

Summary
After infecting peripheral sites, herpes simplex virus (HSV) invades the nervous system and initiates latent infection in sensory neurons. Establishment and maintenance of HSV latency requires host survival, and entails repression of productive cycle (“lytic”) viral gene expression. We find that a neuron-specific microRNA, miR-138, represses expression of ICP0, a viral transactivator of lytic gene expression. A mutant HSV-1 (M138) with disrupted miR-138 target sites in ICP0 mRNA exhibits enhanced expression of ICP0 and other lytic proteins in infected neuronal cells in culture. Following corneal inoculation, M138-infected mice have higher levels of ICP0 and lytic transcripts in trigeminal ganglia during establishment of latency, and exhibit increased mortality and encephalitis symptoms. After full establishment of latency, the fraction of trigeminal ganglia harboring detectable lytic transcripts is greater in M138-infected mice. Thus, miR-138 is a neuronal factor that represses HSV-1 lytic gene expression, promoting host survival and viral latency.
Introduction
Herpes simplex viruses 1 and 2 (HSV-1 and HSV-2) cause a variety of diseases ranging from common cold sores to rare but fatal brain diseases such as encephalitis. HSV alternates between two distinct infection programs, productive (“lytic”) infection and latent infection. During lytic infection, infectious virus is abundantly produced and many viral genes are highly expressed. Immediate early (IE) genes are expressed first, and activate the expression of subsequently expressed early (E), and late (L) genes. Among the IE gene products is ICP0, which acts as a promiscuous transactivator of genes introduced by transfection or infection and is required for efficient lytic infection in many cell types (Cai and Schaffer, 1992; Chen and Silverstein, 1992). ICP0 is a RING finger E3 ubiquitin ligase (Boutell et al., 2002) that promotes degradation and dispersal of proteins that mediate intrinsic antiviral resistance (reviewed in (Boutell and Everett, 2013)). It antagonizes chromatin-mediated silencing (Cliffe and Knipe, 2008; Ferenczy and DeLuca, 2009, 2011; Gu et al., 2005; Gu and Roizman, 2007; Kalamvoki and Roizman, 2010), and also antagonizes innate immunity by interfering with signaling from pattern recognition receptors that leads to interferon production (Melroe et al., 2004; Mossman et al., 2000; Orzalli et al., 2012; Paladino et al., 2010).
Following inoculation and lytic infection at a peripheral site on a mammalian host, HSV gains entry to axons of sensory neurons and is transported back to cell bodies in sensory ganglia. The virus initiates lytic infection of the ganglia (acute ganglionic replication), but also begins the process of establishing latent infection in sensory neurons. HSV latency entails survival of the host in the face of the ability of the virus to invade the central nervous system (CNS) and cause lethal encephalitis. During latent infection no infectious virus is detected and lytic gene expression is strongly repressed, although low levels of lytic transcripts can be found using sensitive PCR-based assays (Chen et al., 2002; Giordani et al., 2008; Kramer et al., 1998; Kramer and Coen, 1995; Pesola et al., 2005; Tal-Singer et al., 1997). The only abundantly expressed gene products arise from the latency associated transcripts (LAT) locus (Rock et al., 1987; Stevens et al., 1987). This locus is important for repressing lytic gene expression during establishment and maintenance of latency in a mouse model (Chen et al., 1997; Garber et al., 1997). Host factors (reviewed in (Roizman et al., 2013)), including elements of innate and acquired immunity (e.g. (Knickelbein et al., 2008; Luker et al., 2003)), and the neuronal milieu (e.g. (Hafezi et al., 2012; Kolb and Kristie, 2008; Yordy et al., 2012)) are also thought to contribute to repression of lytic gene expression, survival, and latency. However, the underlying mechanisms are still largely undefined.
MicroRNAs (miRNAs) are ~22 nucleotide long non-coding RNAs that target mRNA sequences, particularly those complementary to nucleotides 2–8 (seed region) of the miRNA. miRNAs are incorporated into the RNA-induced silencing complex (RISC), which guides the recruitment of the target mRNA for degradation and/or translational repression (reviewed in (Carthew and Sontheimer, 2009)). miRNAs regulate many cellular and developmental processes, and changes in their levels are associated with human diseases including virus infections (reviewed in (Cullen, 2011; Mendell and Olson, 2012)).. Notably, the LAT locus encodes several miRNAs (Flores et al., 2013; Jurak et al., 2010; Tang et al., 2008; Tang et al., 2009; Umbach et al., 2008), which are differentially expressed during acute infection and latent infection (Kramer et al., 2011; Tang et al., 2008; Umbach et al., 2008) and have been hypothesized to play a role in repressing lytic gene expression. Some host miRNAs have also been suggested to play a role in HSV infection (Hill et al., 2009; Mulik et al., 2012; Zheng et al., 2011). However, repression of HSV gene expression by host miRNAs has not been demonstrated.
Because HSV latent infection occurs in neurons, we wondered whether a neuron-specific miRNA might repress lytic gene expression. In a previous deep sequencing study of miRNAs expressed during latency in mice (Umbach et al., 2008), we noticed that reads of miR-138 were highly represented in trigeminal ganglia (TG). Earlier studies had found abundant expression of miR-138 in brain or neuroblastoma cell lines relative to other tissues or cell lines (Landgraf et al., 2007; Obernosterer et al., 2006). This miRNA was later found to regulate dendritic spine morphogenesis in the mouse brain by modulating APT1 (Banerjee et al., 2009; Siegel et al., 2009) and its expression in mouse sensory ganglia was reduced when the gene encoding the miRNA processing enzyme Dicer was disabled in a subset of sensory neurons (Zhao et al., 2010).
The neuronal expression of miR-138 prompted us to search HSV-1 and HSV-2 genomes for complementary sequences, and we identified ICP0 mRNA as a potential target. Notably, ICP0 is required for efficient acute ganglionic replication and reactivation from latency (Cai et al., 1993; Halford and Schaffer, 2001; Leib et al., 1989; Thompson and Sawtell, 2006). We hypothesized that miR-138 might repress ICP0 and thereby lytic gene expression in neurons thus facilitating latency. Here we show that miR-138 directly downregulates ICP0 and lytic gene expression in cell culture. Moreover, mutating miR-138 target sites in ICP0 mRNA enhances lytic viral gene expression in neuronal cells in culture and in TG, and makes the virus more virulent in a mouse model. Therefore, we identify a neuronal factor that promotes HSV-1 latency by directly targeting a viral gene.
Results
Neuron specific miR-138 can repress ICP0 expression
To investigate the neuronal specificity of miR-138 expression, we quantified miR-138 in mouse brain, mouse trigeminal ganglion (TG), primary neurons from rat superior cervical ganglion (SCG), and four different cultured cells, neuronal Neuro-2A cells (from mouse brain), and non-neuronal cells, 293T, Vero, and human foreskin fibroblast (HFF) using stem-loop qRT-PCR. miR-138 levels in mouse brain and TG, rat SCG neurons, and Neuro-2A cells were much higher than those in Vero, 293T, and HFF cells, and levels of miR-138 in Neuro-2A cells remained high following infection by HSV-1 (Figure 1A). These data quantitatively confirm that miR-138 is enriched in neuronal tissues and cells.
Figure 1.

Neuron-specific miR-138 can target HSV ICP0. (A) qRT-PCR analysis. miR-138 copy numbers normalized to amounts of total RNA in the various tissues and cells indicated below the graph. Neuro-2A + HSV-1; MOI = 50, and 16 hpi. Assays were performed in triplicate with standard deviations shown as error bars. (B) Schematic diagram showing candidate miR-138 target sites in the 3′ UTRs of HSV-1 and HSV-2 ICP0 mRNAs. White boxes indicate the coding sequences. Candidate miR-138 target sites are shown in hatched and grey boxes. (C) Sequences of candidate miR-138 target sites. Below each candidate target site is the sequence of miR-138 (conserved in mouse, rat, monkey and human). Underlined are miR-138 seed sequences. Solid and dashed vertical lines signify Watson-Crick and G-U wobble base pairs, respectively. Arrows point to nucleotide substitutions in mutant forms of target sites and miR-138 with altered bases in italics. (D) miR-138 mimic reduces ICP0 mRNA and protein levels. 293T cells were co-transfected with miR-138 or miR-M138 mimics and an ICP0 WT plasmid. Left, ICP0 mRNA levels normalized to the levels of human GAPDH as measured by qRT-PCR in triplicate. The error bar indicates the standard deviation. Right, ICP0 protein levels analyzed by Western blot in the upper panel and actin in the lower panel as a loading control. (E) miR-138 expressed from a expression plasmid downregulates ICP0 protein expression. 293T cells were mock-transfected or co-transfected with pcDNA-miR-138 plasmid, a WT or M138 ICP0 expression plasmid, and a GFP expression plasmid (control), as indicated above the images. ICP0 (top panel) and GFP (bottom panel) protein levels were assessed using Western blots.
Sequence alignments revealed that the seed region of miR-138 is completely complementary to two potential target sites in the 3′ untranslated region (UTR) of HSV-1 ICP0 mRNA (Figure 1B, C) and to one potential target site in the 3′ UTR of HSV-2 ICP0 mRNA. One of the sites in HSV-1 also shows high overall complementarity with miR-138 (Figure 1C). To test whether miR-138 can repress ICP0 expression, we transfected a miR-138 mimic or a variant mimic bearing three nucleotide changes in the seed region (miR-M138; Figure 1C) together with an ICP0 expression plasmid. The miR-138 mimic resulted in reduced ICP0 protein and mRNA expression relative to the variant mimic (Figure 1D). In a second co-transfection experiment, miR-138 expressed from a plasmid downregulated ICP0 expressed from a plasmid encoding a wild type (WT) 3′ UTR but not from one (ICP0 M138) containing 3 nucleotide substitutions at each binding site of the miR-138 seed region (Figure 1 C, E). Taken together, these results show that miR-138 can repress ICP0 expression through its target sites in ICP0 mRNA in a seed region-dependent manner.
Identification of miR-138 target sites by PAR-CLIP
To investigate whether miR-138 binds to its candidate target sites on ICP0 mRNA in HSV-infected cells, we performed a photoactivatable ribonucleoside-enhanced crosslinking and immunoprecipitation (PAR-CLIP) experiment, in which mRNAs were labeled with 4-thiouridine, UV-crosslinked to RISCs, immunoprecipitated using an antibody against Argonaute (Ago) proteins, treated with RNase T1, and converted to cDNA for deep sequencing analysis. 293 cells were stably transduced either with a control vector or a miR-138 expression vector. miR-138 transduced cells expressed 7-fold higher levels of miR-138 than did control transduced cells (Figure S1A). They also exhibited stronger down-regulation of reporter gene expression from a plasmid containing either two perfect miR-138 target sequences or the ICP03′ UTR sequence in the 3′ UTR of the reporter gene than did the control transduced cells (Figure S1B). Both the control and the miR-138 transduced cells were infected with HSV-1 and subjected to PAR-CLIP. While cells transduced with both vectors showed clusters of reads of ICP0 mRNA at sites complementary to HSV-1 miR-H2, as previously observed (Flores et al., 2013), only cells transduced with miR-138 showed clusters of reads aligned to the two predicted miR-138 target sites (sites 1 and 2; Figure 1B, C; Figure 2). The clusters in site 1 showed many more reads than those in site 2, which could be due to the lack of a U in site 2, or the lower overall complementarity of site 2 with miR-138 relative to site 1. These results indicate that miR-138 can bind to its target sites on ICP0 mRNA in HSV-infected cells.
Figure 2.

Binding of miR-138 to ICP0 mRNA. At the top is a schematic diagram of HSV-1 genome with long (UL) and short (US) unique sequences indicated and long (TRL and IRL) and short (IRS and TRS) repeat sequences shown in hatched boxes. Below is an expanded view of the ICP0 gene with the transcription start site indicated as a black dot, introns as zigzags, and the candidate target sites of miR-138 and HSV-1 miR-H2 as arrows. The two bottom panels show aligned Illumina sequence reads from control (upper) and miR-138 transduced (lower) 293 cells. Each point represents a cluster of reads starting at the same 5′ start site. The vertical lines in these panels indicate the positions of the candidate miR-138 and miR-H2 binding sites. See also Figure S1
miR-138 downregulates lytic gene expression in HSV-1-infected cells
As an initial test of whether miR-138 can affect lytic gene expression in HSV-1-infected cells, we transfected 293T cells with a miR-138 mimic together with either an oligonucleotide inhibitor complementary to miR-138 or a control scrambled oligonucleotide inhibitor, and infected with HSV-1. We observed higher expression of ICP0, the L protein gC, and, to a lesser extent, the IE protein ICP4, in the presence of the miR-138 inhibitor than with the control oligonucleotide (Fig. 3A).
Figure 3.
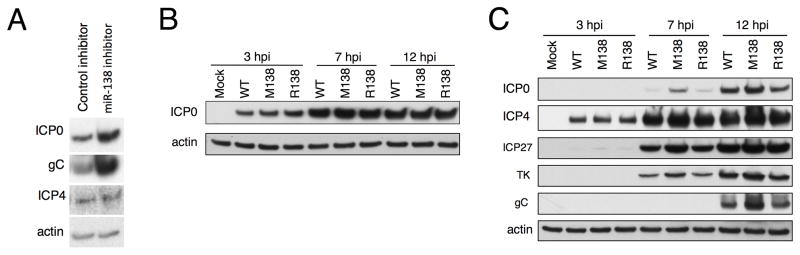
miR-138 downregulates viral gene expression in HSV-1 infected cells. (A) 293T cells were transfected with miR-138 mimic together with either a complementary oligonucleotide inhibitor or a control inhibitor before being infected with WT HSV-1 at an MOI of 0.1, and harvested at 48 hpi. (B, C) Vero (B) and Neuro-2A (C) cells were mock-infected (Mock) or infected with the indicated viruses at an MOI of 10 and harvested at the times shown. In all 3 panels, protein expression was analyzed by Western blot. The proteins analyzed are indicated to the left of the panels with actin serving as a loading control. See also Figure S2.
To investigate further, we used bacterial artificial chromosome (BAC) technology to generate a mutant virus (M138) expressing ICP0 mRNA with the aforementioned M138 mutations in the 3′ UTR in the background of WT HSV-1 strain KOS (Figure 1C). To ensure that differences between WT and M138 are due to the engineered mutations, we also generated a rescued derivative (R138), in which WT ICP0 sequences were restored. The M138 mutations do not change ICP0 protein coding sequences. They are located within sequences encoding the stable LAT intron, but ~500 bp away from the splicing branch point (Krummenacher et al., 1997), and do not affect expression of stable LATs (see below). Because there are two copies of the ICP0 gene in the HSV-1 genome, mutations were introduced into the BACs using two different drug resistance genes as selection markers and two-step Red-mediated recombination (Figure S2A, S2B).
WT, M138, and R138 viruses replicated with similar kinetics in Vero cells and in Neuro-2A cells (Figure S2C). Following infection at an MOI of 10, expression of ICP0 was similar for all three viruses in Vero cells (Figure 3B), where miR-138 is poorly expressed (Figure 1A). However, in infected Neuro-2A cells where miR-138 is abundantly expressed (Figure 1A), the M138 mutations increased the expression of ICP0 between 2- and 4-fold, the IE protein ICP27, and the L protein gC ~ 2 fold, and the expression of the IE protein ICP4 and the E protein TK, but less than 2 fold, at 7 and 12 hours post-infection (hpi) (Figures 3C, S2D). We observed similar levels of ICP4 in cells infected with the three viruses at 3 hpi, indicating that the cells were equally infected. At an MOI of 0.5, the M138 mutations also increased ICP0 levels at 24 hpi (data not shown).
As an additional test, we transfected the miR-138 mimic or the mutant miR-M138 mimic (Figure 1C) into 293T cells where endogenous miR-138 levels are low (Figure 1A), and infected the cells with either WT or M138 virus 8 hours post-transfection at a low MOI. Notably, the miR-M138 mimic contains 3 nucleotide changes that compensate for the changes in ICP0 mRNA in the M138 virus. Transfection of the miR-138 mimic reduced levels of ICP0 in WT-infected cells relative to transfection of the mutant mimic, miR-M138, whereas transfection of miR-M138 reduced levels of ICP0 in M138 virus infected cells relative to transfection of miR-138, particularly at 48 hpi (Figure S2E).
These results taken together indicate that high levels of endogenous or introduced miR-138 downregulate lytic viral gene expression in cells infected with HSV-1, dependent on miR-138 target sites in ICP0 mRNA.
The M138 mutations increase expression of lytic genes during establishment of latency
We next investigated whether miR-138 repression of ICP0 expression affects lytic gene expression during establishment of latency in mouse TG. Mice were inoculated on the cornea with WT or M138, and virus replication was assessed at that site by titrating eye swabs on 1, 3, 5, and 7 days post infection (dpi), and in the TG by titrating ganglionic homogenates at 3, 5, and 7 dpi. The M138 mutant exhibited WT eye swab and ganglionic titers (Figure 4A, B). Eye swab titers decreased from 1 to 3 dpi but increased from 3 to 5 dpi, which might suggest re-infection of the cornea from the TG, before decreasing again to very low levels at 7 dpi. TG titers decreased from 3 dpi to very low levels at 7 dpi (Figure 4B), suggesting that at 7 dpi acute ganglionic replication is waning and latency is being established. Eye swab and ganglionic titers were also similar between M138- and R138-infected mice (data not shown).
Figure 4.

Virus replication and gene expression during acute infection of mice. (A) Virus replication in the eye. Each point represents the geometric mean titer of at least 15 eye swabs. (B) Virus replication in the TG. Each point represents the geometric mean titer of at least 10 TG. There are no statistically significant differences between the titers of WT and M138 viruses in panels (A) and (B). (C) Viral genome and transcript levels in TG at 7 dpi. Each point represents a value from one TG. Virus names are shown at the bottom, and the molecules assayed are labeled at the top of each graph. Vertical axes show logarithmic values, and RNA levels were normalized to viral genome levels. The horizontal lines represent the geometric means (any undetectable values were included in the calculations of means as logs of the detection limits). P values for the two viruses compared are shown on top of the brackets. (D) Correlations between transcript levels in TG infected by the viruses indicated at the top of each plot at 7 dpi. The level of gC transcripts or LATs were plotted against the level of ICP0 transcripts in the same TG. One TG that exhibited undetectable levels of gC and ICP0 transcripts was excluded from the plots. R2 and P values for each correlation are shown at the upper left corners of the plots. See also Figure S3 and Table S1.
Next we analyzed TG samples obtained at 7 dpi from mice infected with WT, M138, or R138 for viral DNA levels by qPCR, and RNA levels by qRT-PCR. The M138 mutations had little or no effect on viral genome levels or levels of the stable LAT intron (Figure 4C). However, they significantly increased the levels of ICP0 transcripts (Figure 4C). Considering ICP0’s role in activating lytic genes we also analyzed the levels of other lytic transcripts, namely ICP4 (IE), ICP27 (IE), tk (E) and gC (L). The M138 mutations increased levels of all four of these lytic transcripts by about 3-fold (2.3–3.8 fold; Figure 4C), and all of these differences were statistically significant (P<0.05), except the difference between the ICP4 levels of WT and M138 (P = 0.06). There were no significant differences in transcript levels in ganglia infected with WT vs. R138 viruses (Figure 4C), indicating that the increases in transcript levels in M138-infected mice were due to the engineered mutations. We performed an independent replicate of this experiment, which yielded very similar results (Figure S3). We also found that ICP0 transcript levels were significantly correlated with levels of each of the other lytic transcripts assayed but not with LATs, consistent with previous studies suggesting divergent pathways for acute and latent infections (Margolis et al., 1992; Speck and Simmons, 1991). Notably, the correlation between ICP0 and the other lytic transcripts was increased by the M138 mutations (Figure 4D; Table S1). Taken together, the results show that the miR-138 target site mutations in ICP0 mRNA increase lytic gene expression during establishment of latency in mouse TG.
The M138 mutations increase morbidity and mortality
Infection of CD-1 mice by HSV-1 strain KOS usually results in low mortality and thus efficient establishment of latency (Leib et al., 1989). To examine the effect of the M138 mutations on morbidity and mortality, we followed infected mice for 32 days in each of the two replicate experiments. Pooling the data from the two experiments, the percentage of mice infected with M138 that died (or in one case became moribund and was euthanized) was nearly four times higher than that for WT or R138 (28% vs. 7.7% or 7.5%; Figures 5A, S4A). The differences in survival were statistically significant, with P values of 0.019 and 0.016, respectively. Most deaths occurred between 9 and 13 dpi (Figure 5A). At 17 and 25 dpi, the surviving mice were observed for partial paralysis or lethargy (impaired mobility), which are sequelae of HSV encephalitis. A higher percentage of mice infected with M138 showed impaired mobility than did those infected with WT or R138 (Figure 5B). To determine whether the increased morbidity and mortality were due to increased spread to the CNS, we measured viral titers in the brain, and found no significant difference between mice infected with WT and M138 (Figure 5C). Interestingly, when M138 was delivered to the brain by a different route -- intracranial inoculation -- it caused little if any increase in mortality relative to R138 (Fig. S4B; see Discussion). Regardless, these results indicate that following corneal inoculation, the miR-138 target site mutations increase morbidity and mortality, and thus decrease survival and latency.
Figure 5.
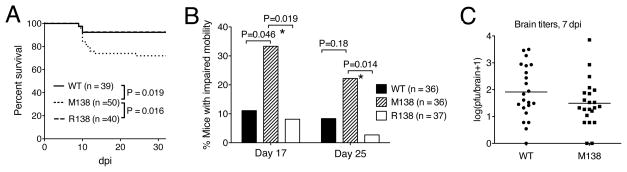
The M138 mutant virus causes increased mortality and disease. (A) Survival curves of mice infected by the viruses indicated in the key. Percentages of live mice as a function of time were plotted. The numbers of mice studied and the P values for the differences between viruses are shown below the curves. (B) Mice showing encephalitis symptoms. The bars represent percentages of mice at 17 or 25 dpi that showed impaired mobility; i.e. partial paralysis or lethargy. The viruses and the numbers of mice studied for each virus are indicated to the right of the graph. P values for statistical comparisons are shown above the brackets, and those with statistical significance are labeled with asterisks below the brackets. Since two Fisher’s Exact tests were performed for each time point, the threshold alpha level below which a P value is considered significant is adjusted from 0.05 to 0.0253 in order to control for multiple comparisons. (C) Titers of WT and M138 viruses in mouse brain at 7 dpi. Each point represents a value from one brain. The horizontal lines represent the geometric means of titers. See also Figure S4.
Effects of M138 mutations on gene expression during maintenance of latency
To study whether miR-138 repression of ICP0 regulates viral gene expression during maintenance of latency, TG were harvested from mice infected with WT, M138, or R138 at 32 dpi. Viral genome levels were analyzed by qPCR, and levels of the stable LAT intron and the lytic ICP0, ICP27, tk, and gC transcripts were analyzed by qRT-PCR. Genome and LAT intron levels were very similar for the three viruses (Figure 6A). Lytic transcript levels were, as expected (Kramer and Coen, 1995), low and detectable in only 30–70% of TG for any given transcript (Figure 6A). Making the assumption that ganglia in which a transcript was undetectable meant that there was no expression of that transcript, then the M138 mutant expressed more lytic transcripts on average for all four genes analyzed than did the WT and R138 viruses (not shown). However, these differences were not statistically significant. Nevertheless, all four lytic transcripts were detectable in a higher fraction of M138-infected TG than in WT- or R138-infected TG (Figure 6A). A separate experiment performed with fewer mice but using identical conditions also resulted in a higher fraction of M138-infected TG having detectable ICP0 and ICP27 transcripts than WT- and R138-infected TG (data not shown). Pooling the data from the two experiments, M138 infection was associated with increased fractions of TG exhibiting detectable transcripts for all four lytic genes, with the increases being statistically significant for ICP27 (Figure 6B). Analyzing only those TG in which both transcripts could be detected, ICP0 levels were uncorrelated with LAT levels, but were significantly correlated with the levels of the other three lytic transcripts in M138-infected TG (Figure 6C). These data suggest that the miR-138 target site mutations may also increase expression of lytic genes during maintenance of latency.
Figure 6.

Viral genome levels and gene expression during latent infection of mice. (A) Viral genome and transcript levels at 32 dpi. Values are presented as in Figure 4C except that mean values are not indicated in the plots for viral lytic transcripts, because the large numbers of undetectable values make difficult meaningful calculations of means. (B) Percentages of TG exhibiting detectable lytic transcripts. Each vertical bar represents the value for each virus (indicated in the key) and each RNA (below the graph). Since two Fisher’s Exact tests were performed for each gene, the threshold alpha level below which a P value is considered significant is adjusted from 0.05 to 0.0253 in order to control for multiple comparisons. Therefore, only P values below 0.0253 have been displayed. (C) Correlation between transcript levels in M138 virus infected TG at 32 dpi. The plots are presented as in Figure 4D. Only TGs exhibiting detectable transcripts for both RNAs being compared are included in the analysis.
Discussion
The mechanisms that promote HSV-1 latency have remained largely obscure, although roles for neuronal factors and for repression of ICP0, which is important for antagonizing host silencing mechanisms and for efficient replication and reactivation in ganglia, have long been suspected (reviewed in (Roizman et al., 2013; Boutell and Everett, 2013). Here we found that a neuron-specific host miRNA, miR-138, binds to two target sites in ICP0 mRNA and can repress ICP0 expression in both transfected and HSV-1 infected cells. This repression is dependent on the miR-138 seed sequence and its target sites in ICP0 mRNA. Importantly, mutations of these miR-138 target sites do not affect ICP0 expression in cells in which miR-138 is not abundant. However, in cells into which exogenous miR-138 is introduced, and in Neuro-2A cells and in mouse TG tissue where endogenous miR-138 is abundant, these mutations result in increased expression of ICP0 and other lytic genes. By far the simplest interpretation of these results is that high levels of miR-138 ordinarily repress ICP0 expression in infected neurons, which, given ICP0’s role as a viral transactivator, results in reduced lytic gene expression. As repression of lytic gene expression is a hallmark of HSV latency, miR-138 targeting of ICP0 expression in infected TG promotes latency. Moreover, the miR-138 target site mutations in ICP0 increase host morbidity and mortality. As host survival is a crucial prerequisite for latency, miR-138 targeting of ICP0 expression also promotes latency by preventing the death of the host.
Thus, miR-138 is a neuronal factor that contributes to HSV-1 latency. We speculate that HSV-1 has evolved to utilize this host miRNA to gain the selective advantage of increased latency, which in turn increases the ability of the virus to spread throughout the population. Consistent with this speculation, HSV-2 ICP0 mRNA also contains a predicted miR-138 target site with full seed complementarity in its 3′ UTR, although we caution that there is only one such site and it is less complementary overall to miR-138 than is either of the HSV-1 ICP0 target sites. It is also possible that miR-138 targets viral (and host) genes other than ICP0 to promote latency.
We found more obvious effects of miR-138 targeting of ICP0 on lytic gene expression during establishment of latency (7 dpi) than during maintenance (32 dpi). This may be due, at least in part, to our assays of gene expression at 32 dpi not being sensitive enough to detect lytic transcripts in a substantial fraction of samples. Alternatively, it is possible that by 32 dpi, mechanisms other than miR-138 targeting of ICP0, such as chromatin-mediated effects (reviewed in (Knipe and Cliffe, 2008)), are more important for repression of lytic gene expression. Regardless, levels of ICP0 transcripts were still positively correlated with those of other lytic transcripts, at least in TG latently infected with M138, consistent with ICP0 being able to activate expression of these other viral genes during latency. Although the M138 mutations do not appear to affect reactivation of infectious virus following explant of TG (unpublished results), and although abundant activation of ICP0 expression by a reactivation stimulus, may overcome any miR-138 repression, it is nevertheless possible that the M138 mutations could affect gene expression during this phase of infection. This possibility is under investigation.
Although there is evidence that miRNAs encoded by herpesviruses including HSV may promote latency by repressing expression of key lytic genes (reviewed in (Cullen, 2011)), and there are examples of host miRNAs that modulate virus production in herpesviruses (Nachmani et al., 2010; Yin et al., 2008), hepatitis C virus (Jopling et al., 2005), and human papillomaviruses (Gunasekharan and Laimins, 2013), here we show that miR-138 is a host miRNA that promotes herpesvirus latency by directly repressing lytic gene expression. We suspect that other herpesviruses may utilize host miRNAs that are abundant in the cell types in which they form latent infections to repress lytic gene expression. A recent paper describes one possible case in which oligonucleotide inhibitors of either of two host miRNAs increased lytic gene expression and virus production in cells that were latently infected with Kaposi’s sarcoma herpesvirus (KSHV) and stimulated with a phorbol ester (Yan et al., 2013). This study also provided evidence from reporter gene assays that these host miRNAs target a KSHV IE gene, but did not examine unstimulated cells and did not examine a viral mutant. Interestingly, it has been reported that human immunodeficiency virus-1 contains binding sites for several host miRNAs that can repress viral gene expression and reactivation in latently infected resting CD4+ T cells, although a virus whose binding sites for one or more of these miRNAs were inactivated was not investigated (Huang et al., 2007).
A key question remaining is what accounts for the role of miR-138 targeting of ICP0 in suppressing viral virulence. Our data argue against the miR-138 target site mutations causing increased spread of virus to the brain to cause encephalitis and death. Although we cannot rule out that we missed an increase in infectious virus due to choice of time points, the mutations also did not result in increased virus production in cell culture or in TG. Perhaps WT levels of ICP0 are sufficient for maximal virus production or perhaps the increase in ICP0 expression occurs too late during the infectious cycle to affect virus production. Regardless, we raise the possibility that over-expression of ICP0 and/or other lytic genes without increased virus production results in increased virulence following corneal inoculation. One possible mechanism, consistent with increased virulence following corneal but not intracranial inoculation, is that increased viral gene expression results in increased pathology due to adaptive immune responses. Another possible mechanism stems from the role of ICP0 in combating innate immunity (Melroe et al., 2004; Mossman et al., 2000; Orzalli et al., 2012; Paladino et al., 2010). Perhaps increased expression of ICP0 results in more inhibition of innate immunity in the brain following spread of virus from the TG. Consistent with this possibility, impaired innate immune signaling has been associated with increased susceptibility to HSV encephalitis in humans (Lafaille et al., 2012).
In summary, we have identified a molecular mechanism by which host neurons provide an environment that promotes HSV latency. We emphasize that as the M138 mutant is still able to establish and maintain latency, this mechanism is just one of likely many that contribute to latent infection. It is possible, for example, that miR-138 acts synergistically with virus-encoded miRNAs such as miR-H2 that target ICP0. Another interesting possibility is that expression of miR-138 may be regulated differentially during lytic infection and the various phases of latent infection (or even in different neuron subtypes), especially as the tissue-specific expression of this miRNA appears to be post-transcriptionally controlled (Obernosterer et al., 2006). It will be important to identify the various mechanisms that contribute to latent infection and to understand how they interact in repressing lytic gene expression and ensuring host survival.
Experimental Procedures
Cells and viruses
Vero, 293T, and HFF cells were maintained as previously described (Pan and Coen, 2012a; Jurak et al., 2012). 293 and Neuro-2A cells (ATCC) were maintained in DMEM supplemented with 10% fetal bovine serum at 37°C in 5% CO2. Bacterial artificial chromosome (BAC)-derived HSV-1 strain KOS (Jurak et al., 2012) and M138 and R138 (see below) were used for all experiments except the PAR-CLIP assay, which used HSV-1 strain 17syn+. Infectious virus was harvested from cell cultures and TG and titrated as described previously (Leib et al., 1989). The derivation of M138 and R138 using BAC technology is outlined in Figure S2A and described in detail in Supplemental Experimental Procedures.
Quantitative PCR and RT-PCR
For miRNA quantification, total RNA from cells or tissues were purified using RNeasy mini kit (Qiagen). RNA from primary neuron cultures isolated from rat SCG was a generous gift from Angus Wilson and Michael O’Brien (New York University). miR-138 and snRNA U6 levels were evaluated using 10 ng of total RNA and TaqMan microRNA assays (Applied Biosystems) according to the manufacturer’s instructions. Previous qPCR or qRT-PCR assays for HSV-1 genome and transcripts of ICP0, tk, the LAT intron and mouse GAPDH (Chen et al., 2002; Kramer et al., 2011; Pesola et al., 2005) were modified and optimized, and new assays were developed for ICP4, ICP27, gC, and human GAPDH. Details are provided in Supplemental Experimental Procedures.
DNA Constructs
The ICP0-WT plasmid (pRS-1) was a generous gift of Rozanne Sandri-Goldin (Sandri-Goldin et al., 1987). This plasmid includes the entire ICP0 gene, with its promoter, cloned into pUC-18. The ICP0-M138 plasmid was created using a Quikchange site directed mutagenesis kit (Agilent) following the manufacturer’s protocol. The seed sequence for both predicted miR-138 binding sites was changed from 5′-ACACCAGC-3′ to 5′-ACAGCTGA-3′. The pre-miR-138 stem-loop was PCR amplified from human genomic DNA derived from SH-SY5Y cells, together with 200bpof the 5′ and 3′ flanking regions, and cloned into pcDNA3 to generate pcDNA-miR138for cotransfection assays. The same region of human DNA was also cloned into the pLEX lentiviral vector for production of lentiviral particles by transfection of293 cells. Sequences of primers used are provided in Supplemental Experimental Procedures.
PAR-CLIP library construction and bioinformatics
293 cells were transduced with either the control, parental pLEX lentiviral vector (OpenBiosystems), lacking an inserted pri-miRNA sequence, or the same vector containing the pri-miR-138 sequence described above, and transductants selected using puromyc in. The selected cells were pulsed with 4SU 16 h prior to infection, then infected with HSV-1 at an MOI of 2 for 4 h, UV cross-linked and collected for PAR-CLIP. The PAR-CLIP library was constructed as described previously (Hafner et al., 2010) with minor modifications, i.e., an Illumina TrueSeq small RNA kit was used for cDNA preparation and an Illumina HiSeq 2000 was used for sequencing. Resulting reads were then processed using the FASTX-toolkit and aligned to the HSV-1 genome using Bowtie (Langmead et al., 2009). Reads of at least 15 nt in length, with minimum 50 reads total, were retained if they mapped to HSV-1 with ≤2 mismatches.
Transfection
For co-transfection using a miR-138 mimic, 293T cells in each well of a 24-well plate were co-transfected with 75 ng of ICP0 (WT) plasmid and 4 pmol of miScript miR-138 or miR-M138 mimic (Qiagen). Cells were harvested at 24 hpi. For co-transfection using a miR-138 expression plasmid, 293T cells in each well of a 6-well plate were co-transfected with 0.5 μg of ICP0 (WT or M138) plasmid and 2.5 μg of pcDNA-miR138, with an additional 1 μg of GFP expression plasmid, which served as a transfection and loading control. Cells were harvested at 48 h post transfection. For transfection-infection, 293T cells in each well of a 24-well plate were transfected with 8 pmol of the miR-138 or miR-M138 mimic with or without 16 pmol of miScript miR-138 inhibitor or a control scrambled inhibitor (Qiagen). After 8 h, the cells were infected with virus at an MOI of 0.1 and lysed for analysis at 48 hpi. All transfection procedures used Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol.
Western blotting
Western blots were performed as previously described (Pan and Coen, 2012b). The following antibodies and dilutions were used for probing the blots: ICP0 mouse monoclonal antibody (Virusys), 1:8000, GFP monoclonal (Santa Cruz), 1:20,000, ICP4 mouse monoclonal (Abcam), 1:1000, ICP27 mouse monoclonal (Virusys), 1:1000, TK mouse monoclonal (a generous gift from Bill Summers, Yale University), 1:1000, gC mouse monoclonal (Fitzgerald), 1:1000, actin antibody (Sigma), 1:10,000, and HRP-conjugated goat anti-mouse antiserum (SouthernBiotech), 1:2000. Images were visualized by either using a Syngene G:Box gel imaging system or film.
Animal procedures
Animal housing and experimental procedures were approved by the Institutional Animal Care and Use Committee of Harvard Medical School in accordance with federal guidelines. Male CD-1 mice (Charles River Laboratories) were anesthetized by intraperitoneal injection of 100 mg/kg ketamine (Fort Dodge Animal Health) and 10 mg/kg xylazine (Lloyd Laboratories). 2×105 pfu of virus in 3 μl was dropped onto each scarified cornea of 7 week old mice. For eye swab collection mice were anesthetized transiently in an induction chamber with isoflurane (3% in oxygen 0.5 ml/min) using an anesthesia machine (Colonial Medical Supply). Both eyes of each mouse were swabbed with cotton-tipped applicators, which were suspended in 1 ml of cell culture media. For TG or brain acquisition mice were sacrificed by cervical dislocation while under inhalation anesthesia with isoflurane as above, and the TG or brain were rapidly removed and placed on dry ice before storing at −80°C. TG and brain were homogenized in cell culture media for titers. Mice were observed daily, and mortality figures include mice that had died and one that was moribund and therefore euthanized. At 17 and 25 dpi each mouse was scored for its mobility (partial paralysis or lethargy). All of these mice were recovering by 32 dpi.
Statistical analysis
Statistical analysis was performed using Prism 6 (GraphPad). Titers and DNA and RNA levels were analyzed by Kruskal-Wallis tests for all virus genotypes, along with Dunn’s post tests for M138 vs WT and M138 vs. R138 (controlling for multiple comparisons). Survival curves were analyzed by the log-rank (Mantel-Cox) test. The number of mice with encephalitis sequelae at 17 and 25 dpi or of TG with detectable transcripts at 32 dpi were analyzed by Fisher’s Exact test. The covariance of transcript levels was evaluated using Pearson correlations.
Supplementary Material
Highlights.
miR-138 is a neuron-specific microRNA.
miR-138 represses expression of the HSV-1 lytic gene transactivator, ICP0.
Mutating miR-138 target sites in ICP0 causes higher lytic gene expression in neurons.
Mutating these miR-138 sites results in increased host morbidity and mortality.
Acknowledgments
We thank Seamus McCarron for technical assistance, Angus Wilson and Michael O’Brien for kindly providing total RNA from rat SCG neurons, Jeremy Kamil and Igor Jurak for advice on the mutagenesis strategy, Martha Kramer for plasmids for transcribing ICP27 and gC mRNA standards, John Carbone for designing the ICP27 primers for qRT-PCR, David Knipe for helpful discussions, and funding from the National Institutes of Health (P01 NS035138, R21 AI105896, and P01 AI098681 (DMC). P01 AI098681 (DAL), R01 AI0973376 (BRC). an NCI training grant T32 CA009111 (OF), and the Geisel School of Medicine Microbiology and Molecular Pathogenesis Program Training Grant, T32 AI007519 (PCR).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Banerjee S, Neveu P, Kosik KS. A coordinated local translational control point at the synapse involving relief from silencing and MOV10 degradation. Neuron. 2009;64:871–884. doi: 10.1016/j.neuron.2009.11.023. [DOI] [PubMed] [Google Scholar]
- Boutell C, Everett RD. Regulation of alphaherpesvirus infections by the ICP0 family of proteins. J Gen Virol. 2013;94:465–481. doi: 10.1099/vir.0.048900-0. [DOI] [PubMed] [Google Scholar]
- Boutell C, Sadis S, Everett RD. Herpes simplex virus type 1 immediate-early protein ICP0 and its isolated RING finger domain act as ubiquitin E3 ligases in vitro. J Virol. 2002;76:841–850. doi: 10.1128/JVI.76.2.841-850.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai W, Astor TL, Liptak LM, Cho C, Coen DM, Schaffer PA. The herpes simplex virus type 1 regulatory protein ICP0 enhances virus replication during acute infection and reactivation from latency. J Virol. 1993;67:7501–7512. doi: 10.1128/jvi.67.12.7501-7512.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai W, Schaffer PA. Herpes simplex virus type 1 ICP0 regulates expression of immediate-early, early, and late genes in productively infected cells. J Virol. 1992;66:2904–2915. doi: 10.1128/jvi.66.5.2904-2915.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carthew RW, Sontheimer EJ. Origins and mechanisms of miRNAs and siRNAs. Cell. 2009;136:642–655. doi: 10.1016/j.cell.2009.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Silverstein S. Herpes simplex viruses with mutations in the gene encoding ICP0 are defective in gene expression. J Virol. 1992;66:2916–2927. doi: 10.1128/jvi.66.5.2916-2927.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SH, Kramer MF, Schaffer PA, Coen DM. A viral function represses accumulation of transcripts from productive-cycle genes in mouse ganglia latently infected with herpes simplex virus. J Virol. 1997;71:5878–5884. doi: 10.1128/jvi.71.8.5878-5884.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SH, Lee LY, Garber DA, Schaffer PA, Knipe DM, Coen DM. Neither LAT nor open reading frame P mutations increase expression of spliced or intron-containing ICP0 transcripts in mouse ganglia latently infected with herpes simplex virus. J Virol. 2002;76:4764–4772. doi: 10.1128/JVI.76.10.4764-4772.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cliffe AR, Knipe DM. Herpes simplex virus ICP0 promotes both histone removal and acetylation on viral DNA during lytic infection. J Virol. 2008;82:12030–12038. doi: 10.1128/JVI.01575-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen BR. Viruses and microRNAs: RISCy interactions with serious consequences. Genes Dev. 2011;25:1881–1894. doi: 10.1101/gad.17352611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferenczy MW, DeLuca NA. Epigenetic modulation of gene expression from quiescent herpes simplex virus genomes. J Virol. 2009;83:8514–8524. doi: 10.1128/JVI.00785-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferenczy MW, DeLuca NA. Reversal of heterochromatic silencing of quiescent herpes simplex virus type 1 by ICP0. J Virol. 2011;85:3424–3435. doi: 10.1128/JVI.02263-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores O, Nakayama S, Whisnant AW, Javanbakht H, Cullen BR, Bloom DC. Mutational inactivation of herpes simplex virus 1 microRNAs identifies viral mRNA targets and reveals phenotypic effects in culture. J Virol. 2013;87:6589–6603. doi: 10.1128/JVI.00504-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garber DA, Schaffer PA, Knipe DM. A LAT-associated function reduces productive-cycle gene expression during acute infection of murine sensory neurons with herpes simplex virus type 1. J Virol. 1997;71:5885–5893. doi: 10.1128/jvi.71.8.5885-5893.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordani NV, Neumann DM, Kwiatkowski DL, Bhattacharjee PS, McAnany PK, Hill JM, Bloom DC. During herpes simplex virus type 1 infection of rabbits, the ability to express the latency-associated transcript increases latent-phase transcription of lytic genes. J Virol. 2008;82:6056–6060. doi: 10.1128/JVI.02661-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu H, Liang Y, Mandel G, Roizman B. Components of the REST/CoREST/histone deacetylase repressor complex are disrupted, modified, and translocated in HSV-1-infected cells. Proc Natl Acad Sci U S A. 2005;102:7571–7576. doi: 10.1073/pnas.0502658102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu H, Roizman B. Herpes simplex virus-infected cell protein 0 blocks the silencing of viral DNA by dissociating histone deacetylases from the CoREST-REST complex. Proc Natl Acad Sci U S A. 2007;104:17134–17139. doi: 10.1073/pnas.0707266104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunasekharan V, Laimins LA. Human papillomaviruses modulate microRNA 145 expression to directly control genome amplification. J Virol. 2013;87:6037–6043. doi: 10.1128/JVI.00153-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafezi W, Lorentzen EU, Eing BR, Muller M, King NJ, Klupp B, Mettenleiter TC, Kuhn JE. Entry of herpes simplex virus type 1 (HSV-1) into the distal axons of trigeminal neurons favors the onset of nonproductive, silent infection. PLoS Pathog. 2012;8:e1002679. doi: 10.1371/journal.ppat.1002679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M, Jr, Jungkamp AC, Munschauer M, et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 2010;141:129–141. doi: 10.1016/j.cell.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halford WP, Schaffer PA. ICP0 is required for efficient reactivation of herpes simplex virus type 1 from neuronal latency. J Virol. 2001;75:3240–3249. doi: 10.1128/JVI.75.7.3240-3249.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill JM, Zhao Y, Clement C, Neumann DM, Lukiw WJ. HSV-1 infection of human brain cells induces miRNA-146a and Alzheimer-type inflammatory signaling. Neuroreport. 2009;20:1500–1505. doi: 10.1097/WNR.0b013e3283329c05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Wang F, Argyris E, Chen K, Liang Z, Tian H, Huang W, Squires K, Verlinghieri G, Zhang H. Cellular microRNAs contribute to HIV-1 latency in resting primary CD4+ T lymphocytes. Nat Med. 2007;13:1241–1247. doi: 10.1038/nm1639. [DOI] [PubMed] [Google Scholar]
- Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science. 2005;309:1577–1581. doi: 10.1126/science.1113329. [DOI] [PubMed] [Google Scholar]
- Jurak I, Kramer MF, Mellor JC, van Lint AL, Roth FP, Knipe DM, Coen DM. Numerous conserved and divergent microRNAs expressed by herpes simplex viruses 1 and 2. J Virol. 2010;84:4659–4672. doi: 10.1128/JVI.02725-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurak I, Silverstein LB, Sharma M, Coen DM. Herpes simplex virus is equipped with RNA- and protein-based mechanisms to repress expression of ATRX, an effector of intrinsic immunity. J Virol. 2012;86:10093–10102. doi: 10.1128/JVI.00930-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalamvoki M, Roizman B. Circadian CLOCK histone acetyl transferase localizes at ND10 nuclear bodies and enables herpes simplex virus gene expression. Proc Natl Acad Sci U S A. 2010;107:17721–17726. doi: 10.1073/pnas.1012991107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knickelbein JE, Khanna KM, Yee MB, Baty CJ, Kinchington PR, Hendricks RL. Noncytotoxic lytic granule-mediated CD8+ T cell inhibition of HSV-1 reactivation from neuronal latency. Science. 2008;322:268–271. doi: 10.1126/science.1164164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knipe DM, Cliffe A. Chromatin control of herpes simplex virus lytic and latent infection. Nat Rev Microbiol. 2008;6:211–221. doi: 10.1038/nrmicro1794. [DOI] [PubMed] [Google Scholar]
- Kolb G, Kristie TM. Association of the cellular coactivator HCF-1 with the Golgi apparatus in sensory neurons. J Virol. 2008;82:9555–9563. doi: 10.1128/JVI.01174-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer MF, Chen SH, Knipe DM, Coen DM. Accumulation of viral transcripts and DNA during establishment of latency by herpes simplex virus. J Virol. 1998;72:1177–1185. doi: 10.1128/jvi.72.2.1177-1185.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer MF, Coen DM. Quantification of transcripts from the ICP4 and thymidine kinase genes in mouse ganglia latently infected with herpes simplex virus. J Virol. 1995;69:1389–1399. doi: 10.1128/jvi.69.3.1389-1399.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer MF, Jurak I, Pesola JM, Boissel S, Knipe DM, Coen DM. Herpes simplex virus 1 microRNAs expressed abundantly during latent infection are not essential for latency in mouse trigeminal ganglia. Virology. 2011;417:239–247. doi: 10.1016/j.virol.2011.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krummenacher C, Zabolotny JM, Fraser NW. Selection of a nonconsensus branch point is influenced by an RNA stem-loop structure and is important to confer stability to the herpes simplex virus 2-kilobase latency-associated transcript. J Virol. 1997;71:5849–5860. doi: 10.1128/jvi.71.8.5849-5860.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafaille FG, Pessach IM, Zhang SY, Ciancanelli MJ, Herman M, Abhyankar A, Ying SW, Keros S, Goldstein PA, Mostoslavsky G, et al. Impaired intrinsic immunity to HSV-1 in human iPSC-derived TLR3-deficient CNS cells. Nature. 2012;491:769–773. doi: 10.1038/nature11583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landgraf P, Rusu M, Sheridan R, Sewer A, Iovino N, Aravin A, Pfeffer S, Rice A, Kamphorst AO, Landthaler M, et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell. 2007;129:1401–1414. doi: 10.1016/j.cell.2007.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leib DA, Coen DM, Bogard CL, Hicks KA, Yager DR, Knipe DM, Tyler KL, Schaffer PA. Immediate-early regulatory gene mutants define different stages in the establishment and reactivation of herpes simplex virus latency. J Virol. 1989;63:759–768. doi: 10.1128/jvi.63.2.759-768.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luker GD, Prior JL, Song J, Pica CM, Leib DA. Bioluminescence imaging reveals systemic dissemination of herpes simplex virus type 1 in the absence of interferon receptors. J Virol. 2003;77:11082–11093. doi: 10.1128/JVI.77.20.11082-11093.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis TP, Sedarati F, Dobson AT, Feldman LT, Stevens JG. Pathways of viral gene expression during acute neuronal infection with HSV-1. Virology. 1992;189:150–160. doi: 10.1016/0042-6822(92)90690-q. [DOI] [PubMed] [Google Scholar]
- Melroe GT, DeLuca NA, Knipe DM. Herpes simplex virus 1 has multiple mechanisms for blocking virus-induced interferon production. J Virol. 2004;78:8411–8420. doi: 10.1128/JVI.78.16.8411-8420.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendell JT, Olson EN. MicroRNAs in stress signaling and human disease. Cell. 2012;148:1172–1187. doi: 10.1016/j.cell.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mossman KL, Saffran HA, Smiley JR. Herpes simplex virus ICP0 mutants are hypersensitive to interferon. J Virol. 2000;74:2052–2056. doi: 10.1128/jvi.74.4.2052-2056.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulik S, Xu J, Reddy PB, Rajasagi NK, Gimenez F, Sharma S, Lu PY, Rouse BT. Role of miR-132 in angiogenesis after ocular infection with herpes simplex virus. Am J Pathol. 2012;181:525–534. doi: 10.1016/j.ajpath.2012.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachmani D, Lankry D, Wolf DG, Mandelboim O. The human cytomegalovirus microRNA miR-UL112 acts synergistically with a cellular microRNA to escape immune elimination. Nat Immunol. 2010;11:806–813. doi: 10.1038/ni.1916. [DOI] [PubMed] [Google Scholar]
- Obernosterer G, Leuschner PJ, Alenius M, Martinez J. Post-transcriptional regulation of microRNA expression. RNA. 2006;12:1161–1167. doi: 10.1261/rna.2322506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orzalli MH, DeLuca NA, Knipe DM. Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Proc Natl Acad Sci U S A. 2012;109:E3008–3017. doi: 10.1073/pnas.1211302109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paladino P, Collins SE, Mossman KL. Cellular localization of the herpes simplex virus ICP0 protein dictates its ability to block IRF3-mediated innate immune responses. PLoS One. 2010;5:e10428. doi: 10.1371/journal.pone.0010428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan D, Coen DM. Net -1 frameshifting on a noncanonical sequence in a herpes simplex virus drug-resistant mutant is stimulated by nonstop mRNA. Proc Natl Acad Sci U S A. 2012a;109:14852–14857. doi: 10.1073/pnas.1206582109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan D, Coen DM. Quantification and analysis of thymidine kinase expression from acyclovir-resistant G-string insertion and deletion mutants in herpes simplex virus-infected cells. J Virol. 2012b;86:4518–4526. doi: 10.1128/JVI.06995-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesola JM, Zhu J, Knipe DM, Coen DM. Herpes simplex virus 1 immediate-early and early gene expression during reactivation from latency under conditions that prevent infectious virus production. J Virol. 2005;79:14516–14525. doi: 10.1128/JVI.79.23.14516-14525.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock DL, Nesburn AB, Ghiasi H, Ong J, Lewis TL, Lokensgard JR, Wechsler SL. Detection of latency-related viral RNAs in trigeminal ganglia of rabbits latently infected with herpes simplex virus type 1. J Virol. 1987;61:3820–3826. doi: 10.1128/jvi.61.12.3820-3826.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roizman B, Knipe DM, Whitley RJ. Herpes simplex virus. In: Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racianello VR, Roizman B, editors. Fields Virology. 6. Philadelphia: Lippincott Williams & Wilkins; 2013. pp. 1823–1897. [Google Scholar]
- Sandri-Goldin RM, Sekulovich RE, Leary K. The alpha protein ICP0 does not appear to play a major role in the regulation of herpes simplex virus gene expression during infection in tissue culture. Nucleic Acids Res. 1987;15:905–919. doi: 10.1093/nar/15.3.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel G, Obernosterer G, Fiore R, Oehmen M, Bicker S, Christensen M, Khudayberdiev S, Leuschner PF, Busch CJ, Kane C, et al. A functional screen implicates microRNA-138-dependent regulation of the depalmitoylation enzyme APT1 in dendritic spine morphogenesis. Nat Cell Biol. 2009;11:705–716. doi: 10.1038/ncb1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speck PG, Simmons A. Divergent molecular pathways of productive and latent infection with a virulent strain of herpes simplex virus type 1. J Virol. 1991;65:4001–4005. doi: 10.1128/jvi.65.8.4001-4005.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens JG, Wagner EK, Devi-Rao GB, Cook ML, Feldman LT. RNA complementary to a herpesvirus alpha gene mRNA is prominent in latently infected neurons. Science. 1987;235:1056–1059. doi: 10.1126/science.2434993. [DOI] [PubMed] [Google Scholar]
- Tal-Singer R, Lasner TM, Podrzucki W, Skokotas A, Leary JJ, Berger SL, Fraser NW. Gene expression during reactivation of herpes simplex virus type 1 from latency in the peripheral nervous system is different from that during lytic infection of tissue cultures. J Virol. 1997;71:5268–5276. doi: 10.1128/jvi.71.7.5268-5276.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang S, Bertke AS, Patel A, Wang K, Cohen JI, Krause PR. An acutely and latently expressed herpes simplex virus 2 viral microRNA inhibits expression of ICP34.5, a viral neurovirulence factor. Proc Natl Acad Sci U S A. 2008;105:10931–10936. doi: 10.1073/pnas.0801845105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang S, Patel A, Krause PR. Novel less-abundant viral microRNAs encoded by herpes simplex virus 2 latency-associated transcript and their roles in regulating ICP34.5 and ICP0 mRNAs. J Virol. 2009;83:1433–1442. doi: 10.1128/JVI.01723-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson RL, Sawtell NM. Evidence that the herpes simplex virus type 1 ICP0 protein does not initiate reactivation from latency in vivo. J Virol. 2006;80:10919–10930. doi: 10.1128/JVI.01253-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umbach JL, Kramer MF, Jurak I, Karnowski HW, Coen DM, Cullen BR. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature. 2008;454:780–783. doi: 10.1038/nature07103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Q, Li W, Tang Q, Yao S, Lv Z, Feng N, Ma X, Bai Z, Zeng Y, Qin D, et al. Cellular microRNAs 498 and 320d regulate herpes simplex virus 1 induction of Kaposi’s sarcoma-associated herpesvirus lytic replication by targeting RTA. PLoS One. 2013;8:e55832. doi: 10.1371/journal.pone.0055832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Q, McBride J, Fewell C, Lacey M, Wang X, Lin Z, Cameron J, Flemington EK. MicroRNA-155 is an Epstein-Barr virus-induced gene that modulates Epstein-Barr virus-regulated gene expression pathways. J Virol. 2008;82:5295–5306. doi: 10.1128/JVI.02380-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yordy B, Iijima N, Huttner A, Leib D, Iwasaki A. A neuron-specific role for autophagy in antiviral defense against herpes simplex virus. Cell Host Microbe. 2012;12:334–345. doi: 10.1016/j.chom.2012.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Lee MC, Momin A, Cendan CM, Shepherd ST, Baker MD, Asante C, Bee L, Bethry A, Perkins JR, et al. Small RNAs control sodium channel expression, nociceptor excitability, and pain thresholds. J Neurosci. 2010;30:10860–10871. doi: 10.1523/JNEUROSCI.1980-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SQ, Li YX, Zhang Y, Li X, Tang H. MiR-101 regulates HSV-1 replication by targeting ATP5B. Antiviral Res. 2011;89:219–226. doi: 10.1016/j.antiviral.2011.01.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.