

Summary
Chemical or traumatic damage to the liver is frequently associated with aberrant healing(fibrosis) that overrides liver regeneration1–5. The mechanism by which hepatic niche cells differentially modulate regeneration and fibrosis during liver repair remains to be defined6–8. Hepatic vascular niche predominantly represented by liver sinusoidal endothelial cells (LSECs), deploys paracrine trophogens, known as angiocrine factors, to stimulate regeneration9–15. Nevertheless, it remains unknown how pro-regenerative angiocrine signals from LSECs is subverted to promote fibrosis16,17. Here, by combining inducible endothelial cell (EC)-specific mouse gene deletion strategy and complementary models of acute and chronic liver injury, we revealed that divergent angiocrine signals from LSECs elicit regeneration after immediateinjury and provoke fibrosis post chronic insult. The pro-fibrotic transition of vascular niche results from differential expression of stromal derived factor-1 (SDF-1) receptors, CXCR7 and CXCR418–21in LSECs. After acute injury, CXCR7 upregulation in LSECs acts in conjunction with CXCR4 to induce transcription factor Id1, deploying pro-regenerative angiocrine factors and triggering regeneration. Inducible deletion of Cxcr7 in adult mouse LSECs (Cxcr7iΔEC/iΔEC) impaired liver regeneration by diminishing Id1-mediated production of angiocrine factors9–11. By contrast, after chronic injury inflicted by iterative hepatotoxin (carbon tetrachloride) injection and bile duct ligation, constitutive FGFR1 signaling in LSECs counterbalanced CXCR7-dependent pro-regenerative response and augmented CXCR4 expression. This predominance of CXCR4 over CXCR7 expression shifted angiocrine response of LSECs, stimulating proliferation of desmin+hepatic stellate-like cells22,23 and enforcing a pro-fibrotic vascular niche. EC-specific ablation of either Fgfr1 (Fgfr1iΔEC/iΔEC) or Cxcr4 (Cxcr4iΔEC/iΔEC) in mice restored pro-regenerative pathway and prevented FGFR1-mediated maladaptive subversion of angiocrine factors. Similarly, selective CXCR7 activation in LSECs abrogated fibrogenesis. Thus, we have demonstrated that in response to liver injury, differential recruitment of pro-regenerative CXCR7/Id1 versus pro-fibrotic FGFR1/CXCR4 angiocrine pathways in vascular niche balances regeneration and fibrosis. These results provide a therapeutic roadmap to achieve hepatic regeneration without provoking fibrosis1,2,4.
Despite the liver's capacity to undergo regeneration, chronic or overwhelming injury often causes liver fibrosis that culminates in cirrhosis and hepatic failure1–7. The integrated process of liver repair includes regeneration and wound healing characterized by synthesis of extracellular matrix (ECM) proteins. Both processes are modulated by dynamic interplay between parenchymal hepatocytes and non-parenchymal cells (NPCs)7,22,24,25, including hepatic stellate cells1,23, inflammatory cells6,8, biliary epithelial cells, and liver sinusoidal endothelial cells (LSECs)9,13,15,16. As such, defining the multi-cellular crosstalk that balances regeneration and dysfunctional (maladaptive) healing5holds promise to design treatment for liver diseases.
LSECs that line liver sinusoidal vasculature induce hepatic organogenesis in manners that extend beyond their passive role for metabolite delivery9,12,14,15. By deploying paracrine growth regulators, which we have defined as angiocrine factors, LSECs trigger regeneration of hepatocytes9–11,25,26. However, aberrant activation of LSECs in the contextof chronic injury provokes fibrosis16,17. This dichotomy of LSEC niche function in mediating liver repair implicates that divergent angiocrine signals balance regeneration and fibrosis11. Therefore, we sought to decipher the mechanisms that subvert pro-regenerative capacity of LSECs to a pro-fibrotic state.
In response to tissue injury, cytokines and chemokines, such as stromal-derived factor (SDF)-1 (Cxcl12), are upregulated to initiate regeneration via its receptors CXCR4 and CXCR718–21. While CXCR4 activation in both hematopoietic and vascular cells modulates angiogenesis and hematopoiesis, expression of another SDF-1 receptor CXCR7 is mainly restricted to endothelial cells (ECs), with its function primarily believed to be pivotal in vascular patterning and tumor neo-angiogenesis. However, elucidating the mechanism by which SDF-1 pathway orchestrates liver repair is hindered by the lack of cell type-specific genetic models in defined settings of liver injuries.
To unravel the divergent role of LSECs in modulating liver repair, we employed single injection of carbon tetrachloride (CCl4) and acetaminophen that cause acute liver injury, as well as chronic injury models of repeated CCl4 injection and bile duct ligation (BDL)(Fig. 1a). Notably, at day 2 after single CCl4 injury, CXCR7 was upregulated specifically in VE-cadherin+ LSECs (Fig. 1b-e,supplementary Fig. 1). By contrast, CXCR4 is broadly expressed by other cell types, with its expression remaining relatively stabl on LSECs after CCl4 injury. Therefore, we have identified CXCR7 as an inducible LSEC-specific SDF-1 receptor in response to liver injury.
Figure 1. After acute liver injury, upregulation of stromal derived factor (SDF)-1 receptor CXCR7 in liversinusoidal endothelial cells (LSECs) elicits angiocrine-mediated regeneration.
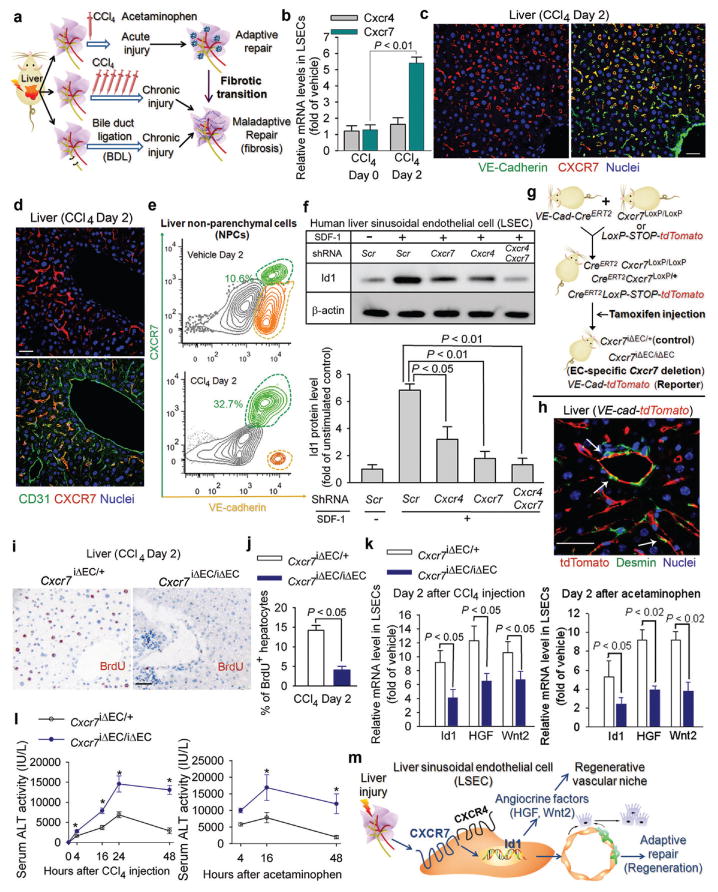
a) Liver injury models for studying the maladaptive transition of pro-regenerative LSEC function to a pro-fibrotic vascular niche.
b-e) CXCR7 is specifically upregulated on VE-cadherin+CD31+LSECs after acute chemical injury. After injection of carbon tetrachloride(CCl4), CXCR7 and CXCR4 were determined in isolated LSECs (b), liver sections (c, d), and non-parenchymal cells (NPCs) (e). CXCR7 was expressed on LSECs but not large vessels;N=5. Scale bar = 50 μm in Figure 1, all data hereafter are presented as mean ± standard error of mean (s.e.m.).
f) SDF-1 stimulation of human LSECs upregulates inhibitor of DNA binding 1 (Id1), a transcription factor inducing production of pro-regenerative angiocrine factors9. Id1 stimulation by SDF-1 in primary human Factor VIII+ LSECs was abrogated by silencing of Cxcr4and Cxcr7 in LSECs; N=5.
g, h) Endothelial cell (EC)-specific inducible deletion ofCxcr7(Cxcr7iΔEC/iΔEC) in mice. Mice harboring loxPsites-flanked Cxcr7were crossed with mouse line with EC-specific VE-cadherinpromoter-driven CreERT2(VE-Cad-CreERT2). Specificity of VE-Cad-CreERT2 was validated in reporter mice carrying tdTomato protein following floxed stop codon. Cxcr7 deletion or tdTomato expression in ECs was induced by tamoxifen injection9.Cxcr7iΔEC/+ mice served as control. Note the specific expression of tdTomato in ECs but not desmin+stellate-like cells (h, white arrows).
i-l) Impaired liver regeneration and enhanced liver injury inCxcr7iΔEC/iΔEC mice after acute liver injury. Cell proliferation was determined by staining for BrdU incorporation (i, j). Expression of Id1 and pro-regenerative angiocrine factors, hepatocyte growth factor (HGF) and Wnt2, in LSECswas measured after CCl4 and acetaminophen administration (k), and serum alanine aminotransferase (ALT) level was assessed to determine the degree of liver injury (l); N=5.
m) CXCR7 activation in LSECs triggers Id1-mediated production of pro-regenerative angiocrine fctors. After acute liver injury, CXCR7 cooperates with CXCR4, induces pro-regenerative Id1 pathway in LSECs, and triggers angiocrine-mediated liver regeneration.
Our group9,10 and others13 have shown that after partial hepatectomy, LSECs by producing angiocrine factors, such as Wnt2 and hepatocyte growth factor (HGF), elicit liver regeneration. Activation of transcription factor Id1 in LSECs was essential for this process9. SDF-1 induced Id1 upregulation in cultured human LSECs, which was abrogated by genetic silencingof either Cxcr7 or Cxcr4 (Fig. 1f,supplementary Fig. 2, 3). Notably, CXCR7-selective agonist TC14102 similarly induced Id1 upregulation. Immunoprecipitation-Western blot (IP-WB) demonstrated that after SDF-1 stimulation, CXCR7 was associated with CXCR4 and β-arrestin in LSECs(supplementary Fig. 4). Therefore, SDF-1 stimulates Id1 induction through enabling cooperation between CXCR7 and CXCR427,28.
To determine the contribution of CXCR7 in LSEC-mediated liver repair, we used a tamoxifen-inducible EC-specific CreERT2 system to knock down Cxcr7 in the ECs of adult mice (Fig. 1g). Mice harboring LoxP site-flanked Cxcr7 were crossed with VE-Cad-CreERT2 mice whereby EC-specific VE-cadherin promoter drivesCreERT2. Mice carrying tdTomato fluorescent protein following floxed stop codon was used to exclude off-target effects of VE-Cad-CreERT2 on other liver cell types. Tamoxifen injection specifically activated CreERT2 activity in ECs but not desmin-expressing stellate-like cells (Fig. 1h,supplementary Fig. 5), demonstrating induced EC-specific deletion of Cxcr7 (Cxcr7iΔEC/iΔEC) in VE-cad-CreERT2Cxcr7loxP/loxP mice. Cxcr7-haplodeficient adult mice (Cxcr7iΔEC/+) were used as control for Cre toxicity.
Compared to control mice, hepatocyte proliferation in Cxcr7iΔEC/iΔEC mice was significantly decreased after CCl4 injury (Fig. 1i ,j). Id1-dependent deployment of angiocrine factors HGF and Wnt2 from LSECs of Cxcr7iΔEC/iΔEC mice was reduced after both CCl4 and acetaminophen-induced liver injuries (Fig. 1k). The extent of liver injury as determined by serum alanine aminotransferase (ALT) level was exacerbated (Fig. 1l,supplementary Fig. 6). Thus, after liverinjury, SDF-1 via activation of CXCR7+LSECs triggers an angiocrine response to initiate liver regeneration (Fig. 1m).
Although hepatocytes regenerate after acute liver injury, chronic liver damage more frequently leads to overt activation of myofibroblasts and causes fibrosis2,3,5. To address how the pro-regenerative angiocrine signals of LSECs are diverted to provoke this maladaptive healing, we employed chronic mouse liver injury model of repeated CCl4injection29 (Fig. 2a, b). Notably, CXCR7-Id1 pathway in LSECs was counterbalanced by CXCR4 upregulation after chronic injury (Fig. 2c-e,supplementary Fig. 7, 8). After repeated CCl4 injection, protein levels of α-smooth muscle actin (SMA) and ECM protein collagen were augmented in Cxcr7iΔEC/iΔEC mice, compared to control mice (Fig. 2f-h,supplementary Fig. 9-10). Notably, injection of CXCR7-specific agonist TC14102 reduced the upregulation of SMA and collagen I in control but not Cxcr7iΔEC/iΔECmice (Fig. 2g-h,supplementary Fig. 11). Therefore, chronic liver injury interferes with pro-regenerative CXCR7-Id1 angiocrine pathway in LSECs and promotes fibrosis.
Figure 2. Iterative hepatotoxic injury perturbs CXCR7 pro-regenerative pathway in LSECs and forces the generation of a pro-fibrotic vascular niche.
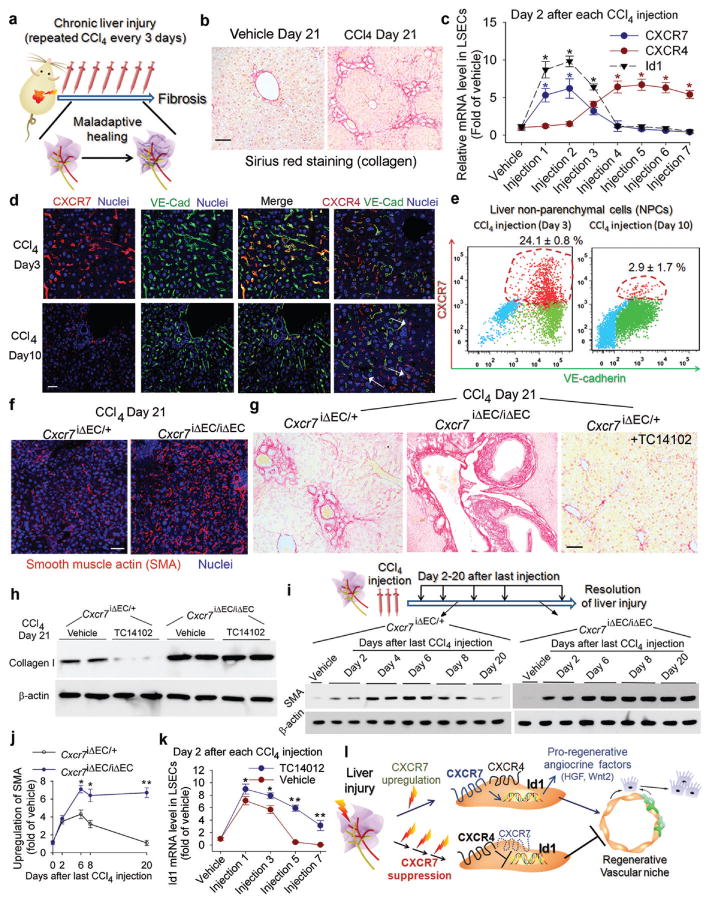
a-b) Mouse liver fibrosis is induced by repeated injection of CCl429. Sirius red staining was used to detect collagen in the injured liver. Scale br= 50 μm in Figure 2.
c-e) Chronic liver injury suppresses CXCR7 pathway and upregulates CXCR4 expression in LSECs. Quantitative PCR (c), immunostaining (d) and flow cytometry (e) showed the abrogationof CXCR7-Id1 pathway in VE-cadherin (VE-Cad)+ LSECs after chronic CCl4 injury. CXCR4 is expressed in both ECs and non-ECs (white arrow). *, P< 0.05 versus vehicle-treated mice; N=8.
f-h) CXCR7 activation in LSECs negates liver fibrosis. The extent of fibrosis was augmented in Cxcr7iΔEC/iΔEC mice, as evidenced by elevated hepatic levels of α-smooth muscle actin (SMA) and collagen I. Notably, CXCR7-selective agonist TC14012 reduced fibrosis in control but not Cxcr7iΔEC/iΔEC mice; N=7.
i, j) Impaired resolution of liver injury inCxcr7iΔEC/iΔEC mice. SMA level in the injured liver was tested to assess the resolution of injury (i). Compared to control mice, SMA level in Cxcr7iΔEC/iΔEC mice was enhanced after last CCl4 injection and remained stable afterwards. Collagen I level was similarly assessed (supplementary Figure 13); *, P < 0.05, **, P < 0.01, versus control mice; N=5.
k) CXCR7 activation restores Id1 induction in chronically injured LSECs. TC14102 prevented Id1 suppression in LSECs during repeated CCl4 injury. *, P < 0.05, **, P < 0.01, compared to vehicle group; N=7.
l) Interference with pro-regenerative CXCR7-Id1 pathway in LSECs causes pro-fibrotic transition of vascular niche. After injury, upregulation of the CXCR7-Id1 pathway in LSECs induces generation of hepatic-active angiocrine factors and stimulates regeneration. Chronic injury perturbs CXCR7-Id1 signaling, counteracting regeneration and provoking fibrogenesis.
The requirement of CXCR7 in LSECs in resolving liver fibrosis was tested6,23. After three CCl4 injections, SMA and collagen protein levels were enhanced in control mice and peaked at da 8 after last injection and approached basal (vehicle-injected group) level at day 20 (Fig. 2i-j,supplementary Fig. 12-13). By contrast, time-dependent resolution of liver injury was impaired in Cxcr7iΔEC/iΔEC mice. Id1 pathway inhibited by repeated injection of CCl4 was induced by CXCR7 agonist TC14102(Fig. 2k). Therefore, in response to injury, CXCR7 pathway in LSECs plays an indispensible role in stimulating regeneration and resolving fibrosis. After iterative stimuli, predominance of CXCR4 pathway over CXCR7-Id1 pathway leads to fibrosis (Fig. 2l).
We then employed bile duct ligation (BDL), a clinically relevant liver cholestasis model, to define how CXCR4 and CXCR7 modulate pro-fibrotic transition of LSECs (Fig. 3a). BDL induces biliary epithelial injury and causes continuous cholestasis an cirrhosis. After BDL, LSECs were invested by perisinusoidal desmin+stellate-likecells (Fig. 3b). Similar to repeated CCl4 injuries, there was temporal upregulation of CXCR4 and suppression of CXCR7-Id1 pathways in LSECs (Fig. 3c). Notably, SMA deposition in the liver of Cxcr7iΔEC/iΔECmice was higher than that of control Cxcr7iΔEC/+ mice (Fig. 3d-f). As such, loss of CXCR7 signaling in LSECs leads to fibrosis during BDL-induced cholestatic injury.
Figure 3. Cholestatic liver injury via FGFR1 overactivation in LSECs shifts CXCR7-dependent pro-regenerative response to a CXCR4-dominated pro-fibrotic vascular niche.
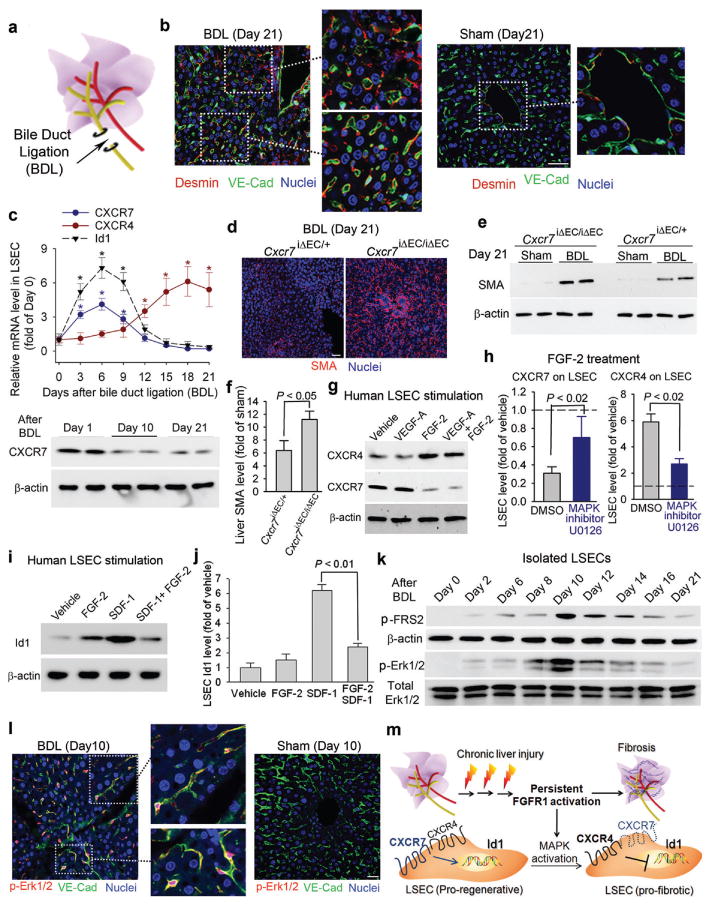
a, b) Pro-fibrotic transition of LSECs caused by bile duct ligation (BDL)-induced cholestatic injury. After BDL-induced cholestatic liver injury (a), majority of VE-cadherin+ LSECs were covered by perisinusoidal desmin+ fibroblasts (b, inset). By contrast, desmin+ stellate-like cells were sparsely distributed in sham-operated liver. Scale bar = 50 μm in Figure 3.
c) BDL suppresses CXCR7-Id1 pathway and upregulates CXCR4 expression in LSECs. Loss of CXCR7 protein in LSECs after BDL is shown at bottom panel; *, P < 0.05, compared with the level at day 0; N=5.
d-f) Liver fibrosis caused by BDL is exacerbated inCxcr7iΔEC/iΔECmice. After BDL, Cxcr7iΔEC/iΔEC mice exhibited higher hepatic level of SMA protein than that of control mice; N=4.
g-j) FGF-2 via MAP kinase activation favors CXCR4 signaling in LSECs, counteracting CXCR7-Id1 pathway. FGF-2, but not VEGF-A, upregulated CXCR4, suppressed CXCR7, and inhibited SDF-1-dependent Id1 induction in LSECs. This FGF-2-mediated predominance of CXCR4 over CXCR7 was attenuated by MAP kinase (MAPK) inhibitor U0126; N=5.
k, l) Activation of FGFR1 and MAPK pathway in LSECs after BDL. There was time-dependent enhancement in phosphorylation/activation of FGFR1 downstream effector FRS2 (p-FRS2) and Erk1/2 (p-Erk1/2) in VE-cadherin+ LSECs after BDL; N=6.
m) Constitutive FGFR1 signaling in LSECs via MAPK activation forces a CXCR4-dominated pro-fibrotic vascular niche. During chronic liver injury, FGFR1-mediated aberrant MAPK activation in LSECs upregulates CXCR4 and perturb CXCR7-Id1 pathway. This predominance of FGFR1/CXCR4 activation in LSECs determines the pathological progression from adaptive (pro-regenerative) to maladaptive (pro-fibrotic) liver repair.
Human LSECs were then stimulated with angiogenic factors VEGF-A and FGF-2 to investigate the mechanism whereby CXCR4 expression is enhanced to dominate over CXCR7 pathway. FGF-2, but notVEGF-A, induced CXCR4 mRNA and protein levels and attenuated CXCR7 expression (Fig. 3g,supplementary Fig. 14). Notably, specific inhibition of MAP Kinase (MAPK blocked FGF-2 driven CXCR4 induction and CXCR7 inhibition in LSECs11(Fig. 3h). As a result, treatment of human LSECs by FGF-2 in conjunction with SDF-1 blocked Id1 induction by SDF-1 alone (Fig. 3i-j,supplementary Fig. 15), suggesting that FGF-2 induced CXCR4 upregulation and CXCR7 suppression in LSECs negate Id1 pro-regenerative pathway.
To test how FGF-2 signaling modulates angiocrine response of LSECs, we examined the activation of the FGF-2 receptor FGFR1 on LSECs after BDL. There was a time-dependent upregulation andactivation of FGFR116,9,30 concomitant with phosphorylation of MAP kinase (Erk1/2) in the injured VE-cadherin+ LSECs (Fig. 3k, l,supplementary Fig. 16-17). Hence, cholestatic injury causes FGFR1-mediated MAPK activation in LSECs, resulting in CXCR4-dominated pro-fibrotic transition of angiocrine response during liver repair (Fig. 3m).
To elucidate the mechanism underlying the pro-fibrotic drift of LSEC niche, we conditionally ablated Fgfr1 and Cxcr4 in ECs of adult mice (Fgfr1iΔEC/iΔEC and Cxcr4iΔEC/iΔEC) (Fig. 4a). After BDL, perisinusoidal expansion of desmin+ cells, deposition of collagen and SMA, MAPK activation, and CXCR7 suppression in LSECs of Fgfr1iΔEC/iΔECmice were all attenuatedcompared to control mice (Fig. 4b-g,supplementary Fig. 18). Notably, CXCR7-dependent angiocrine expression of Wnt2 and HGF was restored in Fgfr1iΔEC/iΔEC mice (Fig. 4e). Therefore, EC-specific deletion of Fgfr1 in adult mice prevented the aberrant transition of LSECs into a pro-fibrotic state by BDL.
Figure 4. FGFR1 activation of CXCR4 in LSECs provokes pro-fibrotic angiocrine signals in liver repair.
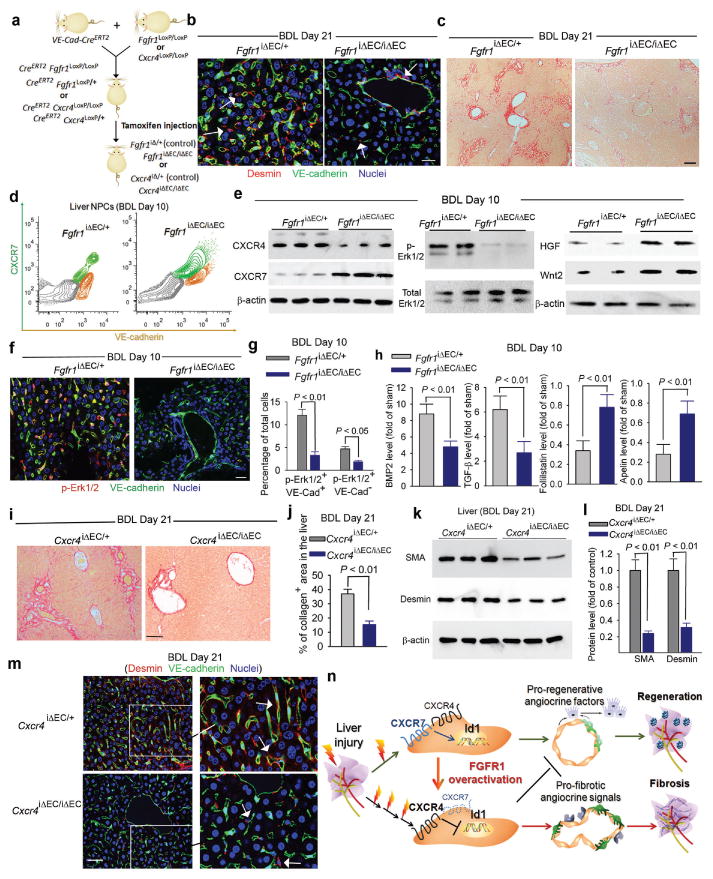
a) EC-specific inducible deletion ofFgfr1and Cxcr4(Fgfr1iΔEC/iΔECand Cxcr4 iΔEC/iΔEC) in adult mice.
b, c) Reduced liver fibrosis in Fgfr1iΔEC/iΔEC mice. Compared to control mice, perisinusoidal enrichment of desmin+ stellate-like cells (b, white arrow) and collagen deposition (c) were diminished in the liver of Fgfr1iΔEC/iΔEC mice after BDL; N=4; Scale bar = 50 μm in Figure 4.
d-g) EC-specific deletion of Fgfr1 in mice prevents CXCR4-mediated maladaptive transition after BDL and restores regenerative angiocrine signals. In BDL-injured LSECs of Fgfr1iΔEC/iEC mice, CXCR7 suppression and CXCR4 upregulation (d) and Erk1/2 activation in VE-cadherin+ LSECs (e-g) were reduced. This was accompanied by restored production of hepatic-active angiocrine factors HGF and Wnt2; N = 4.
h) Pro-fibrotic production of agiocrine factors in LSECs is reduced in Fgfr1iΔEC/iΔEC mice. BDL instigated divergent production of angiocrine factors in LSECs, including upregulation of factors in BMP andTGF-β pathways and suppression of anti-fibrotic genes such as follistatin and apelin (supplementary Fig. 19). This pro-fibrotic drift of angiocrine factor in LSECs after BDL was mitigated i Fgfr1iΔEC/iΔEC mice; N=4.
i-m) Reduction of liver fibrosis in Cxcr4iΔEC/iΔEC mice after BDL. The extent of liver fibrosis after BDL was significantly lower in Cxcr4iΔEC/iΔEC mice than that of control mice, as evidenced by decreased deposition of collagen (i, j),SMA (k, l), and perisinusoidal enrichment of desmin+ stellate-like cells (m, white arrow); N=5.
n) Divergent angiocrine signals from LSECs balance liver regeneration and fibrosis. After acute liver injury, activation of CXCR7-Id1 pathway in LSECsstimulates production of hepatic-active angiocrine factors. By contrast, chronic injury causes overt FGFR1 activation in LSECs that perturbs CXCR7-Id1 pathway and favors a CXCR4-driven pro-fibrotic angiocrine response, thereby provoking liver fibrosis. Therefore, in response to injury, differentially primed LSECs deploy divergent angiocrine signals to balance liver regeneration and fibrosis.
To unravel the altered angiocrine response in chronically injured LSECs, we isolated and analyzed LSECs from BDL and sham-operated mice (Supplementary Fig. 19). In injured LSECs, there was significant upregulation of pro-fibrotic factors, including TGF-β, BMP2, and PDGF-C, concomitant with suppression of anti-fibrotic genes, such as follistatin and apelin. This divergent drift of angiocrine factor production in LSECs after BDL was diminished in Fgfr1iΔEC/iΔEC mice, as evidenced by restoration of anti-fibrotic genes and reduced expression of fibrotic factors (Fig. 4h).
The extent of fibrosis after BDL was further tested in Cxcr4iΔEC/iΔEC mice. Compared to control Cxcr4iΔEC/+mice, hepatic deposition of SMA and collagen, and perisinusoidal accumulation of desmin+ stellate-like cells were reduced in Cxcr4iΔEC/iΔEC mice after BDL (Fig. 4i-m,supplementary Fig. 20). The reduction of liver fibrosis in Cxcr4iΔEC/iΔEC mice implicates that constitutive CXCR4 activation in LSECs by chronic injury establishes a pro-fibrotic vascular niche, activating adjacent myofibroblast cells and provoking fibrogenesis (Fig. 4n).
While liver regeneration after partial hepatectomy proceeds impeccably without fibrosis, liver repair after chronic injury is associated with fibrosis that compromises restoration of liver function. Therefore, identifying the molecular pathways modulating liver regeneration and aberrant healing will open up therapeutic avenues for treatment of liver cirrhosis and failure. We have shown that after 70% partial hepatectomy, activation of the VEGFR2-Id1 pathway in LSECs leads to liver regeneration9. Here, we demonstrate that FGFR1-mediated CXCR4 upregulation and CXCR7 suppression in LSECs counterbalance the pro-regenerative function of LSECs and lead to fibrosis.
We employed complementary acute and chronic injury models to decipher the contribution of LSECs in liver repair (Fig. 1a). In the mouse liver, we have identified a preferential induction of pro-regenerative CXCR7-dependent signaling in LSECs that responds to acute injury (Fig. 1). During chronic liver injury, loss of CXCR7 and upregulation of CXCR4 in LSECs causes progression to fibrosis. Indeed, the critical function of CXCR7 activation in promoting regeneration and counteracting fibrosis is evidenced by the attenuated fibrosis by selective activation of CXCR7 in LSECs, as well as impaired regeneration (Fig. 1) and enhanced fibrogenesis (Fig. 2–3) in Cxcr7iΔEC/iΔECmice.
The shift in SDF-1 signaling from the CXCR7-dependent pro-regenerative response to a CXCR4-dominated pro-fibrotic function in LSECs is due to persistent FGFR1 that drives chronic MAPK activation11,16,29,30 (Fig. 3). We utilized an inducible EC-specific mouse genetic deletion system to demonstrate the contribution of FGFR1-CXCR4 pathway in the pro-fibrotic drift of LSECs. Notably, after chronic liver injury, the enhanced CXCR4 expression relative to CXCR7 in LSECs was prevented in Fgfr1iΔEC/iΔEC mice, and both Fgfr1iΔEC/iΔEC and Cxcr4iΔEC/iΔECmice were resistant to fibrosis (Fig. 4). Therefore, activation of CXCR7-Id1 in LSECs upon injury triggers and safeguards production of hepatogenic angiocrine factors, while overt FGFR1-CXCR4 activation by chronic stimuli converts LSECs to a pro-fibrotic niche. This aberrant angiocrine function of LSECs by FGFR1-CXCR4 pathway causes activation and expansion of desmin+ stellate-like cells. Elucidating how hepatic stellate cells reciprocally modulate phenotypic and functional contributions of LSECs in liver repair remains to be investigated1,23. Moreover, whether activation of CXCR7 and CXCR4 on LSECs regulate inflammatory responses, such as recruitment of macrophages that could potentially modulate liver regeneration and fibrosis6,8, needs to be studied.
In summary, we have found that differentially activated LSECs supply divergent angiocrine signals for liver repair. Selective activation of CXCR7 in LSECs is instrumental in shepherding angiocrine-mediated regeneration. Perturbation of CXCR7 pathway by constitutive FGFR1 activation diverts SDF-1 signaling in LSECs to a maladaptive (pro-fibrotic) angiocrine response. Identifying molecular pathways orchestrating divergent angiocrine responses within the hepatic vascular niche willlay foundation for therapeutic strategy that ensures liver repair without causing fibrosis.
Methods
Endothelial cell (EC)-specific gene deletion strategy
Inducible EC-specific gene deletion was achieved out by treating VE-Cadherin-CreERT2 harboring mice with tamoxifen31. Cxcr4LoxP/loxP mice were previously described32. The Cre+ mice were treated with tamoxifen at a dose of 250 mg/kg i.p. for 6 days interrupted for 3 days after the third dose. After three weeks of tamoxifen treatment, deletion of target genes in LSECs was corroborated by quantitative PCR and immunoblot analysis. All animal experiments were carried out under the guidelines set by Institutional Animal Care and Use Committee, using sex/age/weight matched littermate animals.
Liver injury and fibrosis models
Single and repeated injections of CCl4were used to induce acute and chronic liver injuries, respectively, as previously described29. CCl4 was be diluted in oil to yield a final concentration of 40% (0.64 mg/ml) and injected into mice at 1.6 mg/g body weight. Eight-to 10-week-old mice were subjected to bile duct ligation (BDL). Acute liver injury was also induced in mice by i.p. injection of 400 mg/kg acetaminophen. To perform BDL, mice were subjected to a mid-abdominal incision 3 cm long, under general anesthesia. The common bile duct was ligated in two adjacent positions approximately 1 cm from the porta hepatis. The duct was then severed by incision between the two sites of ligation.
To selectively activate CXCR7, the agonist TC14102 (R&D Systems, MN) was intraperitoneally injected into the mice after CCl4injury or BDL injury every other day at 30 mg/kg. At indicated time points, mice were sacrificed and whole liver tissues were harvested for the analysis of fibrogenesis, including collagen I deposition by Sirius red staining and deposition of SMC and collagen I protein detected by immunoblot (Abcam, CA).
Isolation and culture of mouse liver cells
Liver cells were isolated from mice by a two-step collagenase perfusion technique with modifications, as previously described9. Briefly, the liver was perfused with Liver Perfusion Medium (Invitrogen, CA), and dissociated by Liver Digest Medium (Invitrogen, CA). The non-parenchymal cells (NPCs) were fractionated with percoll gradient centrifugation with 75% stock Percoll solution and 35% stock Percoll solution. LSEC faction was isolated by mouse LSEC binding magnetic beads (Miltenyi, CA) and Dynabeads® Magnetic Beads conjugated with anti mouse-VEGFR3 antibody (Imclone, NY)9. Expression of Id1, CXCR4, CXCR7, and FGFR1 protein and mRNA were determined from isolated LSECs11. For detection of FRS-2 phsophorylation in LSECs, mice were perfused with phosphatase inhibitor before harvesting the tissues (Pierce, CA)11.
Culture and stimulation of human LSECs
Human LSECs were obtained from Science Cell Inc. and cultured following vendor's instruction. The expression of CXCR7, VE-cadherin, vWF and factor VIII was validated by immunostaining or flow cytometric analysis. To selectively knockdown Cxcr4, Cxcr7 in LSECs, shRNA Lentiviruses were generated by cotransfecting 15 μg of shuttle lentiviral vector containing target gene or scrambled shRNA, 3 μg of pENV/VSV-G, 5 μg of pRRE, and 2.5 μg of pRSV-REV in 293T cells by the calcium precipitation method. Viral supernatants were concentrated by ultracentrifugation and used to transduce human LSECs.
To determine the expression of Id1, CXCR4, and CXCR7 in LSEC after cytokine stimulation, 500,000 LSECs were seeded and treated with Cxcr4, Cxcr7, or scrambled shRNA lentiviruses, respectively. After starving in serum-free medium, seeded LSECs were stimulated with 10 ng/ml SDF-1 or 20 ng/ml FGF-2. At various time points, cells were collected for the measurement of Id1 protein and mRNA expression. Treatment of 30 μM U0126 was used to inhibit the activity of MAPK. Activation of MAPK (p-Erk1/2) was assayed by immunoblot using antibodies against p-Erk1/2 and total Erk1/2 (Cell Signaling Technology, MA)11.
For immunoprecipitation (IP)-Western blot (WB), cell lysates were retrieved by RIPA lysis buffer with protease inhibitor舳cocktail舳and舳phosphatase (Pierce) and incubatedwith anti-CXCR7 antibody (R&D Systems, MN) conjugated with Protein A/G beads (Invitrogen, CA). Beads were retrieved by magnet, associated proteins were eluted, and the association of β-arrestin, CXCR4, and CXCR7 was determined by Western blot (Santa Cruz, CA), after normalization to total CXCR7 protein amounts in cell lysates (input).
Flow cytometric analysis of liver NPCs and LSECs
For flow cytometry analysis, retrieved livers from sacrificed animals were minced, digested in liver digestion medium (Invitrogen, CA), and filtered through a 30-μm strainer. Single-cell suspensions were preblocked with Fc block (CD16/CD32; BD Biosciences, CA) and then incubated with the following primary antibodies recognizing mouse LSECs and hematopoietic cells,: rat IgG2aκ and IgG2aβ isotype control; CD31/PECAM-1 (clone MEC 13.3, eBioscience, CA); VE-cadherin/CD144 (clone Bv13, eBioscience, CA); CXCR7 (clone 11G8, R&D Systems, MN). Usually, primary antibodies were directly conjugated to various Alexa Fluor dyes or Quantum Dots using antibody labeling kits (Invitrogen, CA) performed as per the manufacturer's instructions. In the case of Alexa Fluor 750, conjugations were performed using succinimidyl esters and purified over BioSpin P30 Gel (Bio-Rad).
Labeled cell populations were measured by a LSRII flow cytometer (Beckton Dickenson); compensation for multivariate experiments was performed with FACS Diva software. Flow cytometry analysis was performed using a variety of controls such as isotype antibodies, and unstained samples for determining appropriate gates, voltages, and compensations required in multivariate flow cytometry.
Immunostaining and histological analysis of liver cryosection
To harvest tissues for histological analysis, mice were perfused with 4% PFA, cryopreserved, and snap frozen in OCT. For immunofluorescent (IF) microscopy, the liver sections (10 μm) were blocked (5% donkey serum/0.3% Triton X-100) and incubated in primary Abs: anti-VE-cadherin polyclonal Ab (pAb, 2 μg/ml, R&D Systems, MN), anti-CD31 mAb (MEC13.3, 5 μg/ml, BD Biosciences, CA), anti-CXCR7 mAb (11G8, 5 μg/ml, R&D Systems, MN), anti-desmin (pAb, 2 μg/ml, Abcam, CA) and anti-p-Erk1/2 antibody (2 μg/ml, Cell Signaling Technology, MA). After incubation in fluorophore-conjugated secondary antibodies (2.5 μg/ml, Jackson ImmunoResearch, PA), sections were counterstained with TOPRO3 or DAPI (Invitrogen, CA).
Liver cell proliferation in vivowas measured by BrdU uptake. Briefly, mice received a single dose of BrdU (Sigma) intraperitoneally 60 minutes before death (50 mg/kg). Liver lobes were removed, weighed, and further processed. Slices were preincubated with 1 M HCl at room temperature for 1 h, neutralized with 10 mM Tris (pH 8.5) at room temperature for 15 min, and stained using the BrdU Detection System (BD Biosciences, CA) and fluorophore-conjugated secondary antibodies (2.5 μg/ml, Jackson ImmunoResearch, PA). For immunohistochemical (IHC) detection of BrdU, endogenous peroxidase and nonspecific protein block (5% BSA, 10% donkey serum, and 0.02% Tween-20) were performed on liver cryosections and incubated with secondary pAb and streptavidin horseradish peroxidase (Jackson ImmunoResearch, PA).
Image acquisition and analysis
IHC staining of liver slides was captured with Olympus BX51 microscope (Olympus America, NY), and fluorescent images were recorded on AxioVert LSM710 confocal microscope (Zeiss, NY). Co-staining of VE-cadherin with CXCR4 and CXCR7 was also determined.
Gene expression analysis by real-time polymerase chain reaction
Total RNA was extracted from cryopreserved liver tissue or isolated LSECs using RNeasy kit (Qiagen, CA). After isolation, 500 ng of total RNA was transcribed into cDNA by using the superscript reverse transcriptase Kit (Invitrogen, CA). The detection of cDNA expression for the specific genes was performed using quantitative polymerase chain reaction (Applied Biosystems).
Supplementary Material
Acknowledgments
We are grateful to Dr. Timothy Hla for critically reading the manuscript and Drs. Rob Berahovich and Kevin Eggan for their suggestions on CXCR7 signaling for liver repair. B.-S. D. was supported by National Scientist Development Grant from American Heart Association (#12SDG1213004) andDruckenmiller Fellowship from New York Stem Cell Foundation. S.R. is supported by Ansary Stem Cell Institute; Howard Hughes Medical Institute; Empire State Stem Cell Board and New York State Department of Health grants (NYSTEM, C024180, C026438, C026878); NHLBI R01s HL097797, DK095039 and HL119872; Qatar National Priorities Research Foundation NPRP08-663-3-140 and Qatar foundation BioMedical Research Program (BMRP).
Footnotes
Author information:The authors declare no competing financial interests.
Author contribution:B.-S. D. and Z. C. conceived the project, performed experiments, and wrote the paper. R. L., D.N., and P. G. carried out the experiments and analyzed the data. M.E.P, M.S., K.S., and S.Y.R. interpreted the data. S. R. designed the project, analyzed the data and wrote the paper. All authors commented on the manuscript.
References
- 1.Friedman SL, Sheppard D, Duffield JS, Violette S. Therapy for fibrotic diseases: nearing the starting line. Science translational medicine. 2013;5:161sr167. doi: 10.1126/scitranslmed.3004700. [DOI] [PubMed] [Google Scholar]
- 2.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Iredale JP. Models of liver fibrosis: exploring the dynamic nature of inflammation and repair i a solid organ. J Clin Invest. 2007;117:539–548. doi: 10.1172/JCI30542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gurtner GC, Werner S, Barrandon Y, Longaker MT. Wound repair and regeneration. Nature. 2008;453:314–321. doi: 10.1038/nature07039. [DOI] [PubMed] [Google Scholar]
- 5.Wynn TA. Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J Clin Invest. 2007;117:524–529. doi: 10.1172/JCI31487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Duffield JS, et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest. 2005;115:56–65. doi: 10.1172/JCI22675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Diehl AM. Neighborhood watch orchestrates liver regeneration. Nat Med. 2012;18:497–499. doi: 10.1038/nm.2719. [DOI] [PubMed] [Google Scholar]
- 8.Boulter L, et al. Macrophage-derived Wnt opposes Notch signaling to specify hepatic progenitor cell fate in chronic liver disease. Nat Med. 2012;18:572–579. doi: 10.1038/nm.2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ding BS, et al. Inductive angiocrine signals from sinusoidal endothelium are required for liver regeneration. Nature. 2010;468:310–315. doi: 10.1038/nature09493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ding BS, et al. Endothelial-derived angiocrine signals induce and sustain regenerative lung alveolarization. Cell. 2011;147:539–553. doi: 10.1016/j.cell.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kobayashi H, et al. Angiocrine factors from Akt-activated endothelial cells balance self-renewal and differentiation of haematopoietic stem cells. Nat Cell Biol. 2010;12:1046–1056. doi: 10.1038/ncb2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sakaguchi TF, Sadler KC, Crosnier C, Stainier DY. Endothelial signals modulate hepatocyte apicobasal polarization in zebrafish. Curr Biol. 2008;18:1565–1571. doi: 10.1016/j.cub.2008.08.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang L, et al. Liver sinusoidal endothelial cell progenitor cells promote liver regeneration in rats. J Clin Invest. 2012;122:1567–1573. doi: 10.1172/JCI58789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matsumoto K, Yoshitomi H, Rossant J, Zaret KS. Liver organogenesis promoted by endothelial cells prior to vascular function. Science. 2001;294:559–563. doi: 10.1126/science.1063889. [DOI] [PubMed] [Google Scholar]
- 15.LeCouter J, et al. Angiogenesis-independent endothelial protection of liver: role of VEGFR-1. Science. 2003;299:890–893. doi: 10.1126/science.1079562. [DOI] [PubMed] [Google Scholar]
- 16.Huebert RC, et al. Aquaporin-1 facilitates angiogenic invasion in the pathological neovasculature that accompanies cirrhosis. Hepatology. 2010;52:238–248. doi: 10.1002/hep.23628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zeisberg EM, et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med. 2007;13:952–961. doi: 10.1038/nm1613. [DOI] [PubMed] [Google Scholar]
- 18.Miao Z, et al. CXCR7 (RDC1) promotes breast and lung tumor growth in vivo and is expressed on tumor-associated vasculature. Proc Natl Acad Sci USA. 2007;104:15735–15740. doi: 10.1073/pnas.0610444104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu S, Crawford D, Tsuchihashi T, Behrens TW, Srivastava D. The chemokine receptor CXCR7 functions to regulate cardiac valve remodeling. Dev Dyn. 2011;240:384–393. doi: 10.1002/dvdy.22549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sierro F, et al. Disrupted cardiac development but normal hematopoiesis in mice deficient in the second CXCL12/SDF-1 receptor, CXCR7. Proc Natl Acad Sci USA. 2007;104:14759–14764. doi: 10.1073/pnas.0702229104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tachibana K, et al. The chemokine receptor CXCR4 is essential for vascularization of the gastrointestinal tract. Nature. 1998;393:591–594. doi: 10.1038/31261. [DOI] [PubMed] [Google Scholar]
- 22.Armulik A, Genove G, Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell. 2011;21:193–215. doi: 10.1016/j.devcel.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 23.Troeger JS, et al. Deactivation of hepatic stellate cells during liver fibrosis resolution in mice. Gastroenterology. 2012;143:1073–1083. doi: 10.1053/j.gastro.2012.06.036. e1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zaret KS, Grompe M. Generation and regeneration of cells of the liver and pancreas. Science. 2008;322:1490–1494. doi: 10.1126/science.1161431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Woo DH, et al. Direct and indirect contribution of human embryonic stem cell-derived hepatocyte-like cells to liver repair in mice. Gastroenterology. 2012;142:602–611. doi: 10.1053/j.gastro.2011.11.030. [DOI] [PubMed] [Google Scholar]
- 26.Hoehme S, et al. Prediction and validation of cell alignment along microvessels as order principle to restore tissue architecture in liver regeneration. Proc Natl Acad Sci USA. 2010;107:10371–10376. doi: 10.1073/pnas.0909374107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Decaillot FM, et al. CXCR7/CXCR4 heterodimer constitutively recruits beta-arrestin to enhance cell migration. J Biol Chem. 2011;286:32188–32197. doi: 10.1074/jbc.M111.277038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rajagopal S, et al. Beta-arrestin-but not G protein-mediated signaling by the “decoy” receptor CXCR7. Proc Natl Acad Sci USA. 2010;107:628–632. doi: 10.1073/pnas.0912852107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu C, et al. Role of fibroblast growth factor type 1 and 2 in carbon tetrachloride-induced hepatic injury and fibrogenesis. Am J Pathol. 2003;163:1653–1662. doi: 10.1016/S0002-9440(10)63522-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bohm F, et al. FGF receptors 1 and 2 control chemically induced injury and compound detoxification in regenerating livers of mice. Gastroenterology. 2010;139:1385–1396. doi: 10.1053/j.gastro.2010.06.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Y, et al. Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature. 2010;465:483–486. doi: 10.1038/nature09002. [DOI] [PubMed] [Google Scholar]
- 32.Nie Y, et al. The role of CXCR4 in maintaining peripheral B cell compartments and humoral immunity. The Journal of experimental medicine. 2004;200:1145–1156. doi: 10.1084/jem.20041185. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.