

Abstract
In this study we report a series of triazine derivatives that are potent inhibitors of PDE4B. We also provide a series of structure activity relationships that demonstrate the triazine core can be used to generate subtype selective inhibitors of PDE4B versus PDE4D. A high resolution co-crystal structure shows that the inhibitors interact with a C-terminal regulatory helix (CR3) locking the enzyme in an inactive “closed” conformation. The results show that the compounds interact with both catalytic domain and CR3 residues. This provides the first structure-based approach to engineer PDE4B-selective inhibitors.
Keywords: PDE4 inhibitor, PDE4B, PDE4D, Crystallography, triazine
Phosphodiesterases (PDEs) hydrolyze cyclic adenosine monophosphate (cAMP) and cyclic guanosine monophosphate (cGMP) to their cognate 5′-monophosphate derivatives. There are eleven different PDE superfamily members (PDE1–11) and inhibitors have been developed to prolong the effects of physiological processes mediated by cAMP or cGMP. Phosphodiesterase 4 (PDE4) is the primary enzyme that regulates the turnover of cAMP.1 PDE4 is comprised of four genes (PDE4A–D) and each gene has multiple transcripts that can produce three isoforms of the protein termed long, short and super short. Long forms of PDE4 contain two upstream regions known as UCR1 and UCR2. These form a negative regulatory module that is activated by protein kinase A (PKA) phosphorylation of UCR1. Burgin and coworkers were the first to show that the negative regulation results from UCR2 closing over the active site, thereby preventing access of cAMP.2 A third C-terminal control region (CR3) is present in all PDE4 isoforms and was also shown to close over the active site by weakly engaging inhibitors (e.g. PMNPQ; PDB ID: 3G58); however, the significance of this region was not well understood.2 Our recent structural studies reveal that potent and selective PDE4B inhibitors bind CR3 thereby locking the enzyme in a closed conformation.3 PDE4B selectivity is due to a single amino acid polymorphism in CR3 that selects the helical registration of the domain when it closes over the active site. Exchange of a leucine in PDE4B CR3 for a glutamine in PDE4D causes a 70–80 fold shift in inhibitor selectivity. In this report we describe a series of triazine analogs that similarly bind to CR3 thereby resulting in PDE4B selectivity.
PDE4B is a therapeutic target of high interest for central nervous system (CNS), inflammatory and respiratory diseases;4,5 however, it has been extremely difficult to discover selective PDE4B inhibitors due to the high amino sequence conservation of the PDE4 active site.6
We initiated our chemistry effort for discovery of new PDE4B inhibitors that engage PDE4 regulatory domains with in silico screening and docking studies conducted with commercial libraries against known PDE4 structures (3IAD, 3G45 & 3G58).2 Screening of the hits in enzymatic assays using PDE4B1 and PDE4D7 resulted in identification of compound 1 as a novel PDE4 inhibitor, and the 1,3,5-triazine series was selected to begin our synthetic efforts. Although this initial hit does not show PDE4B-subtype selectivity, we noted the similarity to 2, a compound reported by Naganuma (A-33),7 which we confirmed to be highly selective for PDE4B.8 In addition, a series of triazine analogs has recently been reported by others to inhibit PDE4A.9
The general synthesis of the 1,3,5-triazine series is illustrated in scheme 1. Using the procedure of Harris10 a nitrile bearing the AR2 group is converted into the corresponding cyanoamidine. The R1 group is then introduced and the triazine ring formed by reaction of the cyanoamidine with an N,N-dimethylamide. The chlorogroup is then displaced to afford the desired Ar1 products. Detailed experimental procedures are supplied in the supplemental material.
Scheme 1.
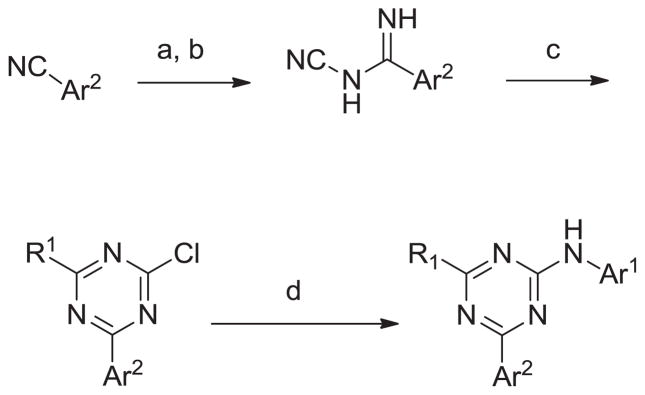
General synthesis of the 1,3,5-triazine series. Reagents and conditions: (a) NaOMe, MeOH (b) NH2CN (c) R1CONMe2, POCl3 CH3CN (d) Ar1NH2, AcOH
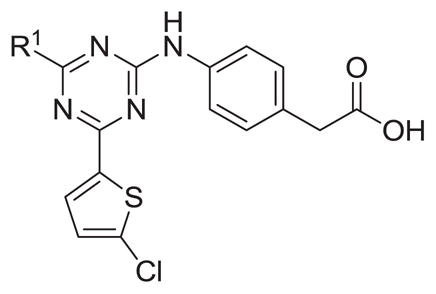
The compounds were screened for their ability to inhibit long isoforms of PDE4D and PDE4B in vitro (see supplemental data), and the results are shown in Tables 1–6. Generally the compounds in this paper are druglike and RO5 compliant.11 The compounds have calculated polar surface areas ranging from 50 to 88 Å2. (see supplemental data),
Table 1.
Inhibitory activity of R1 triazine analogs.
| ||||
|---|---|---|---|---|
| # | R1 | hPDE4B IC50 (nM) | hPDE4D IC50 (nM) | Ratio D/B IC50 |
| 3 | Me | 1287 | NT | |
| 4 | i-Pr | 1945 | 3580 | 1.8 |
| 5 | Et | 383 | 865 | 2.3 |
| 6 | n-Pr | 2447 | NT | |
| 7 |
|
251 | 1489 | 5.9 |
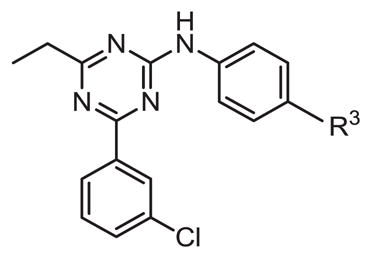
Table 6.
Inhibitory activity of Ar1 substitution.
| ||||
|---|---|---|---|---|
| # | R3 | hPDE4B IC50 (nM) | hPDE4D IC50 (nM) | Ratio D/B IC50 |
| 26 | CH2CO2H | 781 | 681 | 0.9 |
| 27 | -CO2H | 241 | 213 | 0.9 |
| 28 | CN | 12 | 13 | 1.1 |
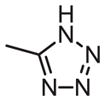
| 29 | CH2CN | 165 | 7 | 0.04 |
| 30 |
|
126 | 132 | 1.0 |
| 31 |
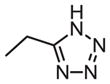
|
69 | 63 | 0.9 |
| 32 |
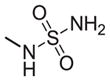
|
402 | 160 | 0.4 |
| 33 |
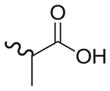
|
1090 | 96 | 0.09 |
| 34 |
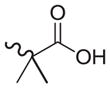
|
1574 | 600 | 0.4 |
Synthesis of various triazine analogs with aliphatic R1 groups revealed ethyl and cyclopropyl substituents to be 4–5 fold more active than methyl-, propyl-, or isopropyl-containing analogs (Table 1). The cyclopropyl containing analog 7 displayed modest selectivity, about six fold, for PDE4B versus PDE4D.
Varying Ar2 revealed that 2-chlorothiophene (7) and 3-chlorophenyl (8) were the preferred substituents for PDE4B selectivity with the 2-chlorothiophene consistently displaying better selectivity. The NH linker between the triazine core and Ar1 is also required for potency against both PDE4B and PDE4D. Analogs with an aminomethylene (14 and 15) or oxygen linker (16) resulted in decreased potency with IC50 values > 1 μM. The inhibitory data is summarized in Tables 2 and 3.
Table 2.
Inhibitory activity of Ar2 analogs.
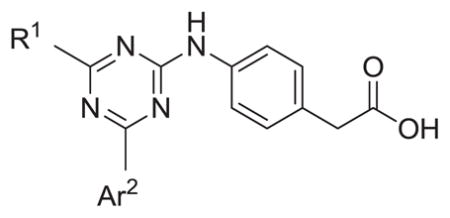
| |||||
|---|---|---|---|---|---|
| # | R1 | Ar2 | hPDE4B IC50 (nM) | hPDE4D IC50 (nM) | Ratio D/B IC50 |
| 7 |
|
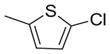
|
251 | 1489 | 5.9 |
| 8 |
|
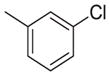
|
237 | 1181 | 5.0 |
| 9 |
|
|
3770 | 5611 | 1.5 |
| 10 | Me |
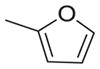
|
18755 | 22260 | 1.2 |
| 11 |
|
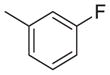
|
1437 | 2107 | 1.5 |
| 12 | Et |
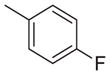
|
1460 | 727 | 0.5 |
| 13 | Et |
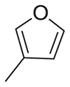
|
2777 | NT | |
Table 3.
Inhibitory activity of linker modified analogs.
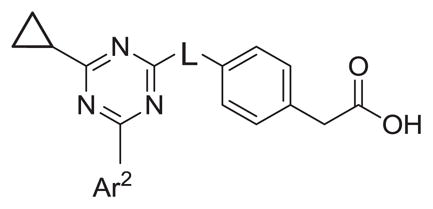
| |||||
|---|---|---|---|---|---|
| # | L | Ar2 | hPDE4B IC50 (nM) | hPDE4D IC50 (nM) | Ratio D/B IC50 |
| 8 | NH |
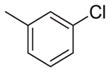
|
237 | 1181 | 5.0 |
| 14 | NHCH2 |
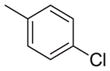
|
2282 | 859 | 0.38 |
| 15 | NHCH2 |
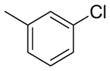
|
845 | 726 | 0.86 |
| 16 | O |
|
1862 | 2268 | 1.2 |
As shown in Tables 4 through 6, the Ar1 equal to CO2H was required for PDE4B selectivity. Analogs containing a tetrazole, which has similar acidity to a carboxylic acid, were potent but lost selectivity for PDE4B, while sulfonamides and imidazolidin-2-ones were inactive as was the methylester (Table 6). Substitution on the benzylic position of Ar1 results in decreased potency (compounds 33 and 34).
Table 4.
Inhibitory activity of Ar1 modified analogs.
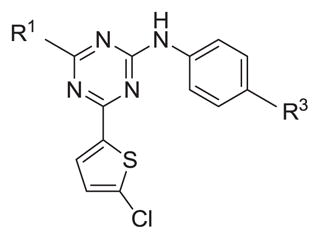
| |||||
|---|---|---|---|---|---|
| # | R1 | R3 | hPDE4B IC50 (nM) | hPDE4D IC50 (nM) | Ratio D/B IC50 |
| 3 | Me | -CH2CO2H | 1287 | NT | |
| 17 | Me | -CO2H | 4048 | NT | |
| 5 | Et | -CH2CO2H | 383 | 865 | 2.3 |
| 18 | Et | -CO2H | 261 | 251 | 1.0 |
| 19 | Et | -F | 1394 | 207 | 0.15 |
| 4 | i-Pr | -CH2CO2H | 1945 | 3580 | 1.8 |
| 20 | i-Pr | -CO2H | 220 | 219 | 1.0 |
| 21 | i-Pr | -F | 8815 | 886 | |
To understand the basis for PDE4B selectivity, we pursued co-crystallization studies with compound 8 and the catalytic domain of PDE4B containing CR3. We identified ligand-dependent crystallization conditions, obtained a complete diffraction dataset, and solved the resulting structure by molecular replacement. Analysis of the refined structure model shows that the central triazine ring stacks between Phe618 and Ile582 (P clamp) and makes a hydrogen bond to Gln615 (Q switch) in the active site demonstrating how the triazine core can function as a general PDE4 inhibitor (Fig 2).6 The cyclopropyl group has good shape complimentarily and fills the Q1 hydrophobic pocket in the active site explaining why substituents at this position are important for potency (Table 1). The amine in the Ar1 linker region is in position to make a hydrogen bond to a conserved water molecule explaining why modifications at this position also affect potency (Table 3). A similar water-bridge was observed in the A-33 PDE4B structure (PDB ID: 4MYQ).3 Also consistent with the A-33 structure, both Ar1 and Ar2 groups, which we show above modulate PDE4B vs. PDE4D selectivity in the triazine series (Tables 2, 4 and 5), reach out of the active site and engage the CR3 regulatory helix. The chlorophenyl Ar2 group on the triazine is in position to make hydrophobic interactions with CR3 Phe678 and Leu674, and the carboxylic acid group of Ar1 hydrogen bonds to multiple water molecules which engage CR3.
Figure 2.

X-ray co-crystal structure of 8 in PDE4B (PDB: 4NW7).
Table 5.
Inhibitory activity of Ar1 substitution.
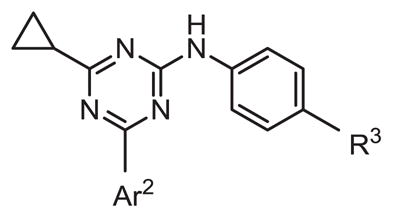
| |||||
|---|---|---|---|---|---|
| # | Ar2 | R3 | hPDE4B IC50 (nM) | hPDE4D IC50 (nM) | Ratio D/B IC50 |
| 8 |
|
-CH2CO2H | 237 | 1181 | 5.0 |
| 22 |
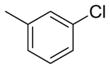
|
-CO2H | 46 | 20 | 0.4 |
| 9 |
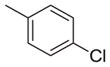
|
-CH2CO2H | 770 | 5611 | 7.3 |
| 23 |
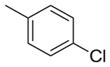
|
-CO2H | 135 | 268 | 2.0 |
| 24 |
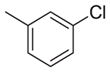
|
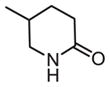
|
135 | 268 | 2.0 |
| 25 |
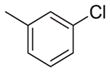
|
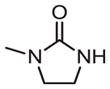
|
inactive | inactive | |
The new triazine co-crystal structure is similar to 4MYQ with CR3 clearly captured over the active site locking the enzyme in an inactive “closed” conformation.3 In both cases, the ligand provides good shape complementarity allowing the CR3 helix to be stabilized over the active site. The structure activity relationships (SAR) of a series of triazine PDE4 inhibitors in combination with co-crystal structure data revealed that the CR3 region can be exploited to generate PDE4B-selective inhibitors.
Supplementary Material
Figure 1.

Structure of lead compound 1 and A-33 (2).
Acknowledgments
This study was funded by the National Institute of Mental Health award 1R43MH091791 to M.E.G.
Footnotes
Supplementary data associated with this article can be found, in the online version doi…
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Houslay MD, Schafer P, Zhang KY. Drug Discov Today. 2005;10:1503. doi: 10.1016/S1359-6446(05)03622-6. [DOI] [PubMed] [Google Scholar]
- 2.Burgin AB, Magnusson OT, Singh J, Witte P, Staker BL, Bjornsson JM, Thorsteinsdottir M, Hrafnsdottir S, Hagen T, Kiselyov AS, Stewart LJ, Gurney ME. Nat Biotechnol. 2010;28:63. doi: 10.1038/nbt.1598. [DOI] [PubMed] [Google Scholar]
- 3.Fox D, 3rd, Burgin AB, Gurney ME. Cell Signal. 2014;26:657. doi: 10.1016/j.cellsig.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jin SL, Goya S, Nakae S, Wang D, Bruss M, Hou C, Umetsu D, Conti M. J Allergy Clin Immunol. 2010;126:1252. doi: 10.1016/j.jaci.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kumar N, Goldminz AM, Kim N, Gottlieb AB. BMC Med. 2013;11:96. doi: 10.1186/1741-7015-11-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Card GL, Blasdel L, England BP, Zhang C, Suzuki Y, Gillette S, Fong D, Ibrahim PN, Artis DR, Bollag G, Milburn MV, Kim SH, Schlessinger J, Zhang KY. Nat Biotechnol. 2005;23:201. doi: 10.1038/nbt1059. [DOI] [PubMed] [Google Scholar]
- 7.Naganuma K, Omura A, Maekawara N, Saitoh M, Ohkawa N, Kubota T, Nagumo H, Kodama T, Takemura M, Ohtsuka Y, Nakamura J, Tsujita R, Kawasaki K, Yokoi H, Kawanishi M. Bioorganic & Medicinal Chemistry Letters. 2009;19:3174. doi: 10.1016/j.bmcl.2009.04.121. [DOI] [PubMed] [Google Scholar]
- 8.Gurney ME, Burgin AB, Fox D. Manuscript in preparation. 2013 [Google Scholar]
- 9.Gewald R, Grunwald C, Egerland U. Bioorg Med Chem Lett. 2013;23:4308. doi: 10.1016/j.bmcl.2013.05.099. [DOI] [PubMed] [Google Scholar]
- 10.Harris RLN. Synthesis. 1980;1980:841. [Google Scholar]
- 11.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Adv Drug Deliv Rev. 2001;46:3. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.