

Aberrant accumulation of collagen IV defines diabetic nephropathy. It is shown here that high glucose increases fibronectin matrix assembly by activating integrin receptors on kidney cells. Collagen IV accumulation depends on this fibronectin matrix. Targeting fibronectin matrix may be a useful therapy to stem matrix accumulation in the diabetic kidney.
Abstract
The filtration unit of the kidney is the glomerulus, a capillary network supported by mesangial cells and extracellular matrix (ECM). Glomerular function is compromised in diabetic nephropathy (DN) by uncontrolled buildup of ECM, especially type IV collagen, which progressively occludes the capillaries. Increased levels of the ECM protein fibronectin (FN) are also present; however, its role in DN is unknown. Mesangial cells cultured under high glucose conditions provide a model system for studying the effect of elevated glucose on deposition of FN and collagen IV. Imaging of mesangial cell cultures and analysis of detergent-insoluble matrix show that, under high glucose conditions, mesangial cells assembled significantly more FN matrix, independent of FN protein levels. High glucose conditions induced protein kinase C–dependent β1 integrin activation, and FN assembly in normal glucose was increased by stimulation of integrin activity with Mn2+. Collagen IV incorporation into the matrix was also increased under high glucose conditions and colocalized with FN fibrils. An inhibitor of FN matrix assembly prevented collagen IV deposition, demonstrating dependence of collagen IV on FN matrix. We conclude that high glucose induces FN assembly, which contributes to collagen IV accumulation. Enhanced assembly of FN might facilitate dysregulated ECM accumulation in DN.
INTRODUCTION
Diabetic nephropathy (DN) is the leading cause of kidney failure in the world. This disease primarily affects the glomerulus, the functional filtration unit of the kidney, which consists of a specialized capillary array within an open three-dimensional capsule (Kwoh et al., 2006; Pozzi et al., 2009; Kolset et al., 2012). The capillary network is given structural support and maintained by the mesangium, composed of mesangial cells and the mesangial extracellular matrix (ECM). Between the capillary lumina and the surrounding space is a filtration barrier that consists of an inner fenestrated endothelial cell layer, an outer layer of visceral epithelial cells (podocytes), and an intervening glomerular basement membrane (GBM) made of ECM components. Diagnostic features of diabetic nephropathy are expansion of the mesangial ECM, thickening of the GBM, and gradual occlusion of glomerular arterioles.
Mesangial expansion is the most important prognostic feature of DN, since functional decline occurs as the urinary space and the glomerular capillaries are gradually eclipsed by aberrant ECM accumulation (Kolset et al., 2012). The pathogenesis of DN is not well understood, but previous studies implicated a host of factors that mediate high glucose–induced ECM production. High glucose culture conditions induce expression of ECM proteins by mesangial cells in vitro (Ayo et al., 1990; Köppel et al., 2011; Huang et al., 2012; Taniguchi et al., 2013). Increased reactive oxygen species (ROS) generation is a hallmark of the diabetic milieu (Brownlee, 2001), and ROS-induced production of profibrotic growth factors, such as transforming growth factor-β (TGFβ), is well documented (Pozzi et al., 2009; Abboud, 2012). High glucose–induced activation of protein kinase C (PKC) has pleiotropic effects in mesangial cells, including a role in increased ECM production (Brownlee, 2001; Abboud, 2012). Activation of these pathways leads to excessive accumulation of ECM proteins in vivo, and this buildup represents a final common pathway in mesangial expansion.
Under physiologic conditions, the mesangial ECM consists of collagens (including types I and IV), laminin, fibronectin (FN), and proteoglycans (Haraldsson et al., 2008; Kolset et al., 2012). The predominant matrix protein found in healthy and diabetic glomeruli is type IV collagen (Zent et al., 2006). Significant increases in FN have been detected in several glomerulopathies, including DN (Dixon et al., 1980; Sumi et al., 2011), and FN is one of the first ECM proteins to increase in the early stages of DN (Falk et al., 1983). However, the role of FN in the progression of DN is unknown. Although there is temporal overlap in the buildup of FN and type IV collagen, no molecular connection between these two proteins has been made in DN.
Much work has been done to understand the roles of collagen-binding integrins in DN (Pozzi and Zent, 2013). Integrin α1β1, the primary type IV collagen receptor, negatively regulates collagen synthesis (Gardner et al., 1999), and integrin α1–null diabetic mice exhibit exacerbated mesangial ECM accumulation (Zent et al., 2006). In contrast, deletion or inhibition of integrin α2β1 limits glomerular damage after renal injury (Borza et al., 2012). α5β1 integrin, the receptor responsible for FN matrix assembly, is expressed by mesangial cells and is present in the glomerulus (Kerjaschki et al., 1989; Kagami et al., 1993). α5β1 expression is up-regulated in mesangial cells in response to TGFβ (Weston et al., 2003), and high glucose has been shown to up-regulate α5β1 expression in podocytes, as well as FN expression in mesangial cells (Kitsiou et al., 2003; Yang et al., 2013). FN matrix assembly is initiated when FN dimers bind to α5β1 integrins, which promotes FN–FN interactions to form fibrils (Singh et al., 2010; Leiss et al., 2008). Other ECM proteins, including type I and III collagens, depend on a fibrillar FN matrix in order to be incorporated into the ECM in vitro (Sottile and Hocking, 2002; Velling et al., 2002; Li et al., 2003; Kadler et al., 2008; Singh et al., 2010). In addition, cells interacting with a fibrillar FN matrix are stimulated to assemble additional matrix (Mao and Schwarzbauer, 2005), a potentially important phenomenon in a chronic and progressive disease such as DN. A stimulatory effect of high glucose on α5β1 integrin–mediated assembly of FN matrix could feed the accumulation of additional FN and might also play a role in the deposition of type IV collagen by mesangial cells in DN. However, the effect of high glucose on FN matrix assembly and the implications for other matrix proteins remain undetermined.
In this study, we investigate the effect of high glucose on mesangial cell–mediated FN matrix assembly and show that high glucose–induced collagen IV deposition is dependent on FN assembly. We propose a role for FN assembly as an early and essential mediator of high glucose–induced ECM accumulation. Our findings suggest that FN matrix may provide a novel target for therapies that control excess ECM deposition and stem the progression of DN.
RESULTS
High glucose conditions stimulate mesangial cell–mediated FN matrix assembly
To investigate the effects of high glucose on FN matrix assembly, we used a conditionally immortalized mouse mesangial cell line (Chen et al., 2004). Matrix assembled by mesangial cells grown under conditions of normal (5 mM) or high (30 mM) glucose was analyzed both visually and biochemically in order to compare levels of fibrillar FN matrix (Figure 1). After 4 d of growth under normal or high glucose conditions, mesangial cells and the endogenously produced matrix were fixed and FN was visualized by indirect immunofluorescence. Images of representative fields reveal that mesangial cells cultured in high glucose assembled more FN matrix than cells maintained in normal glucose conditions (Figure 1A), as indicated by the higher average total fluorescence intensity for high glucose samples. Peak frequencies across randomly selected linear regions show that the density of FN fibrils is higher in high glucose cultures than with normal glucose. The increased amplitudes of lines plotted for the high glucose condition reflect higher fluorescence intensity.
FIGURE 1:
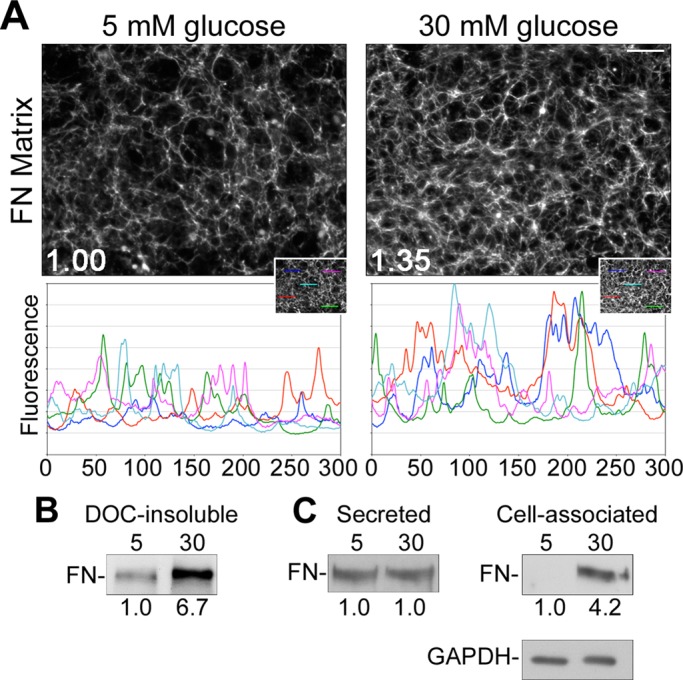
High glucose induces mesangial cell–mediated assembly of FN. Mesangial cells were grown in media containing 5 mM glucose plus 25 mM mannitol (normal glucose) or 30 mM glucose (high glucose). Cells were fixed and stained with R457 anti-FN antiserum after 4 d (A) or lysed in DOC (B) or SDS (C) buffer after 3 d, and FN was isolated from conditioned medium using gelatin–Sepharose beads (C). (A) Total mean fluorescence intensities were measured for 10 randomly selected fields/condition and normalized to DAPI staining. Values were used to calculate the average FN staining intensity, which was 1.35-fold (n = 3, p < 0.05) greater in high than in normal glucose conditions. Line plots display fluorescence intensity per pixel across five randomly selected 300-pixel linear regions (inset). Images are representative of three independent experiments; scale bar, 50 μm. (B) The DOC-insoluble fraction, (C) secreted FN from the conditioned medium, and cell-associated FN from whole-cell SDS lysates were separated by SDS–PAGE and immunoblotted with R457 anti-FN antiserum. Relative densitometry values (below the lanes) are the mean of three experiments normalized to GAPDH from the DOC-soluble fraction. Blots are representative of three independent experiments.
The maturity of a FN matrix can be assessed by the content of deoxycholate (DOC) detergent–insoluble FN protein (McKeown-Longo and Mosher, 1983; Sechler et al., 1996). High glucose conditions stimulated a >6-fold increase in DOC-insoluble FN matrix as compared with normal glucose conditions (Figure 1B). The average increase in the DOC-soluble fraction was more modest at 1.4-fold (1.4 ± 0.05, n = 2). To determine the total amount of FN protein produced by these cells, we examined relative levels of FN secreted into the culture medium and total cell-associated FN in SDS lysates (matrix and intracellular combined) by immunoblot. Amounts of secreted FN were roughly equivalent between the normal and high glucose conditions (Figure 1C). However, total cell-associated FN differed by at least fourfold (Figure 1C). Given the difference in DOC-insoluble FN matrix, it seems likely that the difference in total cell-associated FN results from an enhanced ability to assemble FN matrix under high glucose conditions. Because at least fivefold more FN is found in the secreted than the cell-associated fraction, relative total combined FN protein levels differed by no more than 1.2-fold between high and normal glucose conditions.
If observed differences in FN matrix assembly are due to high glucose–induced changes in FN protein levels, then the addition of exogenous FN to cells cultured in normal glucose should increase the assembly of FN matrix. On the other hand, if differences in matrix assembly persist in the presence of exogenous FN, that would strengthen the case for a distinct effect of high glucose on the ability of mesangial cells to assemble matrix.
To test the effects of FN levels on assembly, we grew mesangial cells under normal or high glucose conditions for 2 d in the presence of exogenous rat FN at a concentration of 10 μg/ml. Cells grown under high glucose conditions assembled exogenous FN into matrix at higher levels than cells grown in normal glucose (Figure 2). This difference can be seen by indirect immunofluorescence using a species-specific monoclonal antibody that recognizes rat, but not mouse, FN (Figure 2A). In line with observations for endogenous FN, increases in average total and regional fluorescence intensities and in fluorescence peak frequencies were measured. Quantification of rat FN in DOC-insoluble matrix confirmed that high glucose had a pronounced effect on assembly, with >10-fold increase in DOC-insoluble rat FN compared with normal glucose conditions (Figure 2B). These data show that increased FN levels do not induce higher levels of assembly by cells cultured in normal glucose.
FIGURE 2:
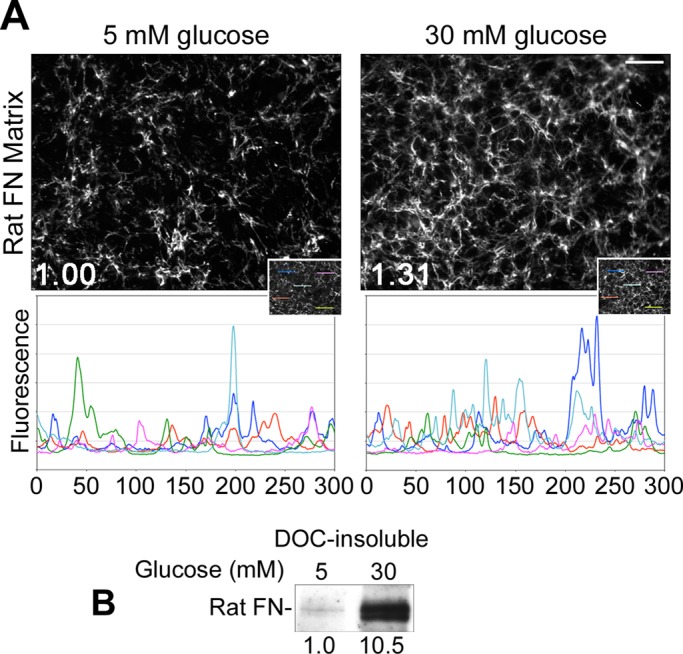
Exogenous FN does not induce assembly in normal glucose. Mesangial cells were grown for 2 d in normal or high glucose media containing 10 μg/ml rat plasma FN. (A) Cells were fixed and stained with IC3 anti-rat FN monoclonal antibody. Total mean fluorescence staining for rat FN measured for 10 randomly selected fields/condition was increased 1.31-fold (n = 3, p < 0.05) in high as compared with normal glucose. Line plots display fluorescence intensity per pixel across five randomly selected 300-pixel linear regions (inset). Images are representative of three independent experiments; scale bar, 50 μm. (B) Cells were lysed in DOC buffer and immunoblotted with IC3 monoclonal antibody. Relative densitometry values (below the lanes) were normalized to GAPDH from the DOC-soluble fraction. Additional replicates had no detectable DOC-insoluble FN in the 5 mM condition. Blot is representative of three independent experiments.
Another mechanism by which cells modulate FN matrix assembly is through levels of the FN receptor, α5β1 integrin, expressed at the cell surface. As such, we measured α5β1 integrin expression levels using flow cytometry. Mesangial cells grown under normal or high glucose conditions for 3 d and then labeled with an anti–α5β1 integrin antibody had equivalent median fluorescence intensities of 3.6-fold over background (Figure 3A). Therefore, in contrast to studies on podocytes (Kitsiou et al., 2003), high glucose conditions had no discernible effect on mesangial cell surface levels of α5β1 integrin, and high glucose–induced FN matrix assembly cannot be explained by changes in the number of cell surface integrins.
FIGURE 3:
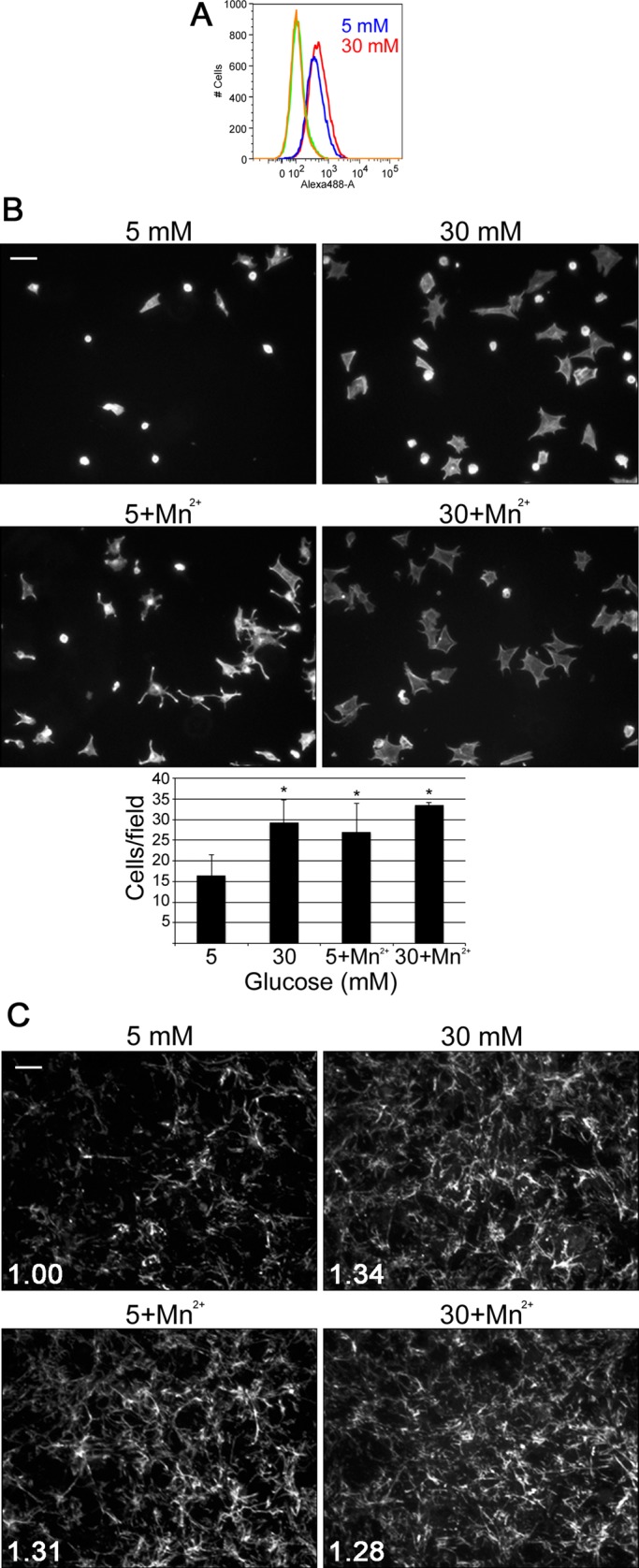
Mn2+ stimulates assembly of FN matrix in normal glucose. Mesangial cells were grown in normal or high glucose media and then labeled with an anti–α5β1 integrin antibody for analysis by flow cytometry (A), replated on FN-coated coverslips (B), or fixed and stained with IC3 anti-rat FN monoclonal antibody (C). (A) The graph is representative of two independent experiments. (B) Cells were incubated for 1 h at 37°C with or without 1.0 mM MnCl2 before unattached cells were washed away. Attached cells were fixed and stained with rhodamine–phalloidin, and 15 fields/condition were counted. Images are representative; scale bar, 100 μm. Bar graph shows the mean of two to four independent experiments ± 1 SD; *p < 0.05 compared with 5 mM glucose. (C) Cells were treated or not, as indicated, with 1.0 mM MnCl2 for the final 4 h of culture and then fixed and stained with IC3 anti-rat FN monoclonal antibody. Images are representative; scale bar, 50 μm. Total mean fluorescence was measured for 10 fields/condition. Values were used to calculate the average FN staining intensity, which was 1.34-fold greater in high glucose, 1.31-fold greater in normal glucose with Mn2+, and 1.28-fold greater in high glucose with Mn2+, all compared with normal glucose conditions (n = 2, p < 0.05).
Integrin stimulation increases mesangial cell adhesion and matrix assembly
The activity of α5β1 integrin has a central role in assembly of FN fibrils and in stabilizing cell interactions with assembled FN matrix (Wierzbicka-Patynowski et al., 2007). We assessed the effect of high glucose on the ability of mesangial cells to bind to FN—a readout for integrin function—using a cell adhesion assay. Mesangial cells grown under normal or high glucose conditions for 3 d were plated in serum-free medium on a FN-coated surface. After 60 min, we observed that about twofold more high glucose–treated cells attached than with the normal glucose control (Figure 3B). High glucose–cultured cells also appeared more spread. To determine whether integrin activation has a role in the differences in adhesion and spreading, we tested cell adhesion in the presence of Mn2+, a known activator of integrins (Gailit and Ruoslahti, 1988). When mesangial cells were allowed to attach to FN in the presence of 1.0 mM MnCl2, normal glucose–conditioned cells attached in equivalent numbers to cells exposed to high glucose (Figure 3B). Therefore activation of mesangial cell integrins with Mn2+ is sufficient to induce robust cell adhesion, independent of whether the cells are cultured in low or high glucose.
To determine whether integrin activation by Mn2+ also affects FN matrix assembly, we grew mesangial cells for 2 d under normal or high glucose conditions in the presence of 10 μg/ml rat FN and added 1.0 mM MnCl2 for the final 4 h of culture (Sechler et al., 1997). FN matrix was then visualized by immunofluorescence, and average total fluorescence intensities were measured. Mesangial cells grown under normal glucose conditions in the presence of Mn2+ assembled FN matrix at levels equivalent to cells grown under high glucose conditions without Mn2+ (Figure 3C). These data show that Mn2+ stimulation of integrin activity under normal glucose conditions is sufficient to promote FN matrix assembly by mesangial cells. Moreover, similar amounts of matrix were obtained whether cells were stimulated with high glucose, Mn2+, or both, suggesting that either treatment is sufficient to achieve maximal assembly under these conditions.
High glucose activates integrins via PKC
Two assays were used to assess the effect of high glucose on integrin activation status: an antibody capture assay and a ligand-binding assay. In the first assay, mesangial cells grown for 3 d under normal or high glucose conditions were tested for binding to surfaces coated with 9EG7, an anti–β1 integrin antibody that recognizes the β1 activated conformation (Bazzoni et al., 1995; Wierzbicka-Patynowski et al., 2007). Cells cultured in high glucose bound significantly more to 9EG7 than did cells grown in normal glucose (Figure 4A). For the second assay, integrin function was assessed by measuring the amount of soluble FN that bound to mesangial cells in suspension (O'Toole et al., 1994). Mesangial cells grown under high glucose conditions bound greater than fivefold more FN than the normal glucose control cells (Figure 4B). Glomerular endothelial cells exposed to high glucose conditions exhibited a similar behavior of increased binding to a 9EG7-coated plate and increased binding of FN in suspension, suggesting that the effect of high glucose on integrins is not unique to mesangial cells (Supplemental Figure S1). PKC activity has been shown to play a role in integrin activation in platelets (Cifuni et al., 2008) and is increased in mesangial cells grown under high glucose (Ayo et al., 1991; Cifuni et al., 2008). Pretreatment of cells with calphostin C, an inhibitor of PKC, abolished the stimulatory effect of high glucose on β1 integrin measured using the 9EG7-binding assay (Figure 4C). These data suggest that high glucose conditions activate β1 integrin in a PKC-dependent manner and implicate changes in integrin activation as a mechanism for high glucose–induced FN matrix assembly.
FIGURE 4:
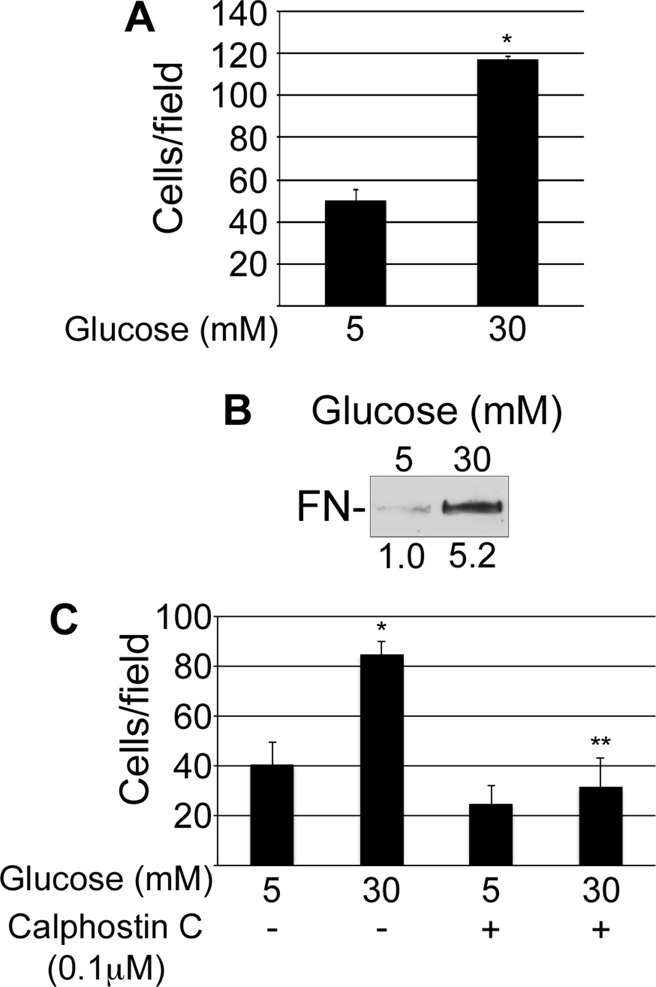
High glucose induces activation of β1 integrins. Mesangial cells were grown for 3 d in normal or high glucose (A, B) or for 2 d in normal or high glucose with dimethyl sulfoxide or 0.1 μM calphostin C (C). (A, C) Cells were then plated on surfaces coated with 9EG7 anti-β1 antibody and incubated for 30 min at 37°C before unattached cells were washed away. Attached cells were fixed, and three wells/condition were counted. Bar graph shows the mean of two independent experiments ± 1 SD; *p < 0.05 compared with 5 mM glucose; **p < 0.05 compared with 30 mM glucose. (B) Cells in suspension were incubated with rat FN for 30 min at room temperature, pelleted through a sucrose cushion, lysed in SDS sample buffer, and immunoblotted with IC3 anti-rat FN monoclonal antibody. Relative densitometry values (below the lanes) are the mean of three experiments.
Type IV collagen binds to FN
Accumulation of FN matrix due to high glucose could serve as an initiating event in DN that affects the accumulation of other ECM proteins found in the glomerulus. Because type IV collagen is the most prominent matrix protein in lesions of glomerulosclerosis, it was relevant to characterize the interaction between FN and collagen IV. In a solid-phase binding assay, purified rat plasma FN was incubated with purified type IV collagen, and bound protein was detected by an enzyme-linked immunoassay. Type IV collagen bound to FN or to a recombinant N-terminal 70-kDa fragment of FN in a concentration-dependent manner (Figure 5). The 70-kDa fragment contains a fibrin-binding/matrix assembly domain and a collagen/gelatin-binding domain (Singh et al., 2010). It is well established that FN's collagen/gelatin domain binds to type I collagen (Hynes, 1990). To address whether this domain also mediates type IV collagen binding to FN, we performed a binding assay in the presence of gelatin as a potential competitive inhibitor. The interaction between collagen IV and the 70-kDa fragment of FN was significantly inhibited in the presence of gelatin (Figure 5). A similar result was obtained with type IV collagen and full-length FN in the presence and absence of gelatin (unpublished data). Therefore type IV collagen binds to FN at the collagen/gelatin-binding domain.
FIGURE 5:
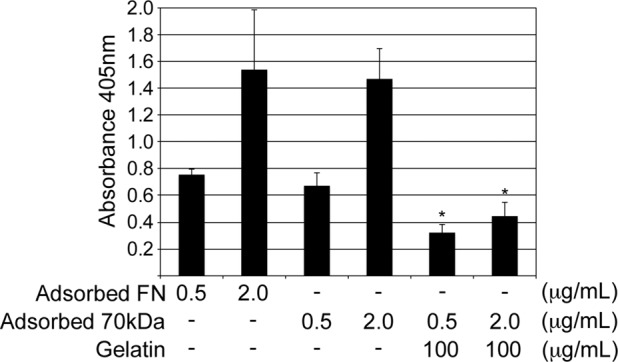
Type IV collagen binds to the gelatin-binding domain of FN. Plates were coated in triplicate with 0.5 and 2.0 μg/ml purified rat plasma FN or a recombinant 70-kDa N-terminal fragment of FN. Purified mouse collagen IV at 30 μg/ml was then bound in the presence or absence of 100 μg/ml gelatin and ELISA performed to detect bound protein. Mean absorbance of two to four independent experiments ±1 SD; *p < 0.05 compared with no gelatin.
Type IV collagen deposition by mesangial cells depends on FN matrix
Early-stage lesions of glomerulosclerosis stain prominently for FN (Dixon et al., 1980; Falk et al., 1983). Because type IV collagen predominates as DN progresses (Zent et al., 2006), we sought to test whether FN matrix serves as a template for subsequent collagen IV deposition. Using a mouse model of DN, we found that FN and collagen IV colocalize to the mesangium in the glomeruli of diabetic mice (Supplemental Figure S2). Mesangial cells grown under normal or high glucose conditions and stained for type IV collagen showed higher levels of matrix collagen in the high glucose condition (Figure 6A). Mesangial cell–assembled matrices costained for both FN and type IV collagen display coalignment of type IV collagen (red) with FN fibrils (green) (Figure 6B). Coalignment is particularly evident in higher-magnification images, in which all collagen staining is closely associated with FN fibrils. These observations suggest that the direct FN–collagen IV interaction observed in vitro might be involved in mesangial cell–mediated ECM assembly.
FIGURE 6:
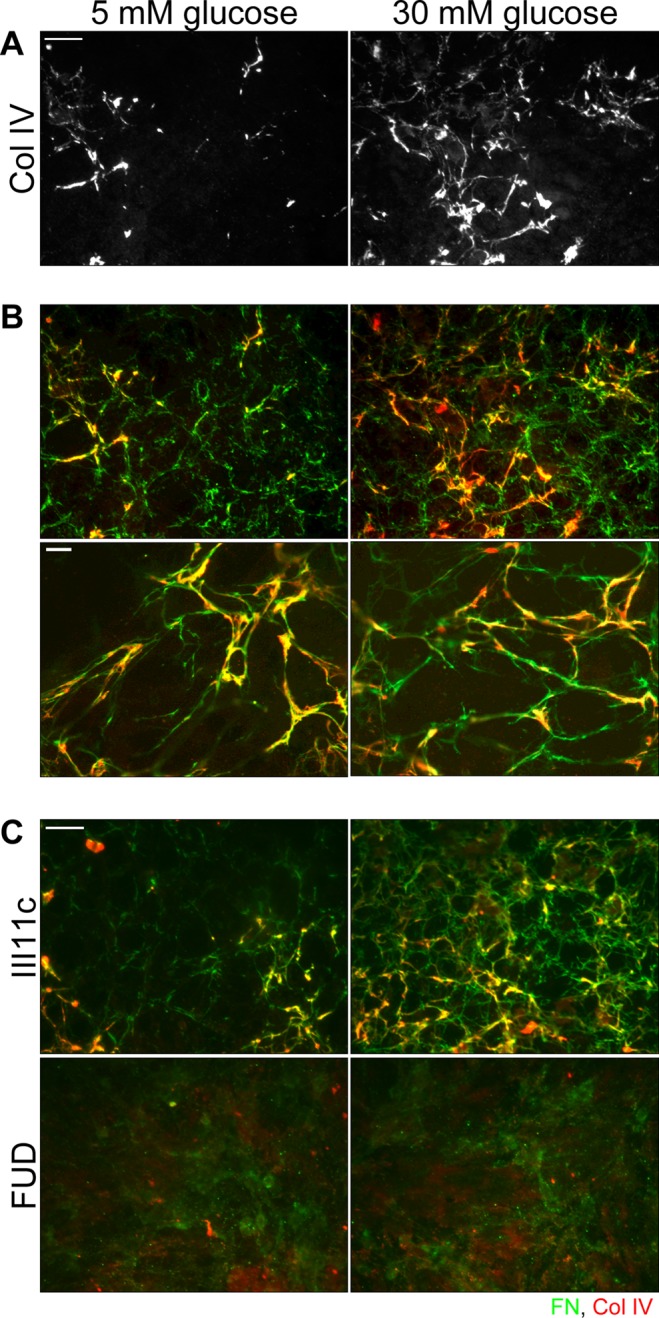
FN and type IV collagen colocalize in mesangial cell–assembled matrix, and inhibition of FN matrix assembly ablates collagen IV matrix. Mesangial cells were grown for 3 d on collagen IV–coated coverslips in normal or high glucose media containing 10 μg/ml rat plasma FN (A–C). We added 2.4 μg/ml III-11C control peptide or FUD, a known inhibitor of FN matrix assembly, with the rat FN (C). Cells were fixed and costained with IC3 anti-rat FN monoclonal antibody (green) and anti-collagen IV antibody (red). (A) Collagen IV staining is shown alone. Scale bar, 50 μm. (B) Yellow staining indicates colocalization of FN and type IV collagen. Images are representative of three independent experiments. Scale bar, 10 μm. (C) Images are representative of two independent experiments; scale bar, 50 μm.
If FN matrix serves as a template for collagen IV deposition, then inhibiting FN matrix assembly should affect the organization of collagen IV. The functional upstream domain (FUD) of the F1 surface protein of Streptococcus pyogenes is a specific and potent inhibitor of FN matrix assembly (Ensenberger et al., 2001). It binds to the matrix assembly domain located at the N-terminus of the FN subunit, thereby preventing the homotypic interactions crucial for fibril assembly (Maurer et al., 2010). The III-11C peptide is commonly used as a control peptide in FUD experiments (Shi et al., 2010). In the presence of III-11C, mesangial cells under both normal and high glucose conditions assembled exogenous FN into fibrils, and type IV collagen organized by these cells colocalizes with those FN fibrils (Figure 6C). In the presence of FUD, not only was FN matrix absent, but also type IV collagen matrix was ablated. Diffuse staining for FN might represent FN that is bound to integrins but prevented from being organized into fibrils by FUD. These results support our hypothesis that FN matrix serves as a template for type IV collagen deposition by mesangial cells. These data, combined with the observation that FN accumulation precedes the buildup of type IV collagen in glomerulosclerosis, point to FN matrix assembly as an important and early event in ECM accumulation during DN.
DISCUSSION
In diabetic nephropathy, dysregulated buildup of ECM proteins results in functional decline of the glomerulus. Our analyses of mesangial cell–mediated matrix assembly show that FN matrix assembly is significantly increased in the presence of elevated glucose levels, suggesting that FN is a potentially important modulator of disease progression. The enhancement of matrix assembly is not due to increases in total FN or cell surface α5β1 integrin, but is recapitulated by stimulation of integrin activity with Mn2+ under normal glucose conditions. Furthermore, high glucose conditions are sufficient to induce β1 integrin activation in a PKC-dependent manner. These data show that integrin activation, downstream of high glucose, increases mesangial cell–mediated matrix assembly. Increased FN matrix assembly correlates with the level of deposition of type IV collagen, the primary constituent of fibrotic ECM in the glomerulus, and both FN and collagen IV are ablated by an inhibitor of FN matrix assembly. These findings show an important role for FN assembly in type IV collagen accumulation and are noteworthy in the context of DN because the FN–collagen IV interaction may prove an appealing therapeutic target to stem ECM protein accumulation in the diabetic glomerulus.
A well-established mechanism for inside-out integrin activation depends on talin binding to the cytoplasmic tail of the β subunit (Tadokoro et al., 2003). Stimulation of talin binding has been shown, in other systems, to be downstream of PKC (Shattil et al., 2010). PKC and a guanine nucleotide exchange factor (GEF), CalDAG-GEFI, regulate Rap1b, a Ras-family small GTPase (Cifuni et al., 2008; Kim et al., 2011). Activated Rap1 binds to Rap1-GTP–interacting adaptor molecule (RIAM), which directly interacts with talin to facilitate its binding to the β integrin cytoplasmic tail (Lee et al., 2009). This pathway has been identified and characterized in Chinese hamster ovary cells expressing activation-dependent αIIbβ3 integrin, but may also be at play in our system. For example, PKC activation by high glucose as a consequence of intracellular accumulation of diacyl glycerol (DAG) is well documented (Brownlee, 2001) and has been shown to result in elevated Rap1b expression and activity in mesangial cells (Lin et al., 2002). CalDAG-GEFIII is expressed specifically in mesangial cells and has been shown to activate Rap1 in both in vitro and in vivo settings (Yamashita et al., 2000). Thus the initial steps in the pathway are present in mesangial cells and may culminate in the Rap1-RIAM complex promoting talin interaction with the β1 integrin cytoplasmic tail. Although this model needs to be tested experimentally, it represents a plausible mechanism for high glucose–induced integrin activation and the consequent increase in mesangial cell–mediated FN matrix assembly.
Before this study, accumulation of FN and the buildup of type IV collagen in the diabetic mesangium have been viewed as concurrent yet independent processes. It is clear from our data that high glucose stimulates integrin-mediated FN assembly. The relevance of this phenomenon to disease progression is heightened by the demonstration of a direct interaction between FN and collagen IV matrix. Previous studies noted soluble FN binding to denatured and native collagen IV (Engvall et al., 1978; Aumailley and Timpl, 1986). Our data demonstrating the dependence of collagen IV matrix on FN assembly suggest a novel role for FN in facilitating the deposition and perhaps also the accumulation of collagen IV in DN. It is possible that FN matrix also serves a more general role by facilitating accumulation of collagen IV as well as other ECM proteins with which it directly interacts, such as perlecan (Hopf et al., 2001). Having localized the relevant binding site for collagen IV on the FN molecule to the collagen/gelatin-binding domain, it will be of interest to determine the impact of inhibiting this interaction on the deposition of type IV collagen matrix. Targeted inhibition of interactions with FN's collagen/gelatin-binding domain prevents type I collagen matrix accumulation (Sottile et al., 2007). Furthermore, inhibition of FN matrix assembly in vivo using FUD has proven to reduce collagen I accumulation in a mouse model of vascular injury (Chiang et al., 2009). We propose that inhibitors of FN assembly, like FUD, might also have efficacy in mouse models of DN.
The role of FN in promoting deposition of other ECM proteins suggests that increases in FN matrix might initiate a positive feedback loop in which high glucose–induced FN matrix assembly begets yet more FN deposition. This idea is supported by the fact that cell interactions with a three-dimensional fibrillar FN matrix stimulate further matrix assembly in vitro (Mao and Schwarzbauer, 2005). Such an effect could be compounded in DN by the established negative regulatory effect of diabetic conditions on matrix turnover, including reduced protease (e.g., MMP-2, MMP-7) activity and increased expression of endogenous protease inhibitors (e.g., TIMP1, PAI-1; Reckelhoff et al., 1993; Hagiwara et al., 2003; Mason and Wahab, 2003; Park et al., 2010). These changes disrupt the balance of matrix assembly and degradation present under physiologic conditions and result in mesangial ECM expansion.
The fibronectin–collagen IV interaction may also have broader implications concerning other forms of glomerulosclerosis, which develops as a consequence of nearly all forms of chronic glomerular disease (Baylis and Corman, 1998; Zhou et al., 2008). Although elevated blood glucose might not play a role in the pathogenesis of these nondiabetic entities, mesangial cell–mediated FN assembly as a general response to injury in chronic disease may be important in collagen IV accumulation. In addition, the effect of high glucose on FN–integrin interactions may be relevant to other forms of diabetes-induced end-organ damage, such as diabetic retinopathy. Retinal endothelial cells exposed to high glucose exhibit a similar improved ability to engage FN to that described here in mesangial cells (Roth et al., 1993). Another study of retinal endothelial cells under high glucose conditions demonstrated the importance of cellular FN levels in modulating production of vascular endothelial growth factor, an important promoter of the neovascularization that defines diabetic retinopathy (Chen et al., 2012 ). In these diseases and others, high glucose–induced, integrin-mediated FN assembly and FN–collagen IV binding may contribute to pathogenesis.
Mesangial cell–mediated ECM deposition represents a final common pathway in DN and all chronic glomerular disease; as the process most directly linked to functional decline, it is a logical point for intervention. Because FN serves as a template for collagen IV, the predominant ECM protein in lesions of DN, it is an optimal target for the development of novel inhibitors such as small molecules that can block FN–collagen IV binding. In addition to serving as useful tools in understanding the importance of this interaction in cell-mediated ECM assembly in vitro and in DN in vivo, such an inhibitor may hold promise as a novel pharmacotherapy for DN.
MATERIALS AND METHODS
Fibronectin, peptides, and antibodies
Fibronectin was purified from fresh-frozen rat plasma by gelatin–Sepharose affinity chromatography (Wilson and Schwarzbauer, 1992). The recombinant 70-kDa fragment of FN was generated using the baculovirus insect cell expression system (Aguirre et al., 1994). FUD and III-11C, expressed in Escherichia coli, were purified as described previously (Hunt et al., 2012). The anti-FN antibodies used were polyclonal antiserum R457 raised against the N-terminal 70-kDa fragment of rat FN (Aguirre et al., 1994) and rat-specific anti-FN monoclonal antibody IC3 from ascites fluid (Sechler et al., 1996). Other antibodies used were against glyceraldehyde-3-phosphate dehydrogenase (GAPDH; 14C10; Cell Signaling Technology, Danvers, MA), collagen IV (AB756P; Millipore, Billerica, MA), β1 integrin (9EG7; BD Biosciences, San Jose, CA), and α5β1 integrin (BMB5; Millipore). Secondary antibodies used were horseradish peroxidase–conjugated (HRP) goat anti-mouse immunoglobulin G (IgG), HRP–goat anti-rabbit IgG (Pierce, Rockford, IL), and Alexa Fluor 488 and 568–conjugated goat anti-mouse, goat anti-rabbit, and goat anti-rat IgG (Life Technologies, Carlsbad, CA).
Cell culture
Conditionally immortalized mesangial and glomerular endothelial cells were isolated from an Immortomouse as previously described (Chen et al., 2007). These cells express a temperature-sensitive SV40 large T antigen when grown in the presence of interferon-γ at 33°C. Cells were maintained at 33°C in the presence of 100 IU/ml interferon-γ (Sigma-Aldrich, St. Louis, MO) and 10% fetal bovine serum (Hyclone, Logan, UT) in DMEM with 1 mM sodium pyruvate, 100 U/ml penicillin, 100 μg/ml streptomycin, and 0.25 μg/ml Fungizone (all Life Technologies). Cells were grown at 37°C in the absence of interferon-γ for at least 4 d and one passage before use in experiments— conditions previously established to result in a phenotype similar to freshly isolated primary mesangial cells (Chen et al., 2007). Cells were propagated in the foregoing medium containing 20 mM glucose and 10 mM mannitol. Cells were seeded for experiments in this same medium, allowed 4 h to adhere, and then serum-starved overnight in media containing 5 mM glucose and 25 mM mannitol. Finally, after 20 h of serum starvation, the medium was changed to DMEM with 5 mM glucose and 25 mM mannitol or 30 mM glucose, plus 10% fetal bovine serum, and maintained for 2–4 d, as indicated. Cells were cultured in fetal bovine serum depleted of plasma FN by gelatin–Sepharose chromatography for experiments to measure total FN levels.
Cell lysis and immunoblotting
Mesangial cells were seeded and serum starved as described and then grown for 2 or 3 d in the presence or absence of rat plasma FN at 10 μg/ml as indicated. Cells were washed with cold phosphate-buffered saline (PBS), and matrix was dissolved in DOC lysis buffer (2% DOC, 20 mM Tris-HCl, pH 8.8, 2 mM EDTA, 2 mM phenylmethylsulfonyl fluoride [PMSF]) with protease inhibitor cocktail (Roche Applied Science, Penzberg, Germany; Sechler et al., 1996). After centrifugation at 14,000 rpm for 10 min at 4°C, the supernatant was separated and the pellet washed once with DOC lysis buffer to ensure removal of all DOC-soluble material. The DOC-insoluble pellet was solubilized in SDS lysis buffer (2% SDS, 20 mM Tris-HCl, pH 8.8, 2 mM EDTA, 2 mM PMSF) with protease inhibitor. Total protein concentrations were measured in the DOC-soluble fraction using a bicinchoninic acid protein assay (Pierce), and equal amounts of DOC-soluble protein or proportional volumes of DOC-insoluble samples were separated by SDS–PAGE on 6% polyacrylamide gel alongside Precision Plus Protein Standard (Bio-Rad, Hercules, CA). Protein was then transferred to nitrocellulose membrane. Antibody incubations were performed in buffer A (25 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.1% Tween-20). Total FN was detected with R457 anti-FN antiserum diluted 1:500, and rat FN was detected with IC3 rat-specific anti-FN ascites fluid diluted 1:10,000. DOC-soluble samples were probed with anti-GAPDH antibody to confirm equal loading. Secondary antibodies were diluted 1:10,000 in buffer A. Blots were developed using SuperSignal West Pico ECL reagents (Pierce). Densitometry was performed on scanned films using ImageJ (National Institutes of Health, Bethesda, MD), and exposures yielding signals within the linear range were quantified. FN levels were normalized to GAPDH.
To measure total FN levels, FN secreted into the cell conditioned medium and cell-associated FN in whole-cell lysates were quantified. Secreted FN was isolated from conditioned medium using gelatin–Sepharose beads. In parallel, cells were lysed in SDS lysis buffer. Samples were reduced and separated by SDS–PAGE on 6% polyacrylamide gel and immunoblotted, and band intensities were quantified as described.
Immunofluorescence and microscopy
Mesangial cells were seeded on glass coverslips and serum starved as indicated. For type IV collagen staining, coverslips were coated with 10 μg/ml purified mouse type IV collagen protein (BD Biosciences) by incubating for 1 h at 37°C. Cells were then grown for 2 or 3 d in the presence or absence of 10 μg/ml rat plasma FN and fixed for 15 min at room temperature in 3.7% (wt/vol) Formalin (Sigma-Aldrich) in PBS supplemented with 0.5 mM MgCl2 (PBS). For R457 staining, antibody was diluted 1:100 in 2% ovalbumin in PBS. IC3 and anti–collagen IV antibodies were diluted 1:1000 and 1:100, respectively, in 5% goat serum in PBS. Secondary antibodies were diluted 1:600 in 2% ovalbumin or 5% goat serum in PBS along with 4′,6-diamidino-2-phenylindole (DAPI) at 1:1000. Coverslips were then mounted using ProLong Gold antifade reagent (Life Technologies). All images were captured using a Nikon Eclipse Ti microscope equipped with a Hamamatsu C10600 ORCA-R2 digital camera. Total mean fluorescence measurements were performed using ImageJ on 10 randomly selected fields per condition; representative fields are shown. Fluorescence fold-change and SD were calculated for the mean of three independent experiments. Five randomly selected 300-pixel linear regions of interest were selected on representative images using ImageJ, and the fluorescence intensities across those regions were plotted to generate line graphs.
Flow cytometry
After 3 d of growth in 5 mM glucose with 25 mM mannitol or 30 mM glucose medium, cells were trypsinized, neutralized in 0.5 mg/ml soybean trypsin inhibitor (Sigma-Aldrich), counted, and then resuspended in cold PBS plus 1% bovine serum albumin (BSA). Anti-α5β1 antibody was added at 1:200, and cells were then pelleted and washed before staining with Alexa Fluor 488–conjugated goat anti-rat IgG at 1:500. One sample was stained with secondary antibody alone to serve as a negative control. Cells were subsequently stained with propidium iodide (PI), and runs were gated on live (PI-negative) cells before measuring Alexa Fluor 488 signal. Flow cytometric analysis was performed using a LSRII flow cytometer (BD Biosciences). Excitation of fluorochromes was at 488 nm, and emission was collected through a 525/50 bandpass filter for Alexa Fluor 488 and a 610/20 bandpass filter for PI. The median fluorescence intensity was determined using FACSDiva software (BD Biosciences) and overlay figures generated using FlowJo, version 9.6.4 (TreeStar, Ashland, OR).
Cell attachment assays
After 3 d of growth in 5 mM glucose with 25 mM mannitol or 30 mM glucose medium, cells were trypsinized, neutralized in 0.5 mg/ml soybean trypsin inhibitor (Sigma-Aldrich), and counted. Glass coverslips were coated with 10 μg/ml rat plasma FN for 1 h at 37°C and then blocked with 1% BSA in PBS for 30 min at room temperature. Cells were seeded in serum-free medium with the corresponding glucose concentration in the presence or absence of 1.0 mM MnCl2 and allowed 1 h to adhere. Unattached cells were washed away with PBS and attached cells fixed for 15 min in 3.7% Formalin, permeabilized in 0.5% NP-40 for 10 min, and then stained with rhodamine–phalloidin (Life Technologies) at 1:500. Images were captured for 15 random fields/condition and cells were counted; representative fields are shown.
Cell binding to 9EG7
A 96-well non–tissue culture–treated plate was coated with 1 μg/well of 9EG7 antibody in PBS for 1 h at 37°C and then blocked with 1% BSA in PBS for 30 min at room temperature. Cells that had been conditioned to 5 mM or 30 mM glucose for 3 d as described were seeded at 1 × 105 cells/well in serum-free medium with the corresponding glucose concentrations in the presence or absence of 1.0 MnCl2 and allowed 30 min to adhere. For experiments in the presence of DMSO or calphostin C (Millipore), cells were conditioned for 2 d. Unattached cells were washed away with PBS and attached cells fixed for 15 min in 3.7% Formalin. Phase-contrast images were captured using a Nikon TMS microscope equipped with a Photometrics CoolSnap camera for each of three wells per condition per experiment, and cells were counted.
Ligand-binding assay
Cells were conditioned to 5 or 30 mM glucose for 3 d as described. Cells were resuspended in modified Tyrode's buffer (150 mM NaCl, 2.5 mM KCl, 2 mM NaHCO3, pH 6.5, 2 mM MgCl2, 2 mM CaCl2, 1 mg/ml BSA, 1 mg/ml dextrose). Each reaction contained 120 μl of cells (2 × 106 cells per condition), 40 μl of rat plasma FN (12.5 μg/ml final concentration), and 40 μl of Tyrode's buffer. Tubes were incubated for 30 min at room temperature and then divided into three 50-μl fractions. Each fraction was layered on 300 μl of 20% sucrose and centrifuged at 12,000 rpm for 3 min (O'Toole et al., 1994). Pellets were resuspended in SDS sample buffer. Samples were then reduced and separated by SDS–PAGE on 6% polyacrylamide gel and immunoblotted, and band intensities were quantified as described.
Solid-phase binding assays
Plates were coated with a solution of rat plasma FN or of the 70-kDa fragment at 0.5 and 2.0 μg/ml in CAPS buffer (10 mM N-cyclohexyl-3-aminopropanesulfonic acid [CAPS], pH 11, 150 mM NaCl) in triplicate by incubating overnight at 4°C. Wells were blocked with 1% BSA in PBS and then incubated with 30 μg/ml mouse type IV collagen (BD Biosciences) for 1 h with or without 100 μg/ml gelatin (Sigma-Aldrich). Bound collagen was detected by enzyme-linked immunosorbent assay (ELISA) with anti-collagen IV antibody and biotinylated goat anti-rabbit IgG (Pierce) at room temperature. Bound antibodies were detected with streptavidin β-galactosidase (Life Technologies) diluted 1:500 in 1% BSA with 1.5 mM MgCl2 and 2 mM β-mercaptoethanol for 1 h at 4°C, followed by nitrophenyl β-d-galactopyranoside (Sigma-Aldrich) at 1 mg/ml in substrate buffer (50 mM Na3PO4, pH 7.2, 1.5 mM MgCl2) for 50 min at room temperature. The reaction was stopped with 0.5 M Na2CO3, and absorbance at 405 nm was measured using an iMark Microplate Reader (Bio-Rad).
Supplementary Material
Acknowledgments
We thank Purva Singh for helpful discussions, Christina DeCoste of the Molecular Biology Flow Cytometry Facility for help with α5β1 analyses, and Mahesha Gangadhariah and Xiwu Chen for guidance in culturing mesangial cells. This research was funded by National Cancer Institute Grants R01-CA160611 to J.E.S. and R01-CA162433 to A.P.; National Institute of Diabetes and Digestive and Kidney Diseases Grants R01-DK095761 to A.P., R01-DK083187 to R.Z., R01-DK075594 to R.Z., and R01-DK383069221 to R.Z.; a National Institute of Diabetes and Digestive and Kidney Diseases National Research Service Award Predoctoral Fellowship (F30-DK095515) to C.G.M.; and VA Merit Reviews 1I01BX002025 to A.P. and 1I01BX002196 to R.Z.
Abbreviations used:
- DN
diabetic nephropathy
- DOC
deoxycholate
- ECM
extracellular matrix
- FN
fibronectin
- FUD
functional upstream domain
- GBM
glomerular basement membrane
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E14-03-0800) on June 18, 2014.
REFERENCES
- Abboud HE. Mesangial cell biology. Exp Cell Res. 2012;318:979–985. doi: 10.1016/j.yexcr.2012.02.025. [DOI] [PubMed] [Google Scholar]
- Aguirre KM, McCormick RJ, Schwarzbauer JE. Fibronectin self-association is mediated by complementary sites within the amino-terminal one-third of the molecule. J Biol Chem. 1994;269:27863–27868. [PubMed] [Google Scholar]
- Aumailley M, Timpl R. Attachment of cells to basement membrane collagen type IV. J Cell Biol. 1986;103:1569–1575. doi: 10.1083/jcb.103.4.1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayo SH, Radnik RA, Garoni JA, Glass WF, Kreisberg JI. High glucose causes an increase in extracellular matrix proteins in cultured mesangial cells. Am J Pathol. 1990;136:1339–1348. [PMC free article] [PubMed] [Google Scholar]
- Ayo SH, Radnik R, Garoni JA, Troyer DA, Kreisberg JI. High glucose increases diacylglycerol mass and activates protein kinase C in mesangial cell cultures. Am J Physiol. 1991;261:F571–F577. doi: 10.1152/ajprenal.1991.261.4.F571. [DOI] [PubMed] [Google Scholar]
- Baylis C, Corman B. The aging kidney: insights from experimental studies. J Am Soc Nephrol. 1998;9:699–709. doi: 10.1681/ASN.V94699. [DOI] [PubMed] [Google Scholar]
- Bazzoni G, Shih DT, Buck CA, Hemler ME. Monoclonal antibody 9EG7 defines a novel beta 1 integrin epitope induced by soluble ligand and manganese, but inhibited by calcium. J Biol Chem. 1995;270:25570–25577. doi: 10.1074/jbc.270.43.25570. [DOI] [PubMed] [Google Scholar]
- Borza CM, et al. Inhibition of integrin α2β1 ameliorates glomerular injury. J Am Soc Nephrol. 2012;23:1027–1038. doi: 10.1681/ASN.2011040367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- Chen S, Chakrabarti R, Keats EC, Chen M, Chakrabarti S, Khan ZA. Regulation of vascular endothelial growth factor expression by extra domain B segment of fibronectin in endothelial cells. Invest Ophthalmol Vis Sci. 2012;53:8333–8343. doi: 10.1167/iovs.12-9766. [DOI] [PubMed] [Google Scholar]
- Chen X, et al. Integrin alpha1beta1 controls reactive oxygen species synthesis by negatively regulating epidermal growth factor receptor-mediated Rac activation. Mol Cell Biol. 2007;27:3313–3326. doi: 10.1128/MCB.01476-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Moeckel G, Morrow JD, Cosgrove D, Harris RC, Fogo AB, Zent R, Pozzi A. Lack of integrin alpha1beta1 leads to severe glomerulosclerosis after glomerular injury. Am J Pathol. 2004;165:617–630. doi: 10.1016/s0002-9440(10)63326-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang H-Y, Korshunov VA, Serour A, Shi F, Sottile J. Fibronectin is an important regulator of flow-induced vascular remodeling. Arterioscler Thromb Vasc Biol. 2009;29:1074–1079. doi: 10.1161/ATVBAHA.108.181081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cifuni SM, Wagner DD, Bergmeier W. CalDAG-GEFI and protein kinase C represent alternative pathways leading to activation of integrin alphaIIbbeta3 in platelets. Blood. 2008;112:1696–1703. doi: 10.1182/blood-2008-02-139733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon AJ, Burns J, Dunnill MS, McGee JO. Distribution of fibronectin in normal and diseased human kidneys. J Clin Pathol. 1980;33:1021–1028. doi: 10.1136/jcp.33.11.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engvall E, Ruoslahti E, Miller EJ. Affinity of fibronectin to collagens of different genetic types and to fibrinogen. J Exp Med. 1978;147:1584–1595. doi: 10.1084/jem.147.6.1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ensenberger MG, Tomasini-Johansson BR, Sottile J, Ozeri V, Hanski E, Mosher DF. Specific interactions between F1 adhesin of Streptococcus pyogenes and N-terminal modules of fibronectin. J Biol Chem. 2001;276:35606–35613. doi: 10.1074/jbc.M105417200. [DOI] [PubMed] [Google Scholar]
- Falk RJ, Scheinman JI, Mauer SM, Michael AF. Polyantigenic expansion of basement membrane constituents in diabetic nephropathy. Diabetes. 1983;32((Suppl 2)):34–39. doi: 10.2337/diab.32.2.s34. [DOI] [PubMed] [Google Scholar]
- Gailit J, Ruoslahti E. Regulation of the fibronectin receptor affinity by divalent cations. J Biol Chem. 1988;263:12927–12932. [PubMed] [Google Scholar]
- Gardner H, Broberg A, Pozzi A, Laato M, Heino J. Absence of integrin alpha1beta1 in the mouse causes loss of feedback regulation of collagen synthesis in normal and wounded dermis. J Cell Sci. 1999;112:263–272. doi: 10.1242/jcs.112.3.263. [DOI] [PubMed] [Google Scholar]
- Hagiwara H, Kaizu K, Uriu K, Noguchi T, Takagi I, Qie YL, Seki T, Ariga T. Expression of type-1 plasminogen activator inhibitor in the kidney of diabetic rat models. Thromb Res. 2003;111:301–309. doi: 10.1016/j.thromres.2003.09.023. [DOI] [PubMed] [Google Scholar]
- Haraldsson B, Nyström J, Deen WM. Properties of the glomerular barrier and mechanisms of proteinuria. Physiol Rev. 2008;88:451–487. doi: 10.1152/physrev.00055.2006. [DOI] [PubMed] [Google Scholar]
- Hopf M, Göhring W, Mann K, Timpl R. Mapping of binding sites for nidogens, fibulin-2, fibronectin and heparin to different IG modules of perlecan. J Mol Biol. 2001;311:529–541. doi: 10.1006/jmbi.2001.4878. [DOI] [PubMed] [Google Scholar]
- Huang K, Liu W, Lan T, Xie X, Peng J, Huang J, Wang S, Shen X, Liu P, Huang H. Berberine reduces fibronectin expression by suppressing the S1P-S1P2 receptor pathway in experimental diabetic nephropathy models. PLoS One. 2012;7:e43874. doi: 10.1371/journal.pone.0043874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt GC, Singh P, Schwarzbauer JE. Endogenous production of fibronectin is required for self-renewal of cultured mouse embryonic stem cells. Exp Cell Res. 2012;318:1820–1831. doi: 10.1016/j.yexcr.2012.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes RO. Fibronectins. New York: Springer-Verlag; 1990. [Google Scholar]
- Kadler KE, Hill A, Canty-Laird EG. Collagen fibrillogenesis: fibronectin, integrins, and minor collagens as organizers and nucleators. Curr Opin Cell Biol. 2008;20:495–501. doi: 10.1016/j.ceb.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagami S, Border WA, Ruoslahti E, Noble NA. Coordinated expression of beta 1 integrins and transforming growth factor-beta-induced matrix proteins in glomerulonephritis. Lab Invest. 1993;69:68–76. [PubMed] [Google Scholar]
- Kerjaschki D, Ojha PP, Susani M, Horvat R, Binder S, Hovorka A, Hillemanns P, Pytela R. A beta 1-integrin receptor for fibronectin in human kidney glomeruli. Am J Pathol. 1989;134:481–489. [PMC free article] [PubMed] [Google Scholar]
- Kim C, Ye F, Ginsberg MH. Regulation of integrin activation. Annu Rev Cell Dev Biol. 2011;27:321–345. doi: 10.1146/annurev-cellbio-100109-104104. [DOI] [PubMed] [Google Scholar]
- Kitsiou PV, Tzinia AK, Stetler-Stevenson WG, Michael AF, Fan W-W, Zhou B, Tsilibary EC. Glucose-induced changes in integrins and matrix-related functions in cultured human glomerular epithelial cells. Am J Physiol Renal Physiol. 2003;284:F671–F679. doi: 10.1152/ajprenal.00266.2002. [DOI] [PubMed] [Google Scholar]
- Kolset SO, Reinholt FP, Jenssen T. Diabetic nephropathy and extracellular matrix. J Histochem Cytochem. 2012;60:976–986. doi: 10.1369/0022155412465073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köppel H, Riedl E, Braunagel M, Sauerhoefer S, Ehnert S, Godoy P, Sternik P, Dooley S, Yard BA. L-carnosine inhibits high-glucose-mediated matrix accumulation in human mesangial cells by interfering with TGF-β production and signalling. Nephrol Dial Transplant. 2011;26:3852–3858. doi: 10.1093/ndt/gfr324. [DOI] [PubMed] [Google Scholar]
- Kwoh C, Shannon MB, Miner JH, Shaw A. Pathogenesis of nonimmune glomerulopathies. Annu Rev Pathol. 2006;1:349–374. doi: 10.1146/annurev.pathol.1.110304.100119. [DOI] [PubMed] [Google Scholar]
- Lee H-S, Lim CJ, Puzon-McLaughlin W, Shattil SJ, Ginsberg MH. RIAM activates integrins by linking talin to ras GTPase membrane-targeting sequences. J Biol Chem. 2009;284:5119–5127. doi: 10.1074/jbc.M807117200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leiss M, Beckmann K, Girós A, Costell M, Fässler R. The role of integrin binding sites in fibronectin matrix assembly in vivo. Curr Opin Cell Biol. 2008;20:502–507. doi: 10.1016/j.ceb.2008.06.001. [DOI] [PubMed] [Google Scholar]
- Li S, Van Den Diepstraten C, D'Souza SJ, Chan BMC, Pickering JG. Vascular smooth muscle cells orchestrate the assembly of type I collagen via alpha2beta1 integrin, RhoA, and fibronectin polymerization. Am J Pathol. 2003;163:1045–1056. doi: 10.1016/s0002-9440(10)63464-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S, Sahai A, Chugh SS, Pan X, Wallner EI, Danesh FR, Lomasney JW, Kanwar YS. High glucose stimulates synthesis of fibronectin via a novel protein kinase C, Rap1b, and B-Raf signaling pathway. J Biol Chem. 2002;277:41725–41735. doi: 10.1074/jbc.M203957200. [DOI] [PubMed] [Google Scholar]
- Mao Y, Schwarzbauer JE. Stimulatory effects of a three-dimensional microenvironment on cell-mediated fibronectin fibrillogenesis. J Cell Sci. 2005;118:4427–4436. doi: 10.1242/jcs.02566. [DOI] [PubMed] [Google Scholar]
- Mason RM, Wahab NA. Extracellular matrix metabolism in diabetic nephropathy. J Am Soc Nephrol. 2003;14:1358–1373. doi: 10.1097/01.asn.0000065640.77499.d7. [DOI] [PubMed] [Google Scholar]
- Maurer LM, Tomasini-Johansson BR, Ma W, Annis DS, Eickstaedt NL, Ensenberger MG, Satyshur KA, Mosher DF. Extended binding site on fibronectin for the functional upstream domain of protein F1 of Streptococcus pyogenes. J Biol Chem. 2010;285:41087–41099. doi: 10.1074/jbc.M110.153692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeown-Longo PJ, Mosher DF. Binding of plasma fibronectin to cell layers of human skin fibroblasts. J Cell Biol. 1983;97:466–472. doi: 10.1083/jcb.97.2.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Toole TE, Katagiri Y, Faull RJ, Peter K, Tamura R, Quaranta V, Loftus JC, Shattil SJ, Ginsberg MH. Integrin cytoplasmic domains mediate inside-out signal transduction. J Cell Biol. 1994;124:1047–1059. doi: 10.1083/jcb.124.6.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Seo JY, Ha H. Plasminogen activator inhibitor-1 antisense oligodeoxynucleotides abrogate mesangial fibronectin accumulation. Korean J Physiol Pharmacol. 2010;14:385–390. doi: 10.4196/kjpp.2010.14.6.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozzi A, Voziyan PA, Hudson BG, Zent R. Regulation of matrix synthesis, remodeling and accumulation in glomerulosclerosis. Curr Pharm Des. 2009;15:1318–1333. doi: 10.2174/138161209787846748. [DOI] [PubMed] [Google Scholar]
- Pozzi A, Zent R. Integrins in kidney disease. J Am Soc Nephrol. 2013;24:1034–1039. doi: 10.1681/ASN.2013010012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reckelhoff JF, Tygart VL, Mitias MM, Walcott JL. STZ-induced diabetes results in decreased activity of glomerular cathepsin and metalloprotease in rats. Diabetes. 1993;42:1425–1432. doi: 10.2337/diab.42.10.1425. [DOI] [PubMed] [Google Scholar]
- Roth T, Podestá F, Stepp MA, Boeri D, Lorenzi M. Integrin overexpression induced by high glucose and by human diabetes: potential pathway to cell dysfunction in diabetic microangiopathy. Proc Natl Acad Sci USA. 1993;90:9640–9644. doi: 10.1073/pnas.90.20.9640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sechler JL, Corbett SA, Schwarzbauer JE. Modulatory roles for integrin activation and the synergy site of fibronectin during matrix assembly. Mol Biol Cell. 1997;8:2563–2573. doi: 10.1091/mbc.8.12.2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sechler JL, Takada Y, Schwarzbauer JE. Altered rate of fibronectin matrix assembly by deletion of the first type III repeats. J Cell Biol. 1996;134:573–583. doi: 10.1083/jcb.134.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shattil SJ, Kim C, Ginsberg MH. The final steps of integrin activation: the end game. Nat Rev Mol Cell Biol. 2010;11:288–300. doi: 10.1038/nrm2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi F, Harman J, Fujiwara K, Sottile J. Collagen I matrix turnover is regulated by fibronectin polymerization. Am J Physiol Cell Physiol. 2010;298:C1265–C1275. doi: 10.1152/ajpcell.00341.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh P, Carraher C, Schwarzbauer JE. Assembly of fibronectin extracellular matrix. Annu Rev Cell Dev Biol. 2010;26:397–419. doi: 10.1146/annurev-cellbio-100109-104020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sottile J, Hocking DC. Fibronectin polymerization regulates the composition and stability of extracellular matrix fibrils and cell-matrix adhesions. Mol Biol Cell. 2002;13:3546–3559. doi: 10.1091/mbc.E02-01-0048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sottile J, Shi F, Rublyevska I, Chiang H-Y, Lust J, Chandler J. Fibronectin-dependent collagen I deposition modulates the cell response to fibronectin. Am J Physiol Cell Physiol. 2007;293:C1934–C1946. doi: 10.1152/ajpcell.00130.2007. [DOI] [PubMed] [Google Scholar]
- Sumi A, Yamanaka-Hanada N, Bai F, Makino T, Mizukami H, Ono T. Roles of coagulation pathway and factor Xa in the progression of diabetic nephropathy in db/db mice. Biol Pharm Bull. 2011;34:824–830. doi: 10.1248/bpb.34.824. [DOI] [PubMed] [Google Scholar]
- Tadokoro S, Shattil SJ, Eto K, Tai V, Liddington RC, de Pereda JM, Ginsberg MH, Calderwood DA. Talin binding to integrin beta tails: a final common step in integrin activation. Science. 2003;302:103–106. doi: 10.1126/science.1086652. [DOI] [PubMed] [Google Scholar]
- Taniguchi K, et al. Inhibition of Src kinase blocks high glucose-induced EGFR transactivation and collagen synthesis in mesangial cells and prevents diabetic nephropathy in mice. Diabetes. 2013;62:3874–3886. doi: 10.2337/db12-1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velling T, Risteli J, Wennerberg K, Mosher DF, Johansson S. Polymerization of type I and III collagens is dependent on fibronectin and enhanced by integrins alpha 11beta 1 and alpha 2beta 1. J Biol Chem. 2002;277:37377–37381. doi: 10.1074/jbc.M206286200. [DOI] [PubMed] [Google Scholar]
- Weston BS, Wahab NA, Mason RM. CTGF mediates TGF-beta-induced fibronectin matrix deposition by upregulating active alpha5beta1 integrin in human mesangial cells. J Am Soc Nephrol. 2003;14:601–610. doi: 10.1097/01.asn.0000051600.53134.b9. [DOI] [PubMed] [Google Scholar]
- Wierzbicka-Patynowski I, Mao Y, Schwarzbauer JE. Continuous requirement for pp60-Src and phospho-paxillin during fibronectin matrix assembly by transformed cells. J Cell Physiol. 2007;210:750–756. doi: 10.1002/jcp.20886. [DOI] [PubMed] [Google Scholar]
- Wilson CL, Schwarzbauer JE. The alternatively spliced V region contributes to the differential incorporation of plasma and cellular fibronectins into fibrin clots. J Cell Biol. 1992;119:923–933. doi: 10.1083/jcb.119.4.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita S, Mochizuki N, Ohba Y, Tobiume M, Okada Y, Sawa H, Nagashima K, Matsuda M. CalDAG-GEFIII activation of Ras, R-ras, and Rap1. J Biol Chem. 2000;275:25488–25493. doi: 10.1074/jbc.M003414200. [DOI] [PubMed] [Google Scholar]
- Yang J, Zeng Z, Wu T, Yang Z, Liu B, Lan T. Emodin attenuates high glucose-induced TGF-β1 and fibronectin expression in mesangial cells through inhibition of NF-κB pathway. Exp Cell Res. 2013;319:3182–3189. doi: 10.1016/j.yexcr.2013.10.006. [DOI] [PubMed] [Google Scholar]
- Zent R, et al. Glomerular injury is exacerbated in diabetic integrin alpha1-null mice. Kidney Int. 2006;70:460–470. doi: 10.1038/sj.ki.5000359. [DOI] [PubMed] [Google Scholar]
- Zhou XJ, Rakheja D, Yu X, Saxena R, Vaziri ND, Silva FG. The aging kidney. Kidney Int. 2008;74:710–720. doi: 10.1038/ki.2008.319. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.