

Abstract
The first examples of the use of crotylation as a stereocontrolled complex fragment coupling strategy are described. Asymmetric aldehyde isoprenylation provides access to 2-substituted-1,3-butadienes that may be subjected to highly regio- and stereoselective 1,4 hydrosilylation with trichlorosilane. After complexation with a chiral diamine, the 2-sub-stituted-cis-crotylsilanes may be employed in highly diastereoselective Sc(OTf)3-catalyzed aldehdye crotylation reactions.
Introduction
Polyketide natural products of the polypropionate/polyacetate type are often possessed of important biological activities, none more so than spongistatin 1. First reported nearly simultaneously by three research groups in 1993,1–3 this extraordinarily complex and exceedingly precious anti-mitotic agent has been shown to have an average IC50 value against the NCI panel of 60 human cancer cell lines of 0.12 nM.4 Not surprisingly, this confluence of scarcity and truly extraordinary potency has elicited an enormous amount of attention from synthetic chemists, and this has thus far resulted in seven groups reporting syntheses of spongistatin 1 and/or 2.5–11 Notably, the Smith group succeeded in synthesizing 1 gram of spongistatin 1 in 2008,7d an amount that is likely all the material that will ever be needed for pre-clinical development. The goal of turning spongistatin 1 or an analog thereof into a cancer drug remains an extremely daunting challenge, however, and will require the development of a synthesis that can both facilitate the rapid production of such analogs and ultimately deliver the quantities needed for clinical development.
Study design
For the synthesis of the ABCD half of spongistatin 1, the Felkin-selective anti-aldol approach pioneered by Evans5c stands as the preeminent fragment coupling method to join the AB and CD spiroketals, and as a means to install the stereocenters at C(15) and C(16) it could scarcely be more effective (Figure 1a). Conversely, it also entails the multi-step synthesis of a sensitive α-chiral-β,γ-unsaturated aldehyde as in 1, and, as we considered the prospect of performing such a fragment coupling on a tens of grams (or larger) scale, we were concerned that this approach might entail an unacceptable level of risk. Our charge was thus clear: develop a new fragment coupling strategy to install the entire C(13)-C(17) array that would require fewer, yet more easily scaled operations, while avoiding the intermediacy of sensitive functionality.
Fig. 1.

a The Evans aldol disconnection strategy for the coupling of the AB and CD spiroketals of spongistatin. b A proposed fragment coupling strategy by tandem crotylsilylation/Tamao oxidation/diastereoselective tautomerization.
To that end, we considered an alternative approach based on a tandem Sc(OTf)3-catalyzed crotylsilylation12/Tamao oxidation/diastereoselective tautomerization13 strategy that would join fragments such as 3 and 4 to produce the entire C(13)-C(17) array of functionality and stereochemistry in an efficient, stereoselective, and scalable way (Figure 1b). As exciting as this prospect seemed, however, its reduction to practice would be of little consequence unless equally efficient and scalable methods for the synthesis of 3 and 4 from simple prercursors could be developed. And while the silylformylation of homopropargylic alcohols such as 5 to produce aldehydes of type 4 is a well precedented14 and highly efficient process, the same cannot be said of the production of complex 2-substituted crotylmetal species such as 3. Indeed, no stereocontrolled complex fragment coupling crotylation has ever been demonstrated,15 largely due to the fact that there is no obvious way to make 2-substituted crotylboranes or boronates of this type.16 With the advent of our crotylsilane methodology,12 however, complex fragment coupling by crotylation might now be within reach due to Tsuji’s remarkably efficient and selective Pd(PPh3)4-catalyzed 1,4-hydrosilylation of 2-substituted-1,3-butadienes with HSiCl3.17 This leads back to diene 6, which we envisioned accessing with an asymmetric aldehyde isoprenylation reaction. Because it seemed possible that this proposed sequence might meet all of our criteria for a step-economical, scalable, and relatively risk- free fragment coupling strategy, we decided to pursue the development of this methodology and report herein the results of that study.
Results and discussion
The first task we faced was the identification or development of an efficient and scalable enantioselective aldehyde isoprenylation reaction. Two such methods are known,18,19 but as it was not clear that either was possessed of the type of practicality, scope, and ready scalability we desired, we decided to examine our strained silane platform in this context. Conversion of 2,3-dibromoprop-1-ene to 720 using the Furuya protocol21 was both efficient (83%) and highly scalable, and was followed by a Stille coupling22 with vinyltributyltin to give 8 in 55% yield (Scheme 1). Complexation with diamine 9 to produce isoprenylsilane (R,R)-10 was straightforward, though – because 10 was not crystalline – it was simply isolated as a solution in CH2Cl2 after filtration of the DBU•HCl salts. This solution could be used in concert with 5 mol% Sc(OTf)3 for the smooth and experimentally straightforward asymmetric isoprenylation of aldehydes. Thus, hydrocinnamaldehyde was converted to 11 in 95% yield and 94% ee, while the vastly more hindered and more relevant aldehyde 12 was converted to 13 in 78% yield (≥15:1 dr).23,24 While the synthesis of 10 requires some effort, we have found that it may readily be prepared on large (~80 g) scale and stored and used as needed, and the payoff in terms of the simplicity and scalability of the isoprenylation reaction more than compensates for this effort, especially with complex and expensive aldehydes.
Scheme 1.

Synthesis and reactions of isoprenylsilane 10.
With ready access to dienes such as 11 and 13 secured, we turned our attention to the diene hydrosilylation, diamine complexation and Sc(OTf)3-catalyzed fragment coupling crotylation reaction sequence. In contrast to the simple crotylation process wherein the crotylation reagent is prepared and isolated separately, neither the crotyltrichlorosilane nor the diamine-complexed crotylsilane may be easily purified when derived from a complex diene, and the efficiency of the fragment coupling crotylation reaction will depend on the cumulative efficiency of all three operations. Another concern at the outset was that we knew little about the compatibility of various functional and protecting groups with the hydrosilylation reaction conditions, and indeed, we quickly found that silyl ether protecting groups (TES, TBS) are cleaved when heated in the presence of HSiCl3. As a result, and because the AB spiroketal (i.e. 6) has two esters (the C(1) carboxyl group and the C(5) OAc group), we focused on the use of acetate groups for alcohol protection. Thus, acetate 14 was prepared and subjected to the Tsuji hydrosilylation reaction17 (Scheme 2). Although it proved necessary to heat this reaction for a reasonable reaction rate, the reaction was otherwise straightforward, efficient, and highly diastereoselective, affording 2-substituted-cis-crotyltrichlorosilane 15. After removal of all volatiles, 15 was treated with diamine 9 and DBU, and following removal of the DBU•HCl, 16 was obtained as an oil. This was immediately employed in the Sc(OTf)3-catalyzed crotylation reaction with dihydrocinnamaldehyde (1.0 equiv relative to 14), and following quench (with n-Bu4NF), workup and purification, 17 was isolated in 79% yield. 1H NMR analysis of the unpurified reaction mixture suggested the presence of small amounts of diastereomers, attributable to the fact that the 14 we used was 94% ee, and/or to a small amount of the diastereomer being produced in the crotylation event. Despite this, 17 was produced with ≥10:1 overall diastereoselectivity. The 3-stage process was repeated with aldehyde 12, leading to 1823 in 74% overall yield and establishing that complex aldehydes may be successfully employed. To establish that the diamine is the dominant source of stereocontrol, the sequence was then performed with the enantiomeric diamine (S,S)-9, and this led to 2023 in 79% yield. That this latter reaction was highly diastereoselective is significant in that it reflects the unique access to the syn-anti-syn array that our crotylsilane methodology provides,12 and in that the relative stereochemistry of 20 matches that of the C(14)-C(16) fragment of spongistatin 1. Finally, more complex diene 21 was subjected to the sequence with (R,R)-9 and aldehyde 12, giving complex polyketide-like array 2223 in 73% yield. These results establish that the entire sequence is reliable (73–79% yields consistently), and applicable in complex settings. This level of overall efficiency is remarkable (the average yield for the three operations is ~91–92%), and the entire sequence requires only a single purification by chromatography at the end.
Scheme 2.

Fragment coupling by crotylation.
The remaining outstanding question with respect to the application of this chemistry to a synthesis of the ABCD half of spongistatin 1 concerned the performance of aldehydes of type 4 in the fragment coupling crotylation process. Such aldehydes are highly deactivated both sterically and electronically, and it seemed prudent to confirm that they would be well behaved with complex crotylsilanes of type 16. For this purpose we employed diene 21, which was converted to crotylsilane 23 as above (Scheme 3). Simultaneously, homopropargylic alcohol 24 was silylated and then silylformylated to give aldehyde 25. Both 23 and 25 were taken directly into the Sc(OTf)3-catalyzed crotylation reaction, and to our delight, this sequence was as efficient as those described in Scheme 2, and led to the isolation of 26 in 76% yield (≥10:1 dr) from 21 and 24. To finish the model sequence for the projected spongistatin 1 AB and CD fragment coupling, 26 was subjected to a Tamao-type oxidation.25 Although the desired product 27 was obtained, the diastereoselectivity of the tautomerization event (at C(10)) was significantly lower (~2–3:1) than we had previously observed (~8:1) when using our “standard” oxidation conditions (H2O2, KF, MeOH, THF).12,13 This was unexpected as it directly implied that remote functionality and stereochemistry can have an impact on the selectivity. After some experimentation, we found that when the oxidation was performed with KHCO3 instead of KF, and with i-PrOH instead of MeOH/THF, the diastereoselectivity could be improved to 5:1. Using these conditions, 27 was isolated as a single diastereomer in 61% yield. Whether or not further optimization proves possible, the fact that the specific structure of the fragments has an impact suggests that such optimization studies will be most effectively carried out on the “real” spongistatin 1 fragments. For now, the 61% yield for the oxidation step – which both installs the C(11) ketone and establishes the C(10) stereocenter – constitutes a convincing proof of concept.
Scheme 3.

Fragment coupling by crotylation and Tamao oxidation/diastereoselective tautomerization.
It is perhaps worthwhile to evaluate in detail the overall transformation typified by the conversion of 21 and 24 to 27. In this series of chemical operations, a 2-substituted-1,3-butadiene, carbon monoxide, and a propynyl group are assembled into a complex array of a 1,1-disubstituted alkene, three stereocenters, and a ketone (Figure 2). Conceptually, as an example of the generation of valuable molecular complexity from truly simple building blocks, this transformation is exceptional. In terms of practicality and scalability, four of the six operations are carried out at ambient temperature, one at 0 °C and one at 70 °C (i.e. no cryogenic (−78 °C) steps). Three transition metal catalysts are employed in this sequence, and the entire list of required reagents is HSiCl3, H2O2, Ph2SiHCl, Et3N, DBU, n-Bu4NF, KHCO3, and (R,R)-9. All but the last of these are commercially available at nominal to moderate cost, and the last is routinely prepared in our lab ~300 g at a time by undergraduate researchers. Contrary to what may be a more or less widely held belief that the use of chiral controllers such as (R,R)-9 in stoichiometric quantities is problematic in practical terms, we can discern no objective justification for this viewpoint – (R,R)-9 is an abundant and inexpensive reagent whose use in stoichiometric quantities presents no significant practical problems, especially in the context of a complex fragment coupling process. We stress this point because if meaningful progress toward the extremely difficult but equally worthwhile goal of being able more rapidly and efficiently to generate large quantities of complex structures such as spongistatin 1 is to be made, it will be important not only to advance new conceptual frameworks at the strategic level, but also to focus on the actual efficiency-limiting problems at the practical level. In that regard, we note that the use of a high-pressure (~500 psi) carbonylation reaction, for example, is less than ideal in practical terms, and we continue to search for ways to obviate its use. Notwithstanding that and other less than ideal aspects of this sequence, we believe the chemistry described here represents both a new conceptual framework and a significant practical advance with respect to the rapid, efficient, and potentially scalable assemblage of complex and precious polyketide natural products such as spongistatin 1.
Fig. 2.
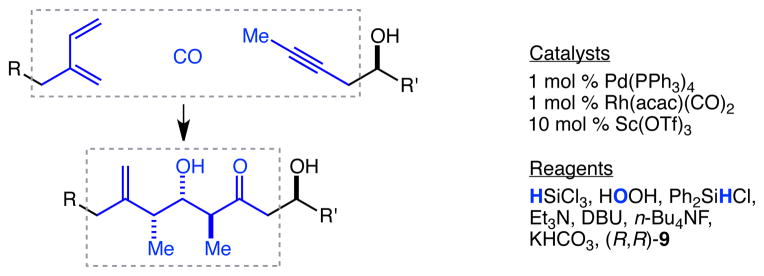
An evaluation of the transformation of a 1,3-butadiene, CO, and a propynyl group into a complex array comprised of a 1,1-disubstituted alkene, three stereocenters, and a ketone.
Conclusions
We have described the first examples of stereocontrolled complex fragment coupling by crotylation. The process is general, applicable in complex settings, and efficient, typically proceeding in 73–79% yield. As an alternative to fragment coupling by aldol addition,26 it may in some cases represent a more enabling tool for polyketide synthesis, and we note as well that the 1,1-disubstituted alkene product may, in principle, be diastereoselectively hydrogenated, epoxidized, or dihydroxylated.27 When applied in concert with the silylformylation and Tamao oxidation chemistry described in Scheme 3, the fragment coupling crotylation methodology can deliver complex polyketide arrays from simple building blocks in an experimentally straightforward, step-economical, and relatively practical fashion. The application of this and other28 new methodology to a step-economical and scalable synthesis and coupling of a diene-bearing AB spiroketal fragment such as 6 is being successfully pursued, and will be reported shortly.
Supplementary Material
Acknowledgments
This work was supported by a grant from the National Institute of General Medical Sciences (GM58133).
Footnotes
Electronic Supplementary Information (ESI) available: Experimental procedures and spectroscopic data. See DOI: 10.1039/b000000x/
References
- 1.(a) Pettit GR, Cichacz ZA, Gao F, Herald CL, Boyd MR, Schmidt JM, Hooper JNA. J Org Chem. 1993;58:1302–1304. [Google Scholar]; (b) Pettit GR, Cichacz ZA, Gao F, Herald CL, Boyd MR. J Chem Soc, Chem Commun. 1993:1166–1168. [Google Scholar]; (c) Pettit GR, Herald CL, Cichacz ZA, Gao F, Schmidt JM, Boyd MR, Christie ND, Boettner FE. J Chem Soc, Chem Commun. 1993:1805–1807. [Google Scholar]; (d) Pettit GR, Cichacz ZA, Herald CL, Gao F, Boyd MR, Schmidt JM, Hamel E, Bai R. J Chem Soc, Chem Commun. 1994:1605–1606. [Google Scholar]
- 2.Fusetani N, Shinoda K, Matsunaga S. J Am Chem Soc. 1993;115:3977–3981. [Google Scholar]
- 3.(a) Kobayashi M, Aoki S, Sakai H, Kawazoe K, Kihara N, Sasaki T, Kitagawa I. Tetrahedron Lett. 1993;34:2795–2798. [Google Scholar]; (b) Kobayashi M, Aoki S, Sakai H, Kihara N, Sasaki T, Kitagawa I. Chem Pharm Bull. 1993;41:989–991. doi: 10.1248/cpb.41.989. [DOI] [PubMed] [Google Scholar]; (c) Kobayashi M, Aoki S, Kitagawa I. Tetrahedron Lett. 1994;35:1243–1246. [Google Scholar]
- 4.Boyd MR. In: Principles and Practices of Oncology Updates. 3. DeVita VT Jr, Hellman S, Rosenberg SA, editors. Vol. 10. 1994. pp. 1–12. [Google Scholar]
- 5.(a) Evans DA, Coleman PJ, Dias LC. Angew Chem Int Ed. 1997;36:2738–2741. [Google Scholar]; (b) Evans DA, Trotter BW, Côté B, Coleman PJ. Angew Chem Int Ed. 1997;36:2741–2744. [Google Scholar]; (c) Evans DA, Trotter BW, Côté B, Coleman PJ, Dias LC, Tyler AN. Angew Chem Int Ed. 1997;36:2744–2747. [Google Scholar]; (d) Evans DA, Trotter BW, Coleman PJ, Côté B, Dias LC, Rajapakse HA, Tyler AN. Tetrahedron. 1999;55:8671–8726. [Google Scholar]
- 6.(a) Guo J, Duffy KJ, Stevens KL, Dalko PI, Roth RM, Hayward MM, Kishi Y. Angew Chem Int Ed. 1998;37:187–192. [Google Scholar]; (b) Hayward MM, Roth RM, Duffy KJ, Dalko PI, Stevens KL, Guo J, Kishi YY. Angew Chem Int Ed. 1998;37:192–196. [Google Scholar]
- 7.(a) Smith AB, III, Doughty VA, Lin Q, Zhuang L, McBriar MD, Boldi AM, Moser WH, Murase N, Nakayama K, Sobukawa M. Angew Chem Int Ed. 2001;40:191–195. doi: 10.1002/1521-3773(20010105)40:1<191::AID-ANIE191>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]; (b) Smith AB, III, Lin Q, Doughty VA, Zhuang L, McBriar MD, Kerns JK, Brook CS, Murase N, Nakayama K. Angew Chem Int Ed. 2001;40:196–199. [PubMed] [Google Scholar]; (c) Smith AB, III, Tomioka T, Risatti CA, Sperry JB, Sfouggatakis C. Org Lett. 2008;10:4359–4362. doi: 10.1021/ol801792k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Smith AB, III, Lin Q, Doughty VA, Zhuang L, McBriar MD, Kerns JK, Boldi AM, Murase N, Moser WH, Brook CS, Bennett CS, Nakayama K, Sobukawa M, Trout REL. Tetrahedron. 2009;65:6470–6488. doi: 10.1016/j.tet.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Smith AB, III, Sfouggatakis C, Risatti CA, Sperry JB, Zhu W, Doughty VA, Tomioka T, Gotchev DB, Bennett CS, Sakamoto S, Atasoylu O, Shirakami S, Bauer D, Takeuchi M, Koyanagi J, Sakamoto Y. Tetrahedron. 2009;65:6489–6509. doi: 10.1016/j.tet.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Paterson I, Chen IDY-K, Coster MJ, Aceña JL, Bach J, Gibson KR, Keown LE, Oballa RM, Trieselmann T, Wallace DJ, Hodgson AP, Norcross RD. Angew Chem Int Ed. 2001;40:4055–4060. [PubMed] [Google Scholar]; (b) Paterson I, Coster MJ, Chen DYK, Oballa RM, Wallace DJ, Norcross RD. Org Biomol Chem. 2005;3:2399–2409. doi: 10.1039/b504146e. [DOI] [PubMed] [Google Scholar]; (c) Paterson I, Coster MJ, Chen DYK, Gibson KR, Wallace DJ. Org Biomol Chem. 2005;3:2410–2419. doi: 10.1039/b504148a. [DOI] [PubMed] [Google Scholar]; (d) Paterson I, Coster MJ, Chen DYK, Aceña JL, Bach J, Keown LE, Trieselmann T. Org Biomol Chem. 2005;3:2420–2430. doi: 10.1039/b504149j. [DOI] [PubMed] [Google Scholar]; (e) Paterson I, Chen DYK, Coster MJ, Aceña JL, Bach J, Wallace DJ. Org Biomol Chem. 2005;3:2431–2440. doi: 10.1039/b504151a. [DOI] [PubMed] [Google Scholar]
- 9.Crimmins MT, Katz JD, Washburn DG, Allwein SP, McAtee LF. J Am Chem Soc. 2002;124:5661–5663. doi: 10.1021/ja0262683. [DOI] [PubMed] [Google Scholar]
- 10.Heathcock CH, McLaughlin M, Medina J, Hubbs JL, Wallace GA, Scott R, Claffey MM, Hayes CJ, Ott GR. J Am Chem Soc. 2003;125:12844–12849. doi: 10.1021/ja030317+. [DOI] [PubMed] [Google Scholar]
- 11.Ball M, Gaunt MJ, Hook DF, Jessiman AS, Kawahara S, Orsini P, Scolaro A, Talbot AC, Tanner HR, Yamanoi S, Ley SV. Angew Chem Int Ed. 2005;44:5433–5438. doi: 10.1002/anie.200502008. [DOI] [PubMed] [Google Scholar]
- 12.Kim H, Ho S, Leighton JL. J Am Chem Soc. 2011;133:6517–6520. doi: 10.1021/ja200712f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spletstoser JT, Zacuto MJ, Leighton JL. Org Lett. 2008;10:5593–5596. doi: 10.1021/ol802489w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Matsuda I, Ogiso A, Sato S, Izumi Y. J Am Chem Soc. 1989;111:2332–2333. [Google Scholar]; (b) Ojima I, Ingallina P, Donovan RJ, Clos N. Organometallics. 1991;10:38–41. [Google Scholar]; (c) Ojima I, Donovan RJ, Shay WR. J Am Chem Soc. 1992;114:6580–6582. [Google Scholar]; (d) Ojima I, Tzamarioudaki M, Tsai CY. J Am Chem Soc. 1994;116:3643–3644. [Google Scholar]; (e) Monteil F, Matsuda I, Alper H. J Am Chem Soc. 1995;117:4419–4420. [Google Scholar]; (f) Ojima I, Vidal E, Tzamarioudaki M, Matsuda I. J Am Chem Soc. 1995;117:6797–6798. [Google Scholar]; (g) Ojima I, McCullagh JV, Shay WR. J Organomet Chem. 1996;521:421–423. [Google Scholar]
- 15.Nakata and coworkers reported a fragment coupling by crotylation approach to the ABCD fragment of spongistatin 1, but the reaction was not stereocontrolled. See: Terauchi T, Tanaka T, Terauchi T, Morita M, Kimijima K, Sato I, Shoji W, Nakamura Y, Tsukada T, Tsunoda T, Hayashi G, Kanoh N, Nakata M. Tetrahedron Lett. 2003;44:7747–7751.
- 16.For the synthesis and use of 2-carboalkoxy-substituted crotylboronates, see: Kennedy JWJ, Hall DG. J Org Chem. 2004;69:4412–4428. doi: 10.1021/jo049773m.Yu S, Ferguson MJ, McDonald R, Hall DG. J Am Chem Soc. 2005;127:12808–12809. doi: 10.1021/ja054171l.Mitra S, Gurrala SR, Coleman RS. J Org Chem. 2007;72:8724–8736. doi: 10.1021/jo701415m.Elford TG, Hall DG. J Am Chem Soc. 2010;132:1488–1489. doi: 10.1021/ja9104478.
- 17.Tsuji J, Hara M, Ohno K. Tetrahedron. 1974;30:2143–2146. [Google Scholar]
- 18.Brown HC, Randad RS. Tetrahedron Lett. 1990;31:455–458. [Google Scholar]
- 19.Yu CM, Jeon M, Lee JY, Jeon J. Eur J Org Chem. 2001:1143–1148. [Google Scholar]
- 20.Nishiyama H, Yokoyama H, Narimatsu S, Itoh K. Tetrahedron Lett. 1982;23:1267–1270. [Google Scholar]
- 21.Furuya N, Sukawa T. J Organomet Chem. 1975;96:C1–C3. [Google Scholar]
- 22.Stille JK. Angew Chem Int Ed Engl. 1986;25:508–524. [Google Scholar]
- 23.These reactions are best quenched with n-Bu4NF which causes cleavage of the TES group (see aldehyde 12) and results in the isolation of the diol products. See the Supporting Information for details.
- 24.The isoprenylation reactions of three additional aldehydes are described in detail in the Supporting Information.
- 25.(a) Tamao K, Ishida N, Tanaka T, Kumada M. Organometallics. 1983;2:1694–1696. [Google Scholar]; (b) Jones GR, Landais Y. Tetrahedron. 1996;52:7599–7662. [Google Scholar]
- 26.(a) Evans DA, Dart MJ, Duffy JL, Rieger DL. J Am Chem Soc. 1995;117:9073–9074. [Google Scholar]; (b) Evans DA, Yang MG, Dart MJ, Duffy JL, Kim AS. J Am Chem Soc. 1995;117:9598–9599. [Google Scholar]; (c) Paterson I, Gibson KR, Oballa RM. Tetrahedron Lett. 1996;37:8585–8588. [Google Scholar]; (d) Evans DA, Coleman PJ, Côté B. J Org Chem. 1997;62:788–789. [Google Scholar]
- 27.Hoveyda AH, Evans DA, Fu GC. Chem Rev. 1993;93:1307–1370. [Google Scholar]
- 28.Chalifoux WA, Reznik SK, Leighton JL. Nature. 2012;487:86–89. doi: 10.1038/nature11189. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.