU gel may restrict phosphate group deprotonation to a minimal. While this partial deprotonation was sufficient for the resolution of hyperphosphorylated H1 (28), phosphorylated H3 only exhibited subtle mobility changes in the TAU gel system. In theory, NUT-PAGE is also compatible with other phosphorylation marks, i.e., phosphotyrosine and the acid-labile phosphohistidine. By the same token, other charge-altering PTMs such as deamination (converting glutamine to glutamate) and sulfation (addition of a sulfate to tyrosine) may also be good subjects for NUT-PAGE.
The effectiveness of NUT-PAGE is limited by two factors intrinsic to proteins: pI and molecular weight. Proteins with a pI near 7.0 will possess very little net charge at pH 7.0, resulting in low or no electrophoretic mobility. Posttranslational modifications that alter the net charge of these proteins may help push the modified species into the gel. Changes made to the current NUT-PAGE recipe, such as a slightly different pH and the addition of Coomassie blue G-250, which, unlike SDS, only adds a limited amount of charges to proteins (29), may broaden the applicability of the current methodology. The effect of molecular weight was first noted when we tried to resolve bovine serum albumin (BSA; molecular weight ca. 65 kD) by NUT-PAGE. BSA displayed limited mobility and resolution (data not shown). A lower percentage gel (e.g., 10%) did provide some improvement, but we have yet to try even lower concentrations due to the increased frailty of the gel. A thicker gel matrix will likely be useful.
NUT-PAGE sometimes runs overnight for a better resolution. An important precaution for overnight electrophoresis is to ensure that the seal between the plate and the top buffer reservoir is intact as buffer leakage may severely impair the overall quality. Additionally, common practices help maintain consistency in gel quality. These include de-aeration and filtering the gel solutions before casting; flushing each well with the running buffer immediately before sample loading; and using minimal volume of the sample. Pre-running with or without the scavenger solution (i.e., the gel loading buffer) did not result in appreciable improvement and sometimes reduces band sharpness (data not shown). Pre-running thus is not recommended.
Lastly, we suspect that certain deviations of the gel recipes are worth testing for improvement. For example, Triton DF-16 and Trixon X-100 have differential effects on histone resolution by TAU gel electrophoresis (30). Varying the concentration of Triton X-100 or switching to Triton DF-16 may be desirable for certain applications. Employment of buffering agents other than MOPS and imidazole may also provide refinement.
Acknowledgments
The authors acknowledge anonymous reviewers for their insightful suggestions and constructive comments. This work was supported by grants from the Rackham Fund, Michigan State University, the NIH (AG039768), and NSF (MCB1050132).
Footnotes
Author Contribution
CB established the immunoblotting procedures for NUT-PAGE. CB and SH were responsible for the preparation of HeLa histones. ML and XD each was responsible for the cloning, expression, isolation, and gel characterization of α-synuclein and histone H3, respectively. RP-HPLC was performed by XD. MHK developed the original NUT-PAGE recipe and coordinated the compilation of this manuscript. The initial setup of the gel system was done by XX.
The authors declare no competing interests.
REFERENCES
- 1.Aka JA, Kim GW, Yang XJ. K-acetylation and its enzymes: overview and new developments. Handb Exp Pharmacol. 2011;206:1–12. doi: 10.1007/978-3-642-21631-2_1. [DOI] [PubMed] [Google Scholar]
- 2.Walsh CT. Posttranslational modification of proteins - expanding nature's inventory. Greenwood Village, CO: Roberts and Company; 2007. [Google Scholar]
- 3.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 4.Vesterberg O, Svensson H. Isoelectric fractionation, analysis, and characterization of ampholytes in natural pH gradients. Acta Chemica Scandanavia. 1966;20:820–834. doi: 10.3891/acta.chem.scand.20-0820. [DOI] [PubMed] [Google Scholar]
- 5.O’Farrell PH. High resolution two–dimensional electrophoresis of proteins. J Biol Chem. 1975;250:4007–4021. [PMC free article] [PubMed] [Google Scholar]
- 6.Shechter D, Dormann HL, Allis CD, Hake SB. Extraction, purification and analysis of histones. Nature Protocols. 2007;2:1445–1457. doi: 10.1038/nprot.2007.202. [DOI] [PubMed] [Google Scholar]
- 7.Johmann CA, Gorovsky MA. Purification and characterization of the histones associated with the macronucleus of Tetrahymena. Biochemistry. 1976;15:1249–1256. doi: 10.1021/bi00651a012. [DOI] [PubMed] [Google Scholar]
- 8.Jackson V. Formaldehyde cross–linking for studying nuclesomal dynamics. Methods. 1999;17:125–139. doi: 10.1006/meth.1998.0724. [DOI] [PubMed] [Google Scholar]
- 9.Thomas JM, Hodes ME. A new discontinuous buffer system for the electrophoresis of cationic proteins at near–neutral pH. Analytical Biochemistry. 1981;118:194–196. doi: 10.1016/0003-2697(81)90178-0. [DOI] [PubMed] [Google Scholar]
- 10.Taber HW, Sherman F. Spectrophotometric analyzers for disc electrophoresis: studies of yeast cytochrome c. Ann N Y Acad Sci. 1964;121:600–615. doi: 10.1111/j.1749-6632.1964.tb14229.x. [DOI] [PubMed] [Google Scholar]
- 11.Hoffmann PJ, Chalkley R. A neutral pH acrylamide gel electrophoretic system for histones and other basic proteins. Analytical Biochemistry. 1976;76:539–546. doi: 10.1016/0003-2697(76)90346-8. [DOI] [PubMed] [Google Scholar]
- 12.Gasparini L, Crowther RA, Martin KR, Berg N, Coleman M, Goedert M, Spillantini MG. Tau inclusions in retinal ganglion cells of human P301S tau transgenic mice: effects on axonal viability. Neurobiology of Aging. 2011;32:419–433. doi: 10.1016/j.neurobiolaging.2009.03.002. [DOI] [PubMed] [Google Scholar]
- 13.Koprinarova M, Markovska P, Iliev I, Anachkova B, Russev G. Sodium butyrate enhances the cytotoxic effect of cisplatin by abrogating the cisplatin imposed cell cycle arrest. BMC Molecular Biology. 2010;11:49. doi: 10.1186/1471-2199-11-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wong C, Sridhara S, Bardwell JCA, Jakob U. Heating greatly speeds Coomassie blue staining and destaining. BioTechniques. 2000;28:426–432. doi: 10.2144/00283bm07. [DOI] [PubMed] [Google Scholar]
- 15.Wray W, Boulikas T, Wray VP, Hancock R. Silver staining of proteins in polyacrylamide gels. Analytical Biochemistry. 1981;118:197–203. doi: 10.1016/0003-2697(81)90179-2. [DOI] [PubMed] [Google Scholar]
- 16.Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marques O, Outeiro TF. Alpha-synuclein: from secretion to dysfunction and death. Cell Death Dis. 2012;3:1–7. doi: 10.1038/cddis.2012.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bellucci A, Zaltieri M, Navarria L, Grigoletto J, Missale C, Spano PF. From α–synuclein to synaptic dysfunctions: new insights into the pathophysiology of Parkinson’s disease. Brain Research. 2012;1476:183–202. doi: 10.1016/j.brainres.2012.04.014. [DOI] [PubMed] [Google Scholar]
- 19.Chen L, Feany MB. α–synuclein phosphorylation controls neurotoxicity and inclusion formation in a drosophila model of Parkinson disease. Nat Neurosci. 2005;8:657–663. doi: 10.1038/nn1443. [DOI] [PubMed] [Google Scholar]
- 20.Cavallarin N, Vicario M, Negro A. The role of phosphorylation in synucleinopathies: focus on Parkinson’s disease. CNS Neurol Disord Drug Targets. 2010;9:471–481. doi: 10.2174/187152710791556140. [DOI] [PubMed] [Google Scholar]
- 21.Okochi M, Walter J, Koyama A, Nakajo S, Baba M, Iwatsubo T, Meijer L, Kahle PJ, Haass C. Constitutive phosphorylation of the Parkinson’s disease associated α–synuclein. J Biol Chem. 2000;275:390–397. doi: 10.1074/jbc.275.1.390. [DOI] [PubMed] [Google Scholar]
- 22.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 23.Zentner GE, Henikoff S. Regulation of nucleosomal dynamics by histone modifications. Nat Struct Mol Biol. 2013;20:259–266. doi: 10.1038/nsmb.2470. [DOI] [PubMed] [Google Scholar]
- 24.Downs JA. Histone H3 K56 acetylation, chromatin assembly, and the DNA damage checkpoint. DNA Repair. 2008;7:2020–2024. doi: 10.1016/j.dnarep.2008.08.016. [DOI] [PubMed] [Google Scholar]
- 25.Weast RC, editor. CRC Handbook of Physics and Chemistry. 47th ed. Cleveland, OH: CRC Press; 1966. [Google Scholar]
- 26.Peacock CJ, Nickless G. The disassociation constants of some phosphorus(V) acids. Z Naturforsche. 1969;24a:245–247. [Google Scholar]
- 27.Wei Y, Yu L, Bowen J, Gorovsky MA, Allis CD. Phosphorylation of histone H3 is required for proper chromosome condensation and segregation. Cell. 1999;97:99–109. doi: 10.1016/s0092-8674(00)80718-7. [DOI] [PubMed] [Google Scholar]
- 28.Ryan CA, Annunziato AT. Separation of histone variants and post–translationally modified isoforms by triton/acetic acid/urea polyacrylamide gel electrophoresis. Curr Protoc Mol Biol. 2001;21:2. doi: 10.1002/0471142727.mb2102s45. [DOI] [PubMed] [Google Scholar]
- 29.Congdon RW, Muth GW, Splittgerber AG. The binding interaction of Coomassie blue with proteins. Analytical Biochemistry. 1993;213:407–413. doi: 10.1006/abio.1993.1439. [DOI] [PubMed] [Google Scholar]
- 30.Alfageme CR, Zweidler A, Mahowald A, Cohen LH. Histones of Drosophila embryos. Journal of Biological Chemistry. 1974;249:3729–3736. [PubMed] [Google Scholar]