

Summary
Tolerance induction and alloreactivity can be applied to the clinic for the transplantation of solid organs and in the treatment of human cancers respectively. Hematopoietic chimerism, the stable coexistence of host and donor blood cells, guarantees that a solid organ from the same donor will be tolerated without a requirement for maintenance immunosuppression, and it also serves as a platform for the adoptive immunotherapy of hematologic malignancies using donor lymphocyte infusions. This review focuses on clinically relevant methods for inducing hematopoietic chimerism and transplantation tolerance, with a special emphasis on reduced intensity transplantation conditioning and high dose, post-transplantation cyclophosphamide to prevent graft rejection and graft-versus-host disease (GVHD). Reduced intensity transplantation regimens permit a transient cooperation between donor and host immune systems to eradicate malignancy without producing GVHD. Their favorable toxicity profile also enables the application of allogeneic stem cell transplantation to treat non-malignant disorders of hematopoiesis and to induce tolerance for solid organ transplantation.
Keywords: transplantation conditioning, transplantation tolerance, cyclophosphamide, graft-versus-leukemia, graft-versus-host disease, hematopoietic chimerism
Introduction
Transplantation of allogeneic tissues is used for either of two clinical indications: (i) the replacement of a depleted, dysfunctional, or non-functional organ with a functional one or (ii) the treatment of cancer, usually a hematologic malignancy. Tissue replacement ranges from red blood cell transfusion to treat anemia, platelet transfusion to treat thrombocytopenia, transplantation of single organs such as kidney, liver, heart, or pancreas, and most recently composite tissue transplantation such as hand or face transplantation. The Holy Grail of solid organ transplantation is a state of permanent transplantation tolerance without maintenance pharmacologic immunosuppression. Clinically, tolerance can be defined as a well-functioning organ without histologic evidence of rejection. Mechanisms of transplantation tolerance include clonal deletion, clonal anergy, suppression, ignorance, or immune deviation. A variety of pharmacologic or cellular approaches have been taken to achieve clinical transplantation tolerance. The mechanism of tolerance achieved may differ between these different approaches.
A major focus of this review is the induction of transplantation tolerance in the clinic. We propose that best guarantee of stable clinical transplantation tolerance is sustained donor hematopoietic macrochimerism, because intrathymic donor chimerism induces the clonal deletion of graft-reactive cells. Clonal deletion guarantees tolerance because dead cells cannot be revived. In contrast, non-deletional mechanisms of tolerance such as anergy, suppression, or exhaustion do not guarantee permanent graft survival because such tolerance can be broken under extreme circumstances such as infection. There is an emerging consensus that sustained donor hematopoietic chimerism induces clonal deletion of donor-reactive host T cells and lifelong tolerance of any organ transplanted from the same donor. Conversely, it is increasingly recognized that non-deletional forms of tolerance, such as anergy, suppression, or immune deviation, may not be stable. Although several transplant recipients have been successfully weaned off pharmacologic immunosuppression and have achieved a state of ‘operational tolerance’ (1, 2), such patients represent the exception rather than the rule, and most individuals without detectable hematopoietic chimerism will require lifelong treatment with immunosuppressive drugs to avoid rejection of the allograft. There is less debate on the benefits of hematopoietic chimerism for transplantation tolerance than there is on the methods to achieve it safely in the clinic. The purpose of this review is to provide a personal account of the evolution of strategies employed to achieve tolerance: first to the male-specific minor histocompatibility antigen, H-Y, next to multiple minor histocompatibility antigens, then to major histocompatibility antigens, and finally the translation of tolerance strategies to the clinic to treat hematologic malignancies and non-malignant disorders of hematopoiesis. The initial strategy was based upon the theory that B cells can present antigen but not costimulatory signals to naive T cells, and proved successful for inducing tolerance to H-Y but nothing more. Subsequent strategies were empirically derived and featured the use of high dose, post-transplantation cyclophosphamide (PT/Cy) for the induction of tolerance to major and minor histocompatibility antigens. The clinical testing ground for the induction of hematopoietic chimerism and transplantation tolerance has been the treatment of patients with hematologic malignancies or non-malignant hematologic disorders with reduced intensity conditioning and allogeneic stem cell transplantation (alloSCT). Patients with hematologic malignancies such as acute leukemia that cannot be cured by standard chemotherapy are candidates for alloSCT, in which donor T cells exert a ‘graft-versus-leukemia’ effect through recognition and destruction of recipient cells bearing disparate histocompatibility antigens. Since alloSCT offers a curative therapy to patients with a generally poor prognosis, this clinical arena forms the crucible for developing and refining methods to safely induce donor hematopoietic chimerism without the morbid and potentially fatal complications of conditioning regimen toxicity, infection, and graft-versus-host disease (GVHD), a pathologic attack by donor T cells on normal tissues such as skin, liver, and gastrointestinal tract. Finally, methods for safely inducing donor hematopoietic chimerism will be exported to the arena of solid organ transplantation for the purpose of inducing transplantation tolerance without maintenance immunosuppression.
A number of outstanding reviews have been published recently on subjects as diverse as the current status and clinical impact of solid organ (3) or hematopoietic stem cell transplantation (4), the biology of the allogeneic response (5), the role of hematopoietic chimerism in tolerance to solid organs (6), regulatory T cells and transplantation tolerance (7, 8), and the status of efforts to translate basic discoveries into the clinic (9). Rather than take these well-travelled paths, this review sets forth a set of design principles that may assist those seeking to achieve transplantation tolerance. The application of these design principles have met with some success in crossing the human leukocyte antigen (HLA) barrier in patients with cancer or with sickle cell anemia. The studies leading to the formulation of these principles are then outlined. Finally, current progress in achieving transplantation tolerance using these principles is described.
The two signal model of T-cell activation and its application to transplantation tolerance induction
A convenient starting point for understanding alloreactivity is the two signal model of T-cell activation proposed by Kevin Lafferty and Alistair Cunningham (10). Their simple model was adapted from Bretscher and Cohn’s two signal model of lymphocyte activation (11) (Fig. 1). In the Lafferty/Cunningham model, signal 1 results from T-cell recognition of antigen on the surface of an antigen-presenting cell (APC) and is an ‘off’ signal to the T cell, unless it simultaneously receives a second, or ‘costimulatory’, signal from the same APC, in which case the T cell is turned on. In a subsequent publication (12), Lafferty et al. noted a correlation between MHC class II gene expression and the ability to deliver the costimulatory signal required for T-cell activation. Based upon experiments demonstrating that donor strain dendritic cells restore the immunogenicity of passenger leukocyte-depleted kidney allografts (13), they nominated the dendritic cell for the physiologic stimulator of allograft rejection.
Fig. 1. The Lafferty/Cunningham ‘two signal’ model of T-cell activation.
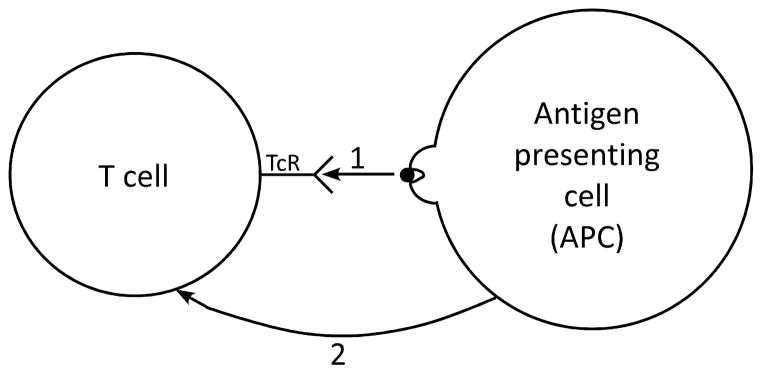
Signal 1 is generated in the T cell when its clonotypic antigen receptor engages an antigenic peptide in the groove of a major histocompatibility complex (MHC) molecule on the surface of an antigen-presenting cell (APC). Signal 2 is a costimulatory signal delivered by the APC to the T cell. According to the model, T-cell activation requires both signal 1 and signal 2, whereas a T cell becomes tolerant if it receives signal 1 in the absence of signal 2.
It is hard to overestimate the influence that the two signal model of T-cell activation has had on immunology theory, the experimental program of immunology, and efforts to induce transplantation tolerance in the clinic. The model wreaked havoc with the existing theories of self/non-self discrimination, because these theories all assumed that adaptive lymphocytes made the discrimination, whereas the Lafferty/Cunningham model ceded control of a T cell’s decision between activation and tolerance to APCs, which do not express clonally distributed receptors for antigen. The crisis generated by the Lafferty/Cunningham model stimulated the development of both the ‘stranger’ (14) and ‘danger’ (15) models of immunity to account for how costimulation by APCs is regulated. The model also provided the theoretical framework (16) for the experimental phenomenon of T-cell clonal anergy (17, 18), which was postulated to result from a T cell seeing antigen in the absence of a costimulatory signal. The idea of a costimulatory signal led to the discovery of the B7-1 (CD80) molecule as a ligand for the CD28 costimulatory receptor on T cells (19) and the clinical development and FDA approval of belatacept, a fusion protein that blocks the interaction of CD28 with its ligands, for the prevention of kidney allograft rejection (20). Finally, the Lafferty/Cunningham model raised the possibility that cells may differ according to their tissue of origin in their capacity to stimulate T cells.
Dendritic cells and B cells are constitutively MHC class II-positive cells that present antigens to CD4+ and CD8+ T cells. A wealth of studies have shown that B cells can activate T cells, but Lassila, Vainio, and Matzinger (21) reasoned that resting B cells should not be able to initiate a T-cell-mediated immune response, because they could then initiate responses against their own immunoglobulin idiotypes. By examining the antibody response to sheep red blood cells (SRBCs) in cyclophosphamide-treated chickens reconstituted with allogeneic B cells, they found that B cells were indeed unable to present antigen to initiate an immune response. Thus, it seems that only ‘professional’ APCs such as dendritic cells are endowed with the capacity to stimulate naive T cells. If B cells can present antigen to naive T cells but cannot activate them, then according to the two signal model they must be inducing tolerance. Fuchs and Matzinger (22) tested this hypothesis by examining the response of unprimed C57BL/6 (B6) female mice to the male-specific minor histocompatibility antigen H-Y. Intravenous injection of purified resting male B cells into syngeneic females induced cytotoxic T-cell tolerance to male spleen cells in vitro and the indefinite survival of male skin grafts placed 1 week after the intravenous injection. In contrast, female recipients of purified syngeneic male dendritic cells generated strong CTL responses against male cells in vitro and rejected male skin grafts more quickly than did previously untreated females. The activation state of the B cell was irrelevant to the outcome of antigen presentation to naive T cells, as tolerance to H-Y was induced by intravenous injection of B cells activated by bacterial lipopolysaccharide or by anti-immunoglobulin plus either interleukin-4 (IL-4) or interferon-γ (IFN-γ). In contrast, when resting male B cells were infused into female mice that had been primed previously to H-Y, tolerance did not occur and the memory CD8+ T-cell response was augmented. These experiments provide support for Lafferty and Cunningham’s two signal model of T-cell activation and establish that T cell’s decision between activation and tolerance upon antigen encounter depends on two parameters: (i) the differentiation state of the T cell (naive versus memory) and (ii) the type of the APC. A naive T cell is activated by recognizing an antigen presented on an activated dendritic cell but is tolerized if it first sees the antigen presented by a B cell. In contrast, a memory T cell is activated upon recognition of antigen presented by a resting B cell. There are two potential explanations for the differential response of naive versus memory T cells to antigens presented by B cells: (i) memory T cells have lower costimulatory signaling requirements for activation or (ii) memory T cells require costimulatory signals to become activated, but can induce them on B cells. The finding that a CD4+ T-cell clone is activated by lightly irradiated resting B cells presenting antigen but is rendered anergic by heavily irradiated B cells presenting the same antigen (23) raises the possibility that T cells can indeed induce costimulatory signal expression on B cells, but only those that have been lightly irradiated. For example, antigen recognition by a memory helper T cell could lead to upregulation of the CD40 ligand, CD154, on the T cell, which ligates CD40 on the B cell, leading to the upregulation of costimulatory signals such as CD80 and CD86 (24).
The fundamental principle that was elucidated by these experiments is that the decision of T cells between activation and tolerance upon encountering antigen is determined primarily, if not exclusively, by two parameters: (i) the differentiation state of the T cell, naive versus experienced and (ii) the type of the APC. The results suggest that the immune system does not discriminate between self and non-self because any antigen, self or foreign, would induce tolerance in an antigen-specific naive T cell if first presented by a B cell. This realization was the first step toward the evolution of the danger model (15, 25), which states that a T-cell response is initiated only in the context of tissue distress or pathologic cell death. Also absent from parameters determining T-cell activation versus tolerance was the age of the animal. Thus, we predicted and subsequently confirmed that male dendritic cells could prime syngeneic neonatal females to the male antigen (26). Thus, in contrast with the assertions of Burnet (27), Medawar (28), and Lederberg (29), the period before or shortly after birth is not a period of unique tolerance susceptibility. Rather, it is a time when there are few T cells, all of which are in the naive state of differentiation. Thus, it is possible that the ability to induce tolerance in neonatal mice with an injection of allogeneic spleen cells results from the small number of alloreactive naive T cells first encountering alloantigen presented by B cells, which vastly outnumber dendritic cells in the injected population. Finally, the experiments demonstrating tolerance induction by B cells support the two signal model of Lafferty and Cunningham, as B cells would be a type of cell that could deliver signal 1 without signal 2, whereas dendritic cells were endowed with the capacity to deliver both signals.
Since male B cells, resting or activated, induced tolerance to male skin grafts in syngeneic females, the next logical step toward inducing tolerance in the clinic was to test the ability of B cells to induce tolerance to cells differing in multiple minor histocompatibility antigens, such as are found between HLA-matched non-identical siblings or possibly between HLA-matched unrelated donors. C57BL/10 (B10) male B cells were purified and injected into syngeneic females, as a control, or into A.BY males, which differ from C57BL/10 males in the expression of multiple minor histocompatibility antigens. Although the B10 male B cells induced tolerance to H-Y in B10 females (Fig. 2, left panel), they actually primed A.BY males to make effective killer cells against B10 cells in vitro (Fig. 2, right panel; unpublished observations). We did not seek to determine why male B cells induce tolerance to H-Y versus prime to multiple minor histocompatibility antigens, but can come up with at least three potential explanations for the differing response. First, the repertoire of T cells reactive to multiple minor histocompatibility antigens may contain some experienced cells, which are boosted by B cells presenting the minor antigens. Second, the purified B cells could have contained a small number of contaminating dendritic cells. Since the frequency of A.BY male T cells reactive against C57BL/10 male cells is likely to be several times higher than the frequency of C57BL/10 female T cells reactive to C57BL/10 male cells, the likelihood that one of the A.BY T cells encounters its antigen first on a B10 male dendritic cell and is primed is also higher. Finally, it is possible that some B10 minor antigens are ‘stronger’ than H-Y; that is, they engage their T cells with higher affinity and so have a lower requirement for costimulation and a lower threshold for activation. Regardless of the explanation, it is clear that the capacity of purified B cells to induce transplantation tolerance was quite limited and not useful for translation to the clinic, as even HLA-matched siblings differ from each other in the expression of multiple minor H antigens.
Fig. 2. Purified male B cells induce cytotoxic T-cell tolerance to the male specific histocompatibility antigen but induce apparent priming to multiple B10 minor histocompatibility antigens.
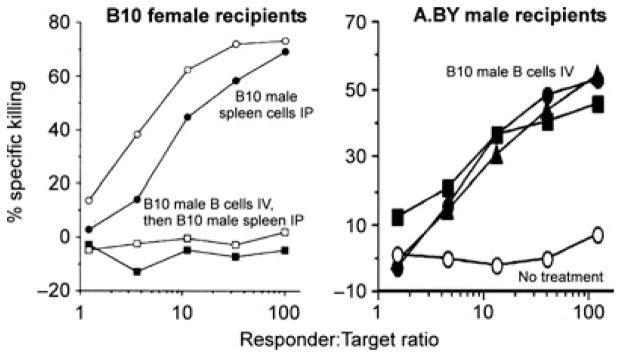
Left panel: naive C57BL/10 (B10; H-2b) female mice received nothing (circles; n = 2) or 10 million purified syngeneic male B cells IV (squares; n = 2). One week later, all mice received 10 million syngeneic male spleen cells IP. Two weeks after spleen cell injection, mice were sacrificed, and spleen cells were cultured with syngeneic male cells and tested for killing of syngeneic male target cells. Right panel: A.BY (H-2b) male mice received nothing (open symbol; n = 1) or 10 million purified B10 male B cells IV (closed symbols; n = 3). Two weeks later, mice were sacrificed, spleen cells were cultured with B10 male cells and tested for killing of B10 male targets.
Cyclophosphamide-induced tolerance to histocompatibility antigens
Fortunately, tolerance to multiple minor histocompatibility antigens can be induced relatively easily using the method of cyclophosphamide-induced tolerance (30), a special case of the phenomenon of drug-induced tolerance first described by Schwartz and Dameshek (31). The principle is simple: antigen-responsive cells are exposed to antigen in immunogenic form, they proliferate, and then they are killed or disabled by an anti-metabolite such as 6-mercaptopurine (31) or methotrexate (32), or an alkylating agent such as cyclophosphamide (30, 33). Berenbaum gave cyclophosphamide to rats either before or after the placement of an MHC-mismatched skin graft and found that the drug was most effective at prolonging skin graft survival when it was administered at any time between shortly after grafting to the fourth day afterward (33). This result suggests that alloantigen-stimulated cells are more sensitive than resting T cells to being killed or disabled by cyclophosphamide. It is also worth noting that post-transplantation Cy (PT/Cy) did not induce complete tolerance in this MHC-mismatched skin transplant model, as median graft survival was prolonged from 12.0 days in untreated control groups to 17.0 days in rats given the drug on day 2.
A series of elegant studies performed by Mayumi et al. (34) established that complete tolerance can be induced in MHC-compatible strain combinations by the intravenous injection of ≥50 million allogeneic spleen cells IV on day 0 and ≥150 mg/kg cyclophosphamide intraperitoneally on day 2. This method worked for all MHC-compatible strain combinations tested, including C3H→AKR, AKR→C3H, and BALB/c→DBA/2, but did not induce tolerance between strains differing in MHC as well as minor antigens, such as C57BL/6 (H-2b) →C3H (H-2k; 35). The dose of spleen cells and cyclophosphamide as well as the timing of drug administration were all critical to the outcome. Tolerance to minor H antigens was not induced with fewer than 50 million donor cells IV, <150 mg/kg of cyclophosphamide, or if the drug was given earlier than 48 h after or later than 96 h after cell infusion (34). Subsequent studies demonstrated three sequential mechanisms of tolerance in this model (36). First, mature peripheral alloreactive T cells were destroyed by the combination of donor cell infusion plus cyclophosphamide treatment. Next, tolerance in the periphery permitted the development of intrathymic donor hematopoietic chimerism and clonal deletion of thymocytes reactive to donor cells. Finally, late after tolerance induction there was a breakdown in the clonal deletion of alloreactive thymocytes and the appearance of tolerogen-specific suppressor T cells. Tolerance was associated with a low but persistently detectable level of donor hematopoietic chimerism.
Since multiple minor histocompatibility antigens are the only immunologic barrier between HLA-matched siblings or between a patient and an HLA-matched unrelated donor, the method of cyclophosphamide-induced tolerance could have clinical relevance. However, the minimum dose of cells required to induce tolerance in the mouse, 50 million, is equivalent to 2.5 × 109 cells/kg in the human, and this cell dose is not feasible to acquire either from the bone marrow or peripheral blood. The minimum number of infused cells required for cyclophosphamide-induced tolerance could be reduced to 5 million by giving a low dose of total body irradiation 1 day prior to cell infusion. In the mouse, stable mixed hematopoietic chimerism in all recipients was achieved by giving 100 cGy TBI, 10 million MHC-matched allogeneic bone marrow cells (equivalent to 5 × 108 cells/kg in the human), and 200 mg/kg cyclophosphamide IP (37). This regimen was not successful in achieving tolerance and chimerism in donor-recipient pairs differing in the expression of MHC antigens. In the absence of any other conditioning, ≥600 cGy TBI is required for the reliable engraftment of MHC-incompatible bone marrow. Post-transplanation cyclophosphamide, 200 mg/kg IP, lowers the dose of radiation required for the stable engraftment of 15 million MHC-incompatible bone marrow cells down to 500 cGy (38). The dose of TBI required for stable engraftment of MHC-mismatched donor bone marrow can be reduced to 300 cGy in mice conditioned on day −3 with anti-lymphocyte globulin and on day +2 with cyclophosphamide 200 mg/kg IP (39). Mayumi and Good (40) developed a radiation-free transplantation regimen comprising anti-T cell antibodies before transplantation, a high dose of MHC- and minor H antigen-mismatched bone marrow and spleen cells, and post-transplantation cyclophosphamide. This regimen was sufficient to achieve long-term, but not permanent, survival of donor strain skin grafts, and only minimal hematopoietic chimerism was observed. In pairs mismatched for MHC +/− minor histocompatibility antigens, we were able to induce tolerance and mixed hematopoietic chimerism with a regimen comprising fludarabine 100–200 mg/kg/day IP from days −6 to −2, 50–200 cGy total body irradiation on day −1, 5–20 million donor bone marrow cells IV on day 0, and cyclophosphamide 200 mg/kg IP on day 0 (41). The degree of donor chimerism was proportional to the dose of radiation therapy and to the dose of donor cells administered. When the dose of TBI was held at 200 cGy, engraftment and stable mixed hematopoietic chimerism was achieved in 4 of 5 recipients of 5 million marrow cells, and in five of five recipients of either 10 or 20 million marrow cells. For a mouse weighing 25 g, these cell doses are equivalent to 2, 4, or 8 × 108 nucleated cells/kg. At our institution, the target nucleated cell dose for a human bone marrow harvest is 4 × 108 marrow nucleated cells/kg of the recipient’s ideal body weight, while a dose of 8 × 108/kg is probably not feasible to acquire if the recipient is an adult. Thus, the doses of 5 or 10 million marrow cells used in the mouse experiment are representative of doses that are feasible to obtain in the clinic. In mice conditioned with fludarabine and 200 cGy TBI, chimerism could not be obtained with as many as 20 million MHC-mismatched bone marrow cells if post-transplantation Cy was not administered, establishing that PT/Cy promotes tolerance of transplanted bone marrow. PT/Cy also diminished the incidence and severity of GVHD in lethally conditioned mice transplanted with marrow plus spleen cells from MHC-mismatched donors, suggesting that the drug promotes bidirectional transplantation tolerance. Finally, the regimen was truly non-myeloablative, as autologous hematopoiesis recovered in all mice that were conditioned with fludarabine, 200 cGy TBI, and PT/Cy but did not receive any marrow cells IV.
Clinical application of cyclophosphamide-induced tolerance
The non-myeloablative platform was translated to the clinic to treat patients with hematologic malignancies who lacked an HLA-matched sibling or unrelated donor (42, 43). Sixty-eight patients were conditioned with a regimen of cyclophosphamide (Cy) 14.5 mg/kg IV on days −6 and −5 prior to transplantation, fludarabine 30 mg/m2/day IV from days −6 to −2, 200 cGy total body irradiation on day −1 prior to receiving T cell-replete bone marrow from an HLA-haploidentical first degree relative on day 0 (Fig. 3). Eligible donors included HLA-haploidentical siblings, parents, or children, but a potential donor was excluded if the patient had significant antibodies against that donor’s HLA molecules. HLA typing was performed for the HLA-A, -B, -Cw, -DRB1, and -DQB1 loci using high resolution techniques. The median mismatch was four loci at the antigen level in both directions. Twenty-eight patients were transplanted in Seattle, Washington, and received cyclophosphamide 50 mg/kg IV on day 3 after transplantation, whereas the 40 patients transplanted in Baltimore, Maryland received Cy 50 mg/kg each on days 3 and 4. All patients were started on mycophenolate mofetil (MMF) and tacrolimus on the day after the last dose of cyclophosphamide, with the MMF continued until day 35 and the tacrolimus continued until day 180 after transplantation. Sixty-six patients were evaluable for donor cell engraftment, with nine patients (13%) experiencing primary graft failure. All but one of these nine patients had autologous recovery of hematopoiesis, indicating that the conditioning regimen is truly non-myeloablative. The median time to recovery of 500 neutrophils/μl of blood was 15 days, whereas the median time to recovery of 20 000 platelets/μl of blood was 24 days. Chimerism studies were performed on cells from the blood or the bone marrow and demonstrated that full donor hematopoietic chimerism was achieved in the vast majority of patients by 30 days after transplantation. Since the conditioning regimen does not eliminate host hematopoiesis, this result suggests that host-reactive T cells of the donor can survive the PT/Cy and mediate a graft-versus-hematopoietic host reaction leading to full donor hematopoietic chimerism. Indeed, acute graft-versus-host disease (GVHD) developed in 34% of all patients and was severe in 6%. However, only two of the 68 patients died of GVHD, which was quite remarkable in light of the >50% incidence of severe GVHD and substantial GVHD-related mortality that were seen in early clinical trials of T-cell-replete, HLA-haploidentical bone marrow transplantation (44). There was no significant difference in the incidence of acute GVHD among recipients of one versus two doses of PT/Cy. However, the incidence of extensive chronic GVHD was significantly lower among patients treated with two doses of PT/Cy (5% versus 25%, P = 0.05). Thirty-one patients died from relapse of the underlying hematologic malignancy, four patients died of infection, two of GVHD, and five died from other causes. The 1 year cumulative incidences of non-relapse mortality and relapse were 15% and 51%, respectively, resulting in a 1-year progression-free survival of 34%. These results illustrate that reduced intensity conditioning, transplantation of T cell-replete, HLA-haploidentical bone marrow, and post-grafting immunosuppression including high dose cyclophosphamide results in acceptably low incidences of fatal graft failure or severe GVHD. Relapse of the underlying hematologic malignancy was the major cause of treatment failure.
Fig. 3. Schema of non-myeloablative conditioning, HLA-haploidentical bone marrow transplantation, and GVHD prophylaxis including high dose, post-transplantation cyclophosphamide (PT/Cy).
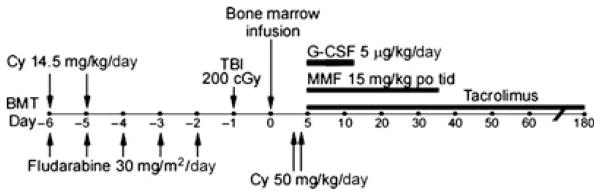
The 28 patients treated in Seattle, WA received Cy 50 mg/kg IV on day 3 only, whereas the 40 patients treated in Baltimore, MD received Cy 50 mg/kg IV each on days 3 and 4 after transplantation.
Historically, increasing HLA mismatch between donor and recipient has been associated with increasing incidences of graft failure, acute GVHD and non-relapse mortality, and decreased overall and event-free survivals (44–46). Kasamon et al. examined the effect of HLA mismatching and outcome among recipients of reduced intensity, HLA-haploidentical BMT with PT/Cy. Interestingly, increasing HLA mismatch in the graft-versus-host direction was not associated with an increased cumulative incidence of acute GVHD (Fig. 4, left panel) or non-relapse mortality, but it was associated with a decreased risk of relapse and an improved event-free survival (Fig. 4, right panel). Each increment of HLA mismatch was associated with a 20% reduction in the risk of an event, either non-relapse mortality or disease progression. The effect of specific HLA mismatches on outcome was also analyzed. Since donors and recipients were HLA-haploidentical to each other, patients and donors had either 0 or 1 allele or antigen-mismatched at any given locus, and there were no double mismatches. Single locus mismatches were in the graft-versus-host direction only, if the recipient was heterozygous and the donor was homozygous at that locus, in the host-versus-graft direction only, if the recipient was homozygous and the donor was heterozygous, or were bidirectional. Compared with donor-recipient pairs that were matched at the HLA-DRB1 locus in the graft-versus-host direction, pairs that were mismatched at the antigen level in the graft-versus-host direction had a significantly improved overall survival (not shown) and event-free survival (Fig. 5, left panel). Mismatched recipients had a 15% absolute reduction in the risk of relapse (P = 0.045 compared with matched pairs; Fig. 5, right panel) and a 5% absolute reduction in the incidence of non-relapse mortality, which was not statistically significant. Although it is possible that the donor-derived, HLA-DRB1-reactive CD4+ T cells conferred these benefits before being killed by PT/Cy, it is also possible that they survived the drug and mediated their clinical benefit afterwards.
Fig. 4. Effects of increasing donor-recipient mismatch on the outcome of reduced intensity, HLA-haploidentical BMT with post-transplantation cyclophosphamide.
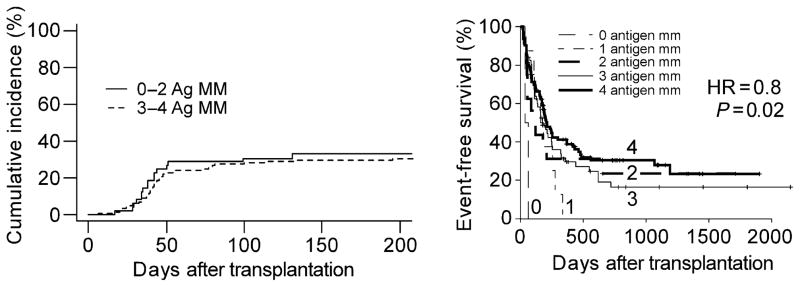
Left panel: Cumulative incidence of acute grades II–IV GVHD according to the number of HLA antigen mismatches (MM) between donor and recipient in the graft-versus-host direction. Right panel: Event-free survival after non-myeloablative, HLA-haploidentical BMT with PT/Cy as a function of the total HLA antigen mismatch between donor in either (GVH or HVG) direction. The hazard ratio (HR) of 0.8 indicates that each increment of HLA mismatch was associated with a 20% reduction in the risk of an event, either relapse or non-relapse mortality. Pairs with 0 HLA antigen mismatches had at least 2 allele mismatches and so were not genotypically HLA-matched.
Fig. 5.

Event-free survival (left panel) and cumulative incidences of relapse and non-relapse mortality (right panel) after reduced intensity, HLA-haploidentical bone marrow transplantation according to the presence or absence of an HLA-DRB1 antigen mismatch between donor and recipient in the graft-versus-host direction.
Separating an anti-tumor effect of alloSCT from graft-versus-host disease: a potential collaboration of donor and host T cells?
The ‘graft-versus-leukemia’ (GVL) effect refers to the capacity of alloreactive donor T cells to reduce the risk of post-transplantation relapse through the recognition and destruction of tumor cells bearing disparate major and/or minor histocompatibility antigens. The GVL effect is a major contributor to the efficacy of alloSCT in the treatment of leukemia, but this effect has been tightly linked to the occurrence of GVHD (47). Whereas mild GVHD is associated with an improvement in event-free survival following alloSCT, severe GVHD typically worsens the outcome of alloSCT because of increased NRM. Intense research has been devoted to dissociating the beneficial GVL effect from the detrimental effects of acute GVHD, but this goal has remained elusive in the clinic. However, the results of Fig. 4 suggest that non-myeloablative, HLA-haploidentical BMT with PT/Cy effectively dissociates the GVL effect from GVHD, because increasing HLA mismatch was associated with a decreased risk of relapse without an increased incidence of acute GVHD. The hypothesis that the regimen shown in Fig. 3 dissociates GVL from GVHD needs to be tested prospectively. But if a prospective analysis supports this hypothesis, then what is the mechanism? We can envision three possibilities. The first is that GVL and GVHD are mediated by distinct populations of donor T cells that differ in their sensitivity to cyclophosphamide. In this case, the GVL effect would be mediated by cyclophosphamide-resistant T cells specific for antigens uniquely expressed by the tumor, whereas GVHD would be mediated by cyclophosphamide-sensitive T cells specific for widely expressed histocompatibility antigens. The second possibility is that the two effects are mediated by the same population of T cells, but the effector functions that produce clinical GVHD are cyclophosphamide-sensitive whereas the effector functions that induce the GVL effect are cyclophosphamide-resistant. Third, the regimen effectively unmasks an anti-tumor cooperation between alloreactive donor CD4+ T cells and tumor-specific, host-derived CD8+ T cells, which cannot mediate GVHD. Inherent in this possibility is the hypothesis that patients with a hematologic malignancy contain a cadre of ‘anergic’, tumor-specific T cells that are awakened by donor-derived, alloreactive CD4+ T cells to become cytotoxic anti-tumor effector cells. The critical piece of evidence in support of this hypothesis would be the demonstration of an increase in the number of functional, tumor-specific CD8+ T cells of recipient origin following reduced intensity, HLA-haploidentical BMT with PT/Cy. Even if host T cells with potent anti-tumor activity are present after alloSCT, they cannot be demonstrated because the graft-versus-hematopoietic host reaction eliminates all host T cells as full donor hematopoietic chimerism is achieved. However, participation of host T cells in the anti-tumor immune response can be inferred in cases when the donor graft is rejected, either unintentionally or by design. There are several reports of clinical responses of hematologic malignancies despite rejection of the donor graft. O’Donnell et al. (42) reported on the first 13 patients to receive HLA-haploidentical BMT with PT/Cy, and two patients with myelodysplastic syndrome obtained complete remissions of their disease despite non-engraftment of the bone marrow. Dey et al. (48) reported on 82 patients treated with reduced intensity conditioning and bone marrow transplantation. Twenty-two patients rejected their grafts, and nine of these patients had partial or complete responses. Based upon animal data showing an anti-tumor effect of recipient-derived lymphocyte infusions (RLI) in established mixed hematopoietic chimeras (49–51), the authors speculated that the host-versus-graft immune response associated with graft rejection mediated the observed clinical responses. These animal models have shown that host-derived CD8+ T cells, IFN-γ, and rejection of the donor cells are critical for the generation of anti-tumor immunity by RLI. Further, invariant natural killer T (iNKT) cells of the recipient are also required for the anti-tumor effect of RLI and for the activation of host dendritic cells and natural killer cells (52). In a different animal model, a graft-versus-host reaction was sufficient to elicit anti-tumor effects, even when the infused lymphocytes were syngeneic to the tumor, and a host-versus-graft response did not prolong survival against a recipient strain tumor (53). Fig. 6 depicts a simple model for how a graft-versus-host reaction by alloreactive donor CD4+ T cells can awaken tumor-specific CD8+ T cells of the recipient. This model borrows from the principle that CD4+ T cells deliver ‘help’ by licensing APCs to stimulate cytotoxic T cells (54–58) and that the helper function of CD4+ T cells is critical to the prevention of CD8+ T-cell exhaustion and the development and maintenance of CD8+ T-cell memory. More recent studies have shown that, in addition to providing the signals required to prevent CD8+ T-cell exhaustion (59), CD4+ T-cell help can act alone or synergize with blockade of PD-1 signaling to restore effector function of exhausted antiviral (60) or anti-tumor (61) CD8+ T cells. In the case of transiently engrafting, histoincompatible lymphocyte infusions, the host APC serves as a bridge between the alloreactive donor CD4+ T cell and the tumor-specific host CD8+ T cell.
Fig. 6. Model for the cooperation of alloreactive donor CD4+ T cells and tumor-specific host CD8+ T cells in producing anti-leukemia immunity after partially HLA-mismatched allogeneic stem cell transplantation.

A donor CD4+ T cell recognizes an alloantigen (red circle) and delivers a signal through the host antigen-presenting cell (APC) via the interaction of CD154 on the T cell with CD40 on the APC. This signal activates the APC, which delivers a signal to a host CD8+ T cell recognizing a leukemia-specific antigen (blue square) cross-presented by the same APC. This signal may result from an interaction of CD70 on the APC with CD27 on the CD8+ T cell, and results in the reversal of T-cell exhaustion and acquisition of cytotoxic effector function.
Results of HLA-haploidentical stem cell transplantation with high dose PT/Cy from various centers are shown in Table 1. Despite significant variation in patient diagnoses, stages of disease treated, and conditioning regimens employed, the outcomes that relate to alloreactivity are reasonably consistent. Graft failure rates after reduced intensity conditioning ranged from 12% to 17% in the studies treating the majority of patients, and 0–6% after myeloablative conditioning. Grades II–IV acute GVHD typically ranged from 30% to 36%, whereas severe acute GVHD occurred in 0–11%. Remarkably, there were very few cases of severe chronic GVHD, and non-relapse mortality was between 15% and 18% independent of the intensity of conditioning.
Table 1.
Graft failure, GVHD, and non-relapse mortality (NRM) after HLA-haploidentical bone marrow transplantation with high dose, post-transplantation cyclophosphamide
| Author
|
n
|
Regimen RIC/Ablative
|
Graft failure (%)
|
aGVHD (%)II–IV/III–IV
|
cGVHD (%)total/severe
|
NRM (%)
|
|---|---|---|---|---|---|---|
| Kasamon (130) | 185 | 185/0 | 16 | 31/NR | 15/NR | 15 |
| El-Cheikh (131) | 69 | 69/0 | 12 | 12/NR | NR/6 | 17 |
| Brunstein (132) | 50 | 50/0 | 2 | 32/0 | 13/5 | 7 |
| Tuve (133) | 18 | 18/0 | 0 | 35/6 | 35/0 | 6 |
| Bilmon (134) | 12 | 12/0 | 17 | 36/9 | 36/0 | 17 |
| Bashey (135) | 53 | 35/18 | 2 | 30/11 | 38/4 | 4 |
| Ciurea (136) | 32 | 6/26 | 6 | 20/5 | 7/0 | 16 |
| Solomon (137) | 20 | 0/20 | 0 | 30/10 | 35/5 | 10 |
| Raiola (138) | 50 | 0/50 | 4 | 12/6 | 26/0 | 18 |
RIC, reduced intensity conditioning.
In summary, reduced intensity, HLA-haploidentical bone marrow transplantation with high-dose, post-transplantation cyclophosphamide is associated with acceptably low risks of fatal graft rejection, severe acute or chronic graft-versus-host disease, and non-relapse mortality. Increasing HLA disparity between donor and recipient was not associated with an increased incidence of acute GVHD or non-relapse mortality but was associated with a decreased incidence of relapse, and consequently improved progression-free survival. Mismatching at the HLA-DRB1 locus in the graft-versus-host direction was associated with a significantly decreased risk of relapse and an improved overall and event-free survival. These results suggest that this transplantation platform effectively separates the graft-versus-tumor effect of allogeneic SCT from GVHD. Since the reduced intensity conditioning does not ablate the patient’s immune system, it is possible that HLA-DRB1-alloreactive donor CD4+ T cells provide helper signals through host APCs to awaken previously exhausted, tumor-specific host CD8+ T cells. This postulated mechanism could account for a graft-versus-tumor effect without GVHD, and is compatible with observed anti-tumor effects of transiently engrafting lymphocytes from partially or fully HLA-mismatched related donors (42, 48, 62–67).
HLA-haploidentical bone marrow transplantation for non-malignant disorders
Allogeneic stem cell transplantation has the potential to cure a variety of non-malignant diseases including inherited (68) or acquired immunodeficiency syndromes (69), autoimmune disease (70), inherited metabolic disorders (71), aplastic anemia (72), and hemoglobinopathies such as thalassemia and sickle cell disease (SCD) (73). The main impediment to the application of alloSCT for these diseases is not the efficacy of the procedure but rather is toxicity and the availability of suitably matched donors. For example, alloSCT from HLA-matched siblings is highly efficacious for patients with SCD, with 5 year overall and event-free survivals of 93–97% and 85–86%, respectively, in the largest reported series (74, 75). Despite these impressive results, less than 1% of SCD patients undergo alloSCT. Only 18% of SCD patients have an unaffected, HLA-matched sibling, and most patients or their parents are unwilling to accept the risks of transplant-related toxicity, especially chronic GVHD (76).
In light of the favorable toxicity profile of non-myeloablative BMT with PT/Cy, a clinical trial of this therapy was conducted for patients with SCD. Getting donor cells to engraft is a much greater challenge in patients with SCD compared with patients with cancer, because the vast majority of SCD patients have been sensitized to HLA alloantigens by transfusion and, unlike cancer patients, their immune systems have not been suppressed by cytotoxic chemotherapy and are relatively intact. Eligibility for transplantation was restricted to patients with clinical features portending a poor prognosis, including history of stroke, acute chest syndrome, recurrent vaso-occlusive crises, stage I or II sickle lung disease, sickle retinopathy, osteonecrosis, red cell allo-immunization, or invasive pneumococcal disease. Fig. 7 shows the sequential conditioning regimens and postgrafting immunosuppression that were applied in this clinical trial. The first two patients received nearly the same regimen as was used in patients with hematologic malignancies (Fig. 3). Full donor hematopoietic chimerism without GVHD was achieved in the first patient, who is now disease-free and off all immunosuppressive medications for more than 5 years (77). The second patient experienced graft rejection with return of sickle hematopoiesis. To improve engraftment, in vivo T-cell depletion with anti-thymocyte globulin was added to the conditioning regimen for the next six patients, but two patients experienced graft rejection and one patient achieved only 6% donor chimerism in the peripheral blood- insufficient to abolish sickle hematopoiesis. Sirolimus was substituted for tacrolimus after two patients developed posterior reversible encephalopathy, a serious but reversible toxicity of calcineurin inhibitors. Two of three patients in this cohort experienced graft failure. Finally, based upon evidence that a high donor cell dose can overcome graft rejection (78) and is required for tolerance induction by post-transplantation cyclophosphamide (34), the last eleven donors were treated with recombinant granulocyte colony stimulating factor (G-CSF) subcutaneously for 5 days prior to bone marrow harvesting. G-CSF treatment of donors increased bone marrow harvest cell yields by over 2.5-fold, and preliminary results for engraftment are encouraging (authors’ unpublished data). Importantly, none of the 22 SCD patients treated with HLA-haploidentical BMT has died or developed chronic GVHD or any serious long-term morbidity.
Fig. 7. Schema for reduced intensity conditioning and HLA-haploidentical bone marrow transplantation for patients with sickle cell disease.

The table refers to sequential modifications that were applied to the conditioning regimen, treatment of the donor, or to post-transplantation prophylaxis of graft-versus-host disease.
The current transplantation regimen for patients with sickle cell disease incorporates three components that have been shown in mouse models to improve engraftment of MHC-disparate hematopoietic stem cell grafts: (i) inclusion of a lymphocyte-depleting antibody in the conditioning regimen (39); (ii) transplantation of a high dose of cells (78, 79); and (iii) post-transplantation cyclophosphamide (40, 41).
Tolerance of solid organ allografts
A final clinical application of hematopoietic stem cell transplantation is in the induction of tolerance to solid organ allografts. Based upon early observations that natural (80) or induced (28, 81) hematopoietic chimeras are tolerant of solid organ allografts from the same donor (82), there has been substantial interest in combining solid organ and hematopoietic stem cell transplantation (HSCT) from the same donor to achieve donor hematopoietic chimerism and durable tolerance of the solid organ. Indeed, patients who develop kidney or liver failure after undergoing allogeneic stem cell transplantation for a hematologic disorder can be successfully transplanted from the same donor and weaned off immunosuppression without experiencing graft rejection (83). However, the morbidity and mortality of lethal conditioning and allogeneic SCT have precluded its application to the induction of chimerism and solid organ allograft tolerance.
A 1998 review of the published literature (84) identified four patients who were treated with allogeneic bone marrow transplantation for a hematologic disorder, developed end-stage renal disease, and then received a kidney allograft from the original stem cell donor (85–87). At the time of reporting, there were no episodes of kidney failure or rejection and all patients were off all immunosuppressive drugs with no evidence of recurrent hematologic disorder or renal dysfunction. These isolated case reports provided the proof of principle that durable donor hematopoietic chimerism permits the safe discontinuation of pharmacologic immunosuppression following transplantation of a kidney from the same donor. Still, the considerable toxicities of HSCT militated against its use solely as an adjunct to SOT to achieve immunosuppression-free tolerance. Spitzer et al. (88–90) performed combined HLA-matched sibling kidney and bone marrow transplants for seven patients with multiple myeloma and myeloma-related end stage renal disease (ESRD). Interestingly, three patients developed tolerance of the transplanted organ and were successfully weaned off all immunosuppression even though donor hematopoietic chimerism was transient. Scandling et al. at Stanford University (91) performed HLA-matched sibling kidney transplants followed by administration of total lymphoid irradiation and anti-thymocyte globulin, infusion of T cells and CD34+ stem cells from the same donor, prophylaxis of GVHD, and graft rejection using mycophenolate mofetil and cyclosporine, and attempted discontinuation of all immunosuppressive drugs as early as 6 months after transplantation. Twelve patients were treated and eight were successfully weaned off all immunosuppressive drugs. Although one of these patients died suddenly while exercising, the remaining seven have normal kidney function with follow-up ranging from 12 to 36 months since discontinuation of anti-rejection drugs. This approach is now being applied to patients receiving combined transplants from an HLA-haploidentical related donor.
The group at Massachusetts General Hospital treated five ESRD patients with non-myeloablative conditioning and combined kidney and bone marrow transplantation from HLA-haploidentical related donors (92). Although donor chimerism was lost in all five patients within the first 21 days after combined transplantation, four were successfully weaned off immunosuppression. The investigators have updated the outcomes of these five patients plus an additional five patients receiving a similar transplantation protocol (93). Nine of the 10 patients developed acute kidney injury, with or without fever and fluid retention, as part of an idiopathic capillary leak syndrome that is similar clinically to engraftment syndrome seen after autologous or allogeneic HSCT (94). As of the most recent reporting, four of the ten patients remain tolerant as defined by a normally functioning transplanted kidney in the absence of maintenance immunosuppression (95). Three patients could not be weaned from immunosuppression and three patients have been returned to mycophenolate mofetil monotherapy, two for chronic rejection developing more than 6 years after transplantation, and one for recurrence of the original disease, membranoproliferative glomerulonephritis (Megan Sykes, personal communication). The most remarkable result is that transplantation tolerance was achieved in several patients who had only transient donor hematopoietic chimerism. Mechanistic studies provided evidence for the generation of CD4+ CD25+ CD127− FOXP3+ regulatory T cells that suppress responses against donor cells for up to 1 year after transplantation, but after then tolerance appears to be mediated by clonal deletion or by anergy (96). If tolerance could be generated reliably without a need for sustained hematopoietic chimerism, it would be the preferred approach for the induction of transplantation tolerance in the clinic because it would not incur the risk of acute or chronic GVHD.
Leventhal et al. (97) developed an approach to combined kidney and stem cell transplantation using an engineered cellular product enriched for hematopoietic stem cells (HSCs) and tolerogenic graft facilitating cells (FCs). Eight patients were conditioned with fludarabine, cyclophosphamide, and 200 centigray total body irradiation, transplanted on the same day with a kidney plus stem cells from a partially mismatched related or unrelated donor, and then received post-transplantation immunosuppression with high dose cyclophosphamide (50 mg/kg IV on day 3), tacrolimus, and mycophenolate mofetil (98). Two patients exhibited only transient chimerism and have been maintained on tacrolimus monotherapy, while one patient developed viral sepsis and renal artery thrombosis. The remaining five patients developed complete donor hematopoietic chimerism and were successfully weaned off immunosuppression by 1 year after transplantation. None of these five patients developed anti-donor HLA antibody, engraftment syndrome, or GVHD. These results represent the first demonstration of durable chimerism and tolerance without maintenance immunosuppression following combined HLA-mismatched kidney and hematopoietic stem cell transplantation from the same donor. The same group provided an update on the original eight patients plus another seven recipients of combined kidney and stem cell transplantation (99). A total of nine patients achieved full donor hematopoietic chimerism; six of these were successfully weaned off immunosuppression, two are being weaned, and one patient developed an atypical viral infection followed by bone marrow failure and kidney graft loss requiring re-transplantation. Five patients had either transient chimerism (n = 4) or stable mixed chimerism on continuing immunosuppression (n = 1), and only one patient had total failure of bone marrow engraftment. The most reliable predictor of clinical tolerance, i.e., the ability to wean the patient off immunosuppression without the development of graft rejection, was peripheral blood T-cell chimerism. All patients with full donor T-cell chimerism demonstrated hyporesponsiveness to both donor and recipient cells by in vitro assays and were fully tolerant by clinical measures. By contrast, in vitro unresponsiveness to donor cells was documented in some patients with transient donor chimerism, but these patients were not clinically tolerant as they could not be weaned off immunosuppression without incurring rejection episodes.
The studies of Leventhal et al. represent a significant milestone on the road to transplantation tolerance, but follow-up is still relatively short (1–26 months) and a number of questions and challenges remain. First, it is not known whether the patients with full donor hematopoietic chimerism will have normally functioning immune systems in the long-term. Some patients received fully HLA-mismatched stem cell and kidney grafts, and some (100, 101) but not all (102) animal studies suggest that fully MHC-mismatched bone marrow chimeras are not competent to respond to infection. Second, it is not entirely clear which components of the transplantation regimen are required for the induction of transplantation tolerance. A major question is whether the engineered cellular product containing graft facilitating cells is required for tolerance induction, or could the same results have been achieved with either unprocessed bone marrow or peripheral blood stem cells. The results clearly suggest that sustained donor hematopoietic chimerism is sufficient for tolerance to the transplanted kidney without maintenance immunosuppression. But is sustained donor hematopoietic chimerism necessary for transplantation tolerance? The results of Kawai et al. would suggest that durable hematopoietic chimerism is not absolutely required for solid organ allograft tolerance, yet it is not clear from their studies why some transient chimeras became tolerant of the donor kidney and some did not. Finally, is post-transplantation cyclophosphamide a critical component of tolerance induction? If so, can solid organ allograft tolerance be induced by PT/Cy in the absence of hematopoietic stem cell transplantation? The Immune Tolerance Network in the United States is sponsoring a clinical trial of combined bone marrow and kidney transplantation in which patients will receive reduced intensity conditioning, unprocessed bone marrow from G-CSF stimulated donors, and GVHD prophylaxis including PT/Cy on day 3 and 4 after transplantation (similar to the last group of patients in Fig. 6, except that tacrolimus will be used instead of sirolimus). Since the graft is not engineered, this trial will address the question of whether an engineered product containing graft facilitating cells is essential to transplantation tolerance induction.
Is hematopoietic chimerism required for transplantation tolerance?
Life would be so much easier if only a transplantation immunologist had a magic wand. She would wave the wand and poof! Her patient, the solid organ transplant recipient, would be a stable mixed hematopoietic chimera with an equal mix of donor and host blood cells. There are many advantages to being a stable mixed hematopoietic chimera. Stable mixed hematopoietic chimerism equates to intrathymic mixed chimerism, clonal deletion of both host- and donor-reactive T cells, and lifelong tolerance of organs transplanted from the same donor without any requirement for maintenance immunosuppression (103–106). Furthermore, mixed chimeras demonstrate superior immunocompetence compared with full donor hematopoietic chimeras (107). Unfortunately, magic wands do not exist, and stable mixed chimerism is very, very hard to induce in the clinic, especially when the donor and recipient are partially or fully HLA-mismatched to each other. Induction of significant donor chimerism (>10% donor cells) requires the delivery of myelotoxic (108) and immunosuppressive conditioning and hematopoietic stem cell transplantation, and this therapy necessarily entails the risks of conditioning regimen toxicity, infection, graft-versus-host disease, and death. For patients undergoing kidney transplantation, these risks can be hard to justify since 1 year patient and graft survivals exceed 95%. It is quite tempting, especially for those without experience in hematopoietic stem cell transplantation, to attempt to induce transplantation tolerance using less toxic methods, such as regulatory T-cell infusion (7, 109, 110) or costimulatory blockade (111–113). But can these methods induce tolerance in alloreactive human T cells and maintain tolerance despite continued thymic output of non-tolerant T cells and the vicissitudes of trauma, danger signaling, intermittent infections, and inflammation?
There is increasing recognition and acceptance that pre-existing memory in the alloreactive repertoire is a major barrier to the induction and maintenance of transplantation tolerance in primates (114), including humans. Rodents exposed to viruses contain virus-specific memory T cells that cross-react against allogeneic cells (115, 116). In humans, approximately 45% of virus-specific T-cell clones react against allogeneic HLA molecules (117). Compared with naive T cells, memory T cells appear to be more resistant to depletion by anti-T cell antibodies (118–120), to tolerance induction by costimulatory blockade (116, 121), or to suppression by regulatory T cells (122). Bacterial or viral infection may generate a cadre of memory cells that cross-react against transplanted alloantigens, which may explain how infection can prevent or abrogate transplantation tolerance (123–125).
If one accepts that the repertoire of alloreactive T cells in humans contains memory cells, then a simple thought experiment should give pause to the clinician who endeavors to induce transplantation tolerance using infusions of alloreactive regulatory T cells. If regulatory T cells could turn off alloreactive memory T cells, then why wouldn’t they turn off T cells against persistent viruses, especially through their capacity to mediate ‘infectious’ tolerance (126–128)? The same considerations apply to tolerance by costimulatory blockade: if such an approach could induce tolerance in alloreactive memory T cells, why does not it also induce tolerance in T cells responsible for protection of the organism against pathogens? These considerations may not figure prominently in experiments involving mice housed in pathogen-free conditions, but indiscriminate tolerance induction, even if it were possible, could wreak havoc in patients by releasing viruses such as herpesviruses (Epstein–Barr, cytomegalovirus, varicella), human papillomavirus, or hepatitis viruses from immune surveillance. For this reason, certain memory T cells should be highly resistant to tolerance induction, and the only way to induce tolerance in such cells would be to kill them with other T cells. And that brings us back to stem cell transplantation and donor hematopoietic chimerism.
In the spirit of healthy debate, permit me to propose the following: (i) the HLA-alloreactive repertoire of most adult humans contains T cells that are effectively implacable: unless they are killed first, they will destroy a transplanted allograft; (ii) immunosuppressive agents such as fludarabine, cyclophosphamide, anti-thymocyte globulin, and total lymphoid or total body irradiation can greatly reduce the number of the patient’s alloreactive T cells but cannot eliminate them entirely (129); (iii) the only agent that can eliminate residual alloreactive host T cells are alloreactive donor T cells; and (iv) pharmacologic immunosuppression such as calcineurin inhibitors, mycophenolate mofetil, and sirolimus may succeed in achieving a suspension of hostilities between host and donor immune systems, but once these drugs are stopped, the graft will be rejected or full donor hematopoietic chimerism will ensue. If these proposals are valid, then the only condition that guarantees tolerance of an HLA-mismatched allograft is full donor hematopoietic chimerism. Fortunately, with advances in reduced intensity conditioning regimens, hematopoietic graft engineering, and post-transplantation immunosuppression, we are now in a position to put these hypotheses to the test.
Acknowledgments
Supported by grant P01 CA15396 from the National Cancer Institute and contract #12086058 from the Immune Tolerance Network.
Footnotes
The authors have no conflicts of interest to declare.
References
- 1.Bishop GA, et al. Approaching the promise of operational tolerance in clinical transplantation. Transplantation. 2011;91:1065–1074. doi: 10.1097/TP.0b013e318215e742. [DOI] [PubMed] [Google Scholar]
- 2.Feng S, et al. Complete immunosuppression withdrawal and subsequent allograft function among pediatric recipients of parental living donor liver transplants. JAMA. 2012;307:283–293. doi: 10.1001/jama.2011.2014. [DOI] [PubMed] [Google Scholar]
- 3.Grinyo JM. Why is organ transplantation clinically important? Cold Spring Harb Perspect Med. 2013;3:1–10. doi: 10.1101/cshperspect.a014985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li HW, Sykes M. Emerging concepts in haematopoietic cell transplantation. Nat Rev Immunol. 2012;12:403–416. doi: 10.1038/nri3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lakkis FG, Lechler RI. Origin and Biology of the Allogeneic Response. Cold Spring Harb Perspect Med. 2013;3:a014993. doi: 10.1101/cshperspect.a014993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kawai T, Sachs DH. Tolerance induction: hematopoietic chimerism. Curr Opin Organ Transplant. 2013;18:402–407. doi: 10.1097/MOT.0b013e328363621d. [DOI] [PubMed] [Google Scholar]
- 7.Wood KJ, Bushell A, Hester J. Regulatory immune cells in transplantation. Nat Rev Immunol. 2012;12:417–430. doi: 10.1038/nri3227. [DOI] [PubMed] [Google Scholar]
- 8.Cobbold SP, Waldmann H. Regulatory cells and transplantation tolerance. Cold Spring Harb Perspect Med. 2013;3:a015545. doi: 10.1101/cshperspect.a015545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Issa F, Wood KJ. Translating tolerogenic therapies to the clinic: where do we stand? Front Immunol. 2012;3:254. doi: 10.3389/fimmu.2012.00254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lafferty KJ, Cunningham AJ. A new analysis of allogeneic interactions. Aust J Exp Biol Med Sci. 1975;53:27–42. doi: 10.1038/icb.1975.3. [DOI] [PubMed] [Google Scholar]
- 11.Bretscher P, Cohn M. A theory of self-nonself discrimination: paralysis and induction involve the recognition of one and two determinants on an antigen, respectively. Science. 1970;169:1042–1049. doi: 10.1126/science.169.3950.1042. [DOI] [PubMed] [Google Scholar]
- 12.Lafferty KJ, Prowse SJ, Simeonovic CJ, Warren HS. Immunobiology of tissue transplantation: a return to the passenger leukocyte concept. Annu Rev Immunol. 1983;1:143–173. doi: 10.1146/annurev.iy.01.040183.001043. [DOI] [PubMed] [Google Scholar]
- 13.Lechler RI, Batchelor JR. Restoration of immunogenicity to passenger cell-depleted kidney allografts by the addition of donor strain dendritic cells. J Exp Med. 1982;155:31–41. doi: 10.1084/jem.155.1.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Janeway CA., Jr Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol. 1989;54:1–13. doi: 10.1101/sqb.1989.054.01.003. [DOI] [PubMed] [Google Scholar]
- 15.Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. 1994;12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015. [DOI] [PubMed] [Google Scholar]
- 16.Mueller DL, Jenkins MK, Schwartz RH. Clonal expansion versus functional clonal inactivation: a costimulatory signalling pathway determines the outcome of T cell antigen receptor occupancy. Annu Rev Immunol. 1989;7:445–480. doi: 10.1146/annurev.iy.07.040189.002305. [DOI] [PubMed] [Google Scholar]
- 17.Lamb JR, Skidmore BJ, Green N, Chiller JM, Feldman M. Induction of tolerance in influenza virus-immune T lymphocyte clones with synthetic peptides of influenza hemagglutinin. J Exp Med. 1983;157:1434–1437. doi: 10.1084/jem.157.5.1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jenkins MK, Schwartz RH. Antigen presentation by chemically modified splenocytes induces antigen-specific T cell unresponsiveness in vitro and in vivo. J Exp Med. 1987;165:302–319. doi: 10.1084/jem.165.2.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Linsley PS, Clark EA, Ledbetter JA. T-cell antigen CD28 mediates adhesion with B cells by interacting with activation antigen B7/BB-1. Proc Natl Acad Sci USA. 1990;87:5031–5035. doi: 10.1073/pnas.87.13.5031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vincenti F, Dritselis A, Kirkpatrick P. Belatacept. Nat Rev Drug Discov. 2011;10:655–656. doi: 10.1038/nrd3536. [DOI] [PubMed] [Google Scholar]
- 21.Lassila O, Vainio O, Matzinger P. Can B cells turn on virgin T cells? Nature. 1988;334:253–255. doi: 10.1038/334253a0. [DOI] [PubMed] [Google Scholar]
- 22.Fuchs EJ, Matzinger P. B cells turn off virgin but not memory T cells. Science. 1992;258:1156–1159. doi: 10.1126/science.1439825. [DOI] [PubMed] [Google Scholar]
- 23.Ashwell JD, Jenkins MK, Schwartz RH. Effect of gamma radiation on resting B lymphocytes. II. Functional characterization of the antigen-presentation defect. J Immunol. 1988;141:2536–2544. [PubMed] [Google Scholar]
- 24.Ranheim EA, Kipps TJ. Activated T cells induce expression of B7/BB1 on normal or leukemic B cells through a CD40-dependent signal. J Exp Med. 1993;177:925–935. doi: 10.1084/jem.177.4.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fuchs E. Two signal model of lymphocyte activation. Immunol Today. 1992;13:462. doi: 10.1016/0167-5699(92)90077-K. [DOI] [PubMed] [Google Scholar]
- 26.Ridge JP, Fuchs EJ, Matzinger P. Neonatal tolerance revisited: turning on newborn T cells with dendritic cells. Science. 1996;271:1723–1726. doi: 10.1126/science.271.5256.1723. [DOI] [PubMed] [Google Scholar]
- 27.Burnet FM, Fenner F. The production of antibodies. London: Macmillan; 1949. pp. 102–105. [Google Scholar]
- 28.Billingham RE, Brent L, Medawar PB. Actively acquired tolerance of foreign cells. Nature. 1953;172:603–606. doi: 10.1038/172603a0. [DOI] [PubMed] [Google Scholar]
- 29.Lederberg J. Genes and antibodies: do antigens bear instructions for antibody specificity or do they select cell lines that arise by mutation? Science. 1959;129:1649–1653. doi: 10.1126/science.129.3364.1649. [DOI] [PubMed] [Google Scholar]
- 30.Mayumi H, Umesue M, Nomoto K. Cyclophosphamide-induced immunological tolerance: an overview. Immunobiol. 1996;195:129–139. doi: 10.1016/S0171-2985(96)80033-7. [DOI] [PubMed] [Google Scholar]
- 31.Schwartz R, Dameshek W. Drug-induced immunological tolerance. Nature. 1959;183:1682–1683. doi: 10.1038/1831682a0. [DOI] [PubMed] [Google Scholar]
- 32.Berenbaum MC, Brown IN. Dose-response relationships for agents inhibiting the immune response. Immunology. 1964;7:65–71. [PMC free article] [PubMed] [Google Scholar]
- 33.Berenbaum MC. Prolongation of homograft survival in mice with single doses of cyclophosphamide. Nature. 1963;200:84. doi: 10.1038/200084a0. [DOI] [PubMed] [Google Scholar]
- 34.Mayumi H, Himeno K, Tokuda N, Nomoto K. Drug-induced tolerance to allografts in mice. VII. Optimal protocol and mechanism of cyclophosphamide-induced tolerance in an H-2 haplotype-identical strain combination. Transplant Proc. 1986;18:363–369. [PubMed] [Google Scholar]
- 35.Mayumi H, Himeno K, Shin T, Nomoto K. Drug-induced tolerance to allografts in mice. VI. Tolerance induction in H-2-haplotype-identical strain combinations in mice. Transplantation. 1985;40:188–194. doi: 10.1097/00007890-198508000-00016. [DOI] [PubMed] [Google Scholar]
- 36.Eto M, Mayumi H, Tomita Y, Yoshikai Y, Nishimura Y, Nomoto K. Sequential mechanisms of cyclophosphamide-induced skin allograft tolerance including the intrathymic clonal deletion followed by late breakdown of the clonal deletion. J Immunol. 1990;145:1303–1310. [PubMed] [Google Scholar]
- 37.Luznik L, Engstrom LW, Iannone R, Fuchs EJ. Posttransplantation cyclophosphamide facilitates engraftment of major histocompatibility complex-identical allogeneic marrow in mice conditioned with low-dose total body irradiation. Biol Blood Marrow Transplant. 2002;8:131–138. doi: 10.1053/bbmt.2002.v8.pm11939602. [DOI] [PubMed] [Google Scholar]
- 38.Colson YL, et al. A nonlethal conditioning approach to achieve durable multilineage mixed chimerism and tolerance across major, minor, and hematopoietic histocompatibility barriers. J Immunol. 1995;155:4179–4188. [PubMed] [Google Scholar]
- 39.Colson YL, Li H, Boggs SS, Patrene KD, Johnson PC, Ildstad ST. Durable mixed allogeneic chimerism and tolerance by a nonlethal radiation-based cytoreductive approach. J Immunol. 1996;157:2820–2829. [PubMed] [Google Scholar]
- 40.Mayumi H, Good RA. Long-lasting skin allograft tolerance in adult mice induced across fully allogeneic (multimajor H-2 plus multiminor histocompatibility) antigen barriers by a tolerance-inducing method using cyclophosphamide. J Exp Med. 1989;169:213–238. doi: 10.1084/jem.169.1.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luznik L, Jalla S, Engstrom LW, Iannone R, Fuchs EJ. Durable engraftment of major histocompatibility complex-incompatible cells after nonmyeloablative conditioning with fludarabine, low-dose total body irradiation, and posttransplantation cyclophosphamide. Blood. 2001;98:3456–3464. doi: 10.1182/blood.v98.12.3456. [DOI] [PubMed] [Google Scholar]
- 42.O’Donnell PV, et al. Nonmyeloablative bone marrow transplantation from partially HLA-mismatched related donors using posttransplantation cyclophosphamide. Biol Blood Marrow Transplant. 2002;8:377–386. doi: 10.1053/bbmt.2002.v8.pm12171484. [DOI] [PubMed] [Google Scholar]
- 43.Luznik L, et al. HLA-haploidentical bone marrow transplantation for hematologic malignancies using nonmyeloablative conditioning and high-dose, posttransplantation cyclophosphamide. Biol Blood Marrow Transplant. 2008;14:641–650. doi: 10.1016/j.bbmt.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Anasetti C, et al. Effect of HLA incompatibility on graft-versus-host disease, relapse, and survival after marrow transplantation for patients with leukemia or lymphoma. Hum Immunol. 1990;29:79–91. doi: 10.1016/0198-8859(90)90071-v. [DOI] [PubMed] [Google Scholar]
- 45.Szydlo R, et al. Results of allogeneic bone marrow transplants for leukemia using donors other than HLA-identical siblings. J Clin Oncol. 1997;15:1767–1777. doi: 10.1200/JCO.1997.15.5.1767. [DOI] [PubMed] [Google Scholar]
- 46.Beatty PG, et al. Marrow transplantation from related donors other than HLA-identical siblings. N Engl J Med. 1985;313:765–771. doi: 10.1056/NEJM198509263131301. [DOI] [PubMed] [Google Scholar]
- 47.Horowitz MM, et al. Graft-versus-leukemia reactions after bone marrow transplantation. Blood. 1990;75:555–562. [PubMed] [Google Scholar]
- 48.Dey BR, et al. Anti-tumour response despite loss of donor chimaerism in patients treated with non-myeloablative conditioning and allogeneic stem cell transplantation. Br J Haematol. 2005;128:351–359. doi: 10.1111/j.1365-2141.2004.05328.x. [DOI] [PubMed] [Google Scholar]
- 49.Rubio MT, Kim YM, Sachs T, Mapara M, Zhao G, Sykes M. Antitumor effect of donor marrow graft rejection induced by recipient leukocyte infusions in mixed chimeras prepared with nonmyeloablative conditioning: critical role for recipient-derived IFN-γ. Blood. 2003;102:2300–2307. doi: 10.1182/blood-2002-12-3949. [DOI] [PubMed] [Google Scholar]
- 50.Rubio MT, Saito TI, Kattleman K, Zhao G, Buchli J, Sykes M. Mechanisms of the antitumor responses and host-versus-graft reactions induced by recipient leukocyte infusions in mixed chimeras prepared with nonmyeloablative conditioning: a critical role for recipient CD4+ T cells and recipient leukocyte infusion-derived IFN-γ-producing CD8+ T Cells. J Immunol. 2005;175:665–676. doi: 10.4049/jimmunol.175.2.665. [DOI] [PubMed] [Google Scholar]
- 51.Saito TI, Rubio MT, Sykes M. Clinical relevance of recipient leukocyte infusion as antitumor therapy following nonmyeloablative allogeneic hematopoietic cell transplantation. Exp Hematol. 2006;34:1270–1276. doi: 10.1016/j.exphem.2006.04.022. [DOI] [PubMed] [Google Scholar]
- 52.Saito TI, Li HW, Sykes M. Invariant NKT cells are required for antitumor responses induced by host-versus-graft responses. J Immunol. 2010;185:2099–2105. doi: 10.4049/jimmunol.0901985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Symons HJ, et al. The allogeneic effect revisited: exogenous help for endogenous, tumor-specific T cells. Biol Blood Marrow Transplant. 2008;14:499–509. doi: 10.1016/j.bbmt.2008.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bennett SR, Carbone FR, Karamalis F, Miller JF, Heath WR. Induction of a CD8+ cytotoxic T lymphocyte response by cross-priming requires cognate CD4+ T cell help. J Exp Med. 1997;186:65–70. doi: 10.1084/jem.186.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bennett SR, Carbone FR, Karamalis F, Flavell RA, Miller JF, Heath WR. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature. 1998;393:478–480. doi: 10.1038/30996. [DOI] [PubMed] [Google Scholar]
- 56.Ridge JP, Di Rosa F, Matzinger P. A conditioned dendritic cell can be a temporal bridge between a CD4+ T- helper and a T-killer cell. Nature. 1998;393:474–478. doi: 10.1038/30989. [DOI] [PubMed] [Google Scholar]
- 57.Schoenberger SP, Toes RE, van der Voort EI, Offringa R, Melief CJ. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature. 1998;393:480–483. doi: 10.1038/31002. [DOI] [PubMed] [Google Scholar]
- 58.Feau S, Garcia Z, Arens R, Yagita H, Borst J, Schoenberger SP. The CD4+ T-cell help signal is transmitted from APC to CD8+ T-cells via CD27-CD70 interactions. Nat Commun. 2012;3:948. doi: 10.1038/ncomms1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zajac AJ, et al. Viral immune evasion due to persistence of activated T cells without effector function. J Exp Med. 1998;188:2205–2213. doi: 10.1084/jem.188.12.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Aubert RD, et al. Antigen-specific CD4 T-cell help rescues exhausted CD8 T cells during chronic viral infection. PNAS. 2011;108:21182–21187. doi: 10.1073/pnas.1118450109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ding ZC, et al. Polyfunctional CD4+ T cells are essential for eradicating advanced B-cell lymphoma after chemotherapy. Blood. 2012;120:2229–2239. doi: 10.1182/blood-2011-12-398321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Colvin GA, et al. Nonengraftment haploidentical cellular immunotherapy for refractory malignancies: tumor responses without chimerism. Biol Blood Marrow Transplant. 2009;15:421–431. doi: 10.1016/j.bbmt.2008.12.503. [DOI] [PubMed] [Google Scholar]
- 63.Medina DJ, et al. A pilot study of allogeneic cellular therapy for patients with advanced hematologic malignancies. Leukemia Res. 2008;32:1842–1848. doi: 10.1016/j.leukres.2008.05.015. [DOI] [PubMed] [Google Scholar]
- 64.Strair RK, et al. Antineoplastic effects of partially HLA-matched irradiated blood mononuclear cells in patients with renal cell carcinoma. J Clin Oncol. 2003;21:3785–3791. doi: 10.1200/JCO.2003.05.094. [DOI] [PubMed] [Google Scholar]
- 65.Guo M, et al. Infusion of HLA-mismatched peripheral blood stem cells improves the outcome of chemotherapy for acute myeloid leukemia in elderly patients. Blood. 2011;117:936–941. doi: 10.1182/blood-2010-06-288506. [DOI] [PubMed] [Google Scholar]
- 66.Guo M, et al. HLA-mismatched stem-cell microtransplantation as postremission therapy for acute myeloid leukemia: long-term follow-up. J Clin Oncol. 2012;30:4084–4090. doi: 10.1200/JCO.2012.42.0281. [DOI] [PubMed] [Google Scholar]
- 67.Schwarzenberg L, Mathe G, Schneider M, Amiel JL, Cattan A, Schlumberger JR. Attempted adoptive immunotherapy of acute leukaemia by leucocyte transfusions. Lancet. 1966;2:365–368. doi: 10.1016/s0140-6736(66)92661-4. [DOI] [PubMed] [Google Scholar]
- 68.Griffith LM, et al. Allogeneic hematopoietic cell transplantation for primary immune deficiency diseases: current status and critical needs. J Allergy Clin Immunol. 2008;122:1087–1096. doi: 10.1016/j.jaci.2008.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hutter G, et al. Long-term control of HIV by CCR5 delta32/delta32 stem-cell transplantation. N Engl J Med. 2009;360:692–698. doi: 10.1056/NEJMoa0802905. [DOI] [PubMed] [Google Scholar]
- 70.Pasquini MC, et al. Transplantation for autoimmune diseases in North and South America: a report of the Center for International Blood and Marrow Transplant Research. Biol Blood Marrow Transplant. 2012;18:1471–1478. doi: 10.1016/j.bbmt.2012.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wynn R. Stem cell transplantation in inherited metabolic disorders. ASH Education Program Book. 2011;2011:285–291. doi: 10.1182/asheducation-2011.1.285. [DOI] [PubMed] [Google Scholar]
- 72.Armand P, Antin JH. Allogeneic stem cell transplantation for aplastic anemia. Biol Blood Marrow Transplant. 2007;13:505–516. doi: 10.1016/j.bbmt.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 73.Lucarelli G, Isgro A, Sodani P, Gaziev J. Hematopoietic stem cell transplantation in thalassemia and sickle cell anemia. Cold Spring Harb Perspect Med. 2012;2:a011825. doi: 10.1101/cshperspect.a011825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bernaudin F, et al. Long-term results of related myeloablative stem-cell transplantation to cure sickle cell disease. Blood. 2007;110:2749–2756. doi: 10.1182/blood-2007-03-079665. [DOI] [PubMed] [Google Scholar]
- 75.Panepinto JA, et al. Matched-related donor transplantation for sickle cell disease: report from the Center for International Blood and Transplant Research. Br J Haematol. 2007;137:479–485. doi: 10.1111/j.1365-2141.2007.06592.x. [DOI] [PubMed] [Google Scholar]
- 76.Chakrabarti S, Bareford D. A survey on patient perception of reduced-intensity transplantation in adults with sickle cell disease. Bone Marrow Transplant. 2007;39:447–451. doi: 10.1038/sj.bmt.1705622. [DOI] [PubMed] [Google Scholar]
- 77.Brodsky RA, Luznik L, Bolanos-Meade J, Leffell MS, Jones RJ, Fuchs EJ. Reduced intensity: HLA-haploidentical BMT with post transplantation cyclophosphamide in nonmalignant hematologic diseases. Bone Marrow Transplant. 2008;42:523–527. doi: 10.1038/bmt.2008.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sykes M, Szot GL, Swenson KA, Pearson DA. Induction of high levels of allogeneic hematopoietic reconstitution and donor-specific tolerance without myelosuppressive conditioning. Nat Med. 1997;3:783–787. doi: 10.1038/nm0797-783. [DOI] [PubMed] [Google Scholar]
- 79.Bachar-Lustig E, Rachamim N, Li HW, Lan F, Reisner Y. Megadose of T cell-depleted bone marrow overcomes MHC barriers in sublethally irradiated mice. Nat Med. 1995;1:1268–1273. doi: 10.1038/nm1295-1268. [DOI] [PubMed] [Google Scholar]
- 80.Owen RD. Immunogenetic consequences of vascular anastomoses between bovine twins. Science. 1945;102:400–401. doi: 10.1126/science.102.2651.400. [DOI] [PubMed] [Google Scholar]
- 81.Sharabi Y, Sachs DH. Mixed chimerism and permanent specific transplantation tolerance induced by a nonlethal preparative regimen. J Exp Med. 1989;169:493–502. doi: 10.1084/jem.169.2.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Anderson D, Billingham RE, Lampkin GH, Medawar PB. The use of skin grafting to distinguish between monozygotic and dizygotic twins in cattle. Heredity. 1951;5:379–397. [Google Scholar]
- 83.Koenecke C, et al. Solid organ transplantation after allogeneic hematopoietic stem cell transplantation: a retrospective, multicenter study of the EBMT. Am J Transplant. 2010;10:1897–1906. doi: 10.1111/j.1600-6143.2010.03187.x. [DOI] [PubMed] [Google Scholar]
- 84.Dey B, Sykes M, Spitzer TR. Outcomes of recipients of both bone marrow and solid organ transplants: a review. Medicine. 1998;77:355–369. doi: 10.1097/00005792-199809000-00005. [DOI] [PubMed] [Google Scholar]
- 85.Helg C, et al. Renal transplantation without immunosuppression in a host with tolerance induced by allogeneic bone marrow transplantation. Transplantation. 1994;58:1420–1422. [PubMed] [Google Scholar]
- 86.Sayegh MH, Fine NA, Smith JL, Rennke HG, Milford EL, Tilney NL. Immunologic tolerance to renal allografts after bone marrow transplants from the same donors. Ann Intern Med. 1991;114:954–955. doi: 10.7326/0003-4819-114-11-954. [DOI] [PubMed] [Google Scholar]
- 87.Sorof JM, Koerper MA, Portale AA, Potter D, DeSantes K, Cowan M. Renal transplantation without chronic immunosuppression after T cell-depleted. HLA-mismatched bone marrow transplantation. Transplantation. 1995;59:1633–1635. [PubMed] [Google Scholar]
- 88.Spitzer TR, et al. Combined histocompatibility leukocyte antigen-matched donor bone marrow and renal transplantation for multiple myeloma with end stage renal disease: the induction of allograft tolerance through mixed lymphohematopoietic chimerism. Transplantation. 1999;68:480–484. doi: 10.1097/00007890-199908270-00006. [DOI] [PubMed] [Google Scholar]
- 89.Fudaba Y, et al. Myeloma responses and tolerance following combined kidney and nonmyeloablative marrow transplantation. In vivo and in vitro analyses. Am J Transplant. 2006;6:2121–2133. doi: 10.1111/j.1600-6143.2006.01434.x. [DOI] [PubMed] [Google Scholar]
- 90.Spitzer TR, et al. Long-term follow-up of recipients of combined human leukocyte antigen-matched bone marrow and kidney transplantation for multiple myeloma with end-stage renal disease. Transplantation. 2011;91:672–676. doi: 10.1097/TP.0b013e31820a3068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Scandling JD, Busque S, Shizuru JA, Engleman EG, Strober S. Induced immune tolerance for kidney transplantation. N Engl J Med. 2011;365:1359–1360. doi: 10.1056/NEJMc1107841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kawai T, et al. HLA-mismatched renal transplantation without maintenance immunosuppression. N Engl J Med. 2008;358:353–361. doi: 10.1056/NEJMoa071074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Farris AB, et al. Acute renal endothelial injury during marrow recovery in a cohort of combined kidney and bone marrow allografts. Am J Transplant. 2011;11:1464–1477. doi: 10.1111/j.1600-6143.2011.03572.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Spitzer TR. Engraftment syndrome following hematopoietic stem cell transplantation. Bone Marrow Transplant. 2001;27:893–898. doi: 10.1038/sj.bmt.1703015. [DOI] [PubMed] [Google Scholar]
- 95.Kawai T, Sachs DH, Sykes M, Cosimi AB. HLA-Mismatched renal transplantation without maintenance immunosuppression. N Engl J Med. 2013;368:1850–1852. doi: 10.1056/NEJMc1213779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Andreola G, et al. Mechanisms of donor-specific tolerance in recipients of haploidentical combined bone marrow/kidney transplantation. Am J Transplant. 2011;11:1236–1247. doi: 10.1111/j.1600-6143.2011.03566.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Huang Y, Bozulic LD, Miller T, Xu H, Hussain LR, Ildstad ST. CD8α+ plasmacytoid precursor DCs induce antigen-specific regulatory T cells that enhance HSC engraftment in vivo. Blood. 2011;117:2494–2505. doi: 10.1182/blood-2010-06-291187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Leventhal J, et al. Chimerism and tolerance without GVHD or engraftment syndrome in hla-mismatched combined kidney and hematopoietic stem cell transplantation. Sci Transl Med. 2012;4:124ra28. doi: 10.1126/scitranslmed.3003509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Leventhal J, et al. Tolerance induction in HLA disparate living donor kidney transplantation by donor stem cell infusion: durable chimerism predicts outcome. Transplantation. 2013;95:169–176. doi: 10.1097/TP.0b013e3182782fc1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zinkernagel RM, Althage A, Callahan G, Welsh RM., Jr On the immunocompetence of H-2 incompatible irradiation bone marrow chimeras. J Immunol. 1980;124:2356–2365. [PubMed] [Google Scholar]
- 101.Zinkernagel RM, Callahan GN, Althage A, Cooper S, Klein PA, Klein J. On the thymus in the differentiation of “H-2 self-recognition” by T cells: evidence for dual recognition? J Exp Med. 1978;147:882–896. doi: 10.1084/jem.147.3.882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Matzinger P, Mirkwood G. In a fully H-2 incompatible chimera, T cells of donor origin can respond to minor histocompatibility antigens in association with either donor or host H-2 type. J Exp Med. 1978;148:84–92. doi: 10.1084/jem.148.1.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lubaroff DM, Silvers WK. The importance of chimerism in maintaining tolerance of skin allografts in mice. J Immunol. 1973;111:65–71. [PubMed] [Google Scholar]
- 104.de Vries-van der Zwan A, Besseling AC, van Twuyver E, Boog CJ, de Waal LP. A substantial level of mixed chimerism is required for the induction of permanent transplantation tolerance. Transpl Immunol. 1996;4:232–240. doi: 10.1016/s0966-3274(96)80023-2. [DOI] [PubMed] [Google Scholar]
- 105.de Vries-van der Zwan A, van der Pol MA, Besseling AC, de Waal LP, Boog CJ. Haematopoietic stem cells can induce specific skin graft acceptance across full MHC barriers. Bone Marrow Transplant. 1998;22:91–98. doi: 10.1038/sj.bmt.1701277. [DOI] [PubMed] [Google Scholar]
- 106.Kanamoto A, Maki T. Chimeric donor cells play an active role in both induction and maintenance phases of transplantation tolerance induced by mixed chimerism. J Immunol. 2004;172:1444–1448. doi: 10.4049/jimmunol.172.3.1444. [DOI] [PubMed] [Google Scholar]
- 107.Ildstad ST, Wren SM, Bluestone JA, Barbieri SA, Sachs DH. Characterization of mixed allogeneic chimeras. Immunocompetence, in vitro reactivity, and genetic specificity of tolerance. J Exp Med. 1985;162:231–244. doi: 10.1084/jem.162.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Tomita Y, Sachs DH, Sykes M. Myelosuppressive conditioning is required to achieve engraftment of pluripotent stem cells contained in moderate doses of syngeneic bone marrow. Blood. 1994;83:939–948. [PubMed] [Google Scholar]
- 109.Hara M, et al. IL-10 is required for regulatory T cells to mediate tolerance to alloantigens in vivo. J Immunol. 2001;166:3789–3796. doi: 10.4049/jimmunol.166.6.3789. [DOI] [PubMed] [Google Scholar]
- 110.Sagoo P, Ali N, Garg G, Nestle FO, Lechler RI, Lombardi G. Human regulatory T cells with alloantigen specificity are more potent inhibitors of alloimmune skin graft damage than polyclonal regulatory T cells. Sci Transl Med. 2011;3:83ra42. doi: 10.1126/scitranslmed.3002076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Pearson TC, et al. CTLA4-Ig plus bone marrow induces long-term allograft survival and donor-specific unresponsiveness in the murine model: evidence for hematopoietic chimerism. Transplantation. 1996;61:997–1004. doi: 10.1097/00007890-199604150-00002. [DOI] [PubMed] [Google Scholar]
- 112.Rostaing L, et al. Long-term belatacept exposure maintains efficacy and safety at 5 years: results from the long-term extension of the BENEFIT study. Am J Transplant. 2013;13:2875–2883. doi: 10.1111/ajt.12460. [DOI] [PubMed] [Google Scholar]
- 113.Lakkis FG, et al. Blocking the CD28-B7 T cell costimulation pathway induces long term cardiac allograft acceptance in the absence of IL-4. J Immunol. 1997;158:2443–2448. [PubMed] [Google Scholar]
- 114.Nadazdin O, et al. Host alloreactive memory T cells influence tolerance to kidney allografts in nonhuman primates. Sci Transl Med. 2011;3:86ra51. doi: 10.1126/scitranslmed.3002093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Finberg R, Burakoff SJ, Cantor H, Benacerraf B. Biological significance of alloreactivity: T cells stimulated by Sendai virus-coated syngeneic cells specifically lyse allogeneic target cells. PNAS. 1978;75:5145–5149. doi: 10.1073/pnas.75.10.5145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Adams AB, et al. Heterologous immunity provides a potent barrier to transplantation tolerance. J Clin Invest. 2003;111:1887–1895. doi: 10.1172/JCI17477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Amir AL, et al. Allo-HLA reactivity of virus-specific memory T cells is common. Blood. 2010;115:3146–3157. doi: 10.1182/blood-2009-07-234906. [DOI] [PubMed] [Google Scholar]
- 118.Ayasoufi K, Yu H, Fan R, Wang X, Williams J, Valujskikh A. Pretransplant antithymocyte globulin has increased efficacy in controlling donor-reactive memory T cells in mice. Am J Transplant. 2013;13:589–599. doi: 10.1111/ajt.12068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Pearl JP, et al. Immunocompetent T-cells with a memory-like phenotype are the dominant cell type following antibody-mediated T-cell depletion. Am J Transplant. 2005;5:465–474. doi: 10.1111/j.1600-6143.2005.00759.x. [DOI] [PubMed] [Google Scholar]
- 120.Gurkan S, et al. Immune reconstitution following rabbit antithymocyte globulin. Am J Transplant. 2010;10:2132–2141. doi: 10.1111/j.1600-6143.2010.03210.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Valujskikh A, Pantenburg B, Heeger PS. Primed allospecific T cells prevent the effects of costimulatory blockade on prolonged cardiac allograft survival in mice. Am J Transplant. 2002;2:501–509. doi: 10.1034/j.1600-6143.2002.20603.x. [DOI] [PubMed] [Google Scholar]
- 122.Yang J, et al. Allograft rejection mediated by memory T cells is resistant to regulation. PNAS. 2007;104:19954–19959. doi: 10.1073/pnas.0704397104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wang T, et al. Infection with the intracellular bacterium, Listeria Monocytogenes, overrides established tolerance in a mouse cardiac allograft model. Am J Transplant. 2010;10:1524–1533. doi: 10.1111/j.1600-6143.2010.03066.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Welsh RM, et al. Virus-induced abrogation of transplantation tolerance induced by donor-specific transfusion and anti-CD154 antibody. J Virol. 2000;74:2210–2218. doi: 10.1128/jvi.74.5.2210-2218.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Pantenburg B, Heinzel F, Das L, Heeger PS, Valujskikh A. T cells primed by Leishmania major infection cross-react with alloantigens and alter the course of allograft rejection. J Immunol. 2002;169:3686–3693. doi: 10.4049/jimmunol.169.7.3686. [DOI] [PubMed] [Google Scholar]
- 126.Qin S, et al. “Infectious” transplantation tolerance. Science. 1993;259:974–977. doi: 10.1126/science.8094901. [DOI] [PubMed] [Google Scholar]
- 127.Kendal AR, et al. Sustained suppression by Foxp3+ regulatory T cells is vital for infectious transplantation tolerance. J Exp Med. 2011;208:2043–2053. doi: 10.1084/jem.20110767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Waldmann H, Adams E, Fairchild P, Cobbold S. Infectious tolerance and the long-term acceptance of transplanted tissue. Immunol Rev. 2006;212:301–313. doi: 10.1111/j.0105-2896.2006.00406.x. [DOI] [PubMed] [Google Scholar]
- 129.Kernan NA, Flomenberg N, Dupont B, O’Reilly RJ. Graft rejection in recipients of T-cell-depleted HLA- nonidentical marrow transplants for leukemia. Identification of host-derived antidonor allocytotoxic T lymphocytes. Transplantation. 1987;43:842–847. [PubMed] [Google Scholar]
- 130.Kasamon YL, et al. Nonmyeloablative HLA-haploidentical bone marrow transplantation with high-dose posttransplantation cyclophosphamide: effect of HLA disparity on outcome. Biol Blood Marrow Transplant. 2010;16:482–489. doi: 10.1016/j.bbmt.2009.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.El-Cheikh J, et al. Comparison of umbilical cord blood and haploidentical donor grafts in adults with high risk hematologic diseases after fludarabine cyclophosphamide and TBI 2 Gy based reduced intensity conditioning regimen stem cell transplantation. ASH Annu Meet Abstr. 2013;122:3288. [Google Scholar]
- 132.Brunstein CG, et al. Alternative donor transplantation: results of parallel phase II trials using HLA-mismatched related bone marrow or unrelated umbilical cord blood grafts. Blood. 2011;118:282–288. doi: 10.1182/blood-2011-03-344853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Tuve S, et al. Haploidentical bone marrow transplantation with post-grafting cyclophosphamide: multicenter experience with an alternative salvage strategy. Leukemia. 2011;25:880–883. doi: 10.1038/leu.2011.11. [DOI] [PubMed] [Google Scholar]
- 134.Bilmon IA, et al. Haploidentical bone marrow transplants for hematological malignancies using non-myeloablative conditioning therapy and post-transplant immunosuppression with cyclophosphamide: results from a single Australian centre. Intern Med J. 2013;43:191–196. doi: 10.1111/j.1445-5994.2012.02843.x. [DOI] [PubMed] [Google Scholar]
- 135.Bashey A, et al. T-cell replete HLA-haploidentical hematopoietic transplantation for hematologic malignancies using post-transplantation cyclophosphamide results in outcomes equivalent to those of contemporaneous hla-matched related and unrelated donor transplantation. J Clin Oncol. 2013;31:1310–1316. doi: 10.1200/JCO.2012.44.3523. [DOI] [PubMed] [Google Scholar]
- 136.Ciurea SO, et al. Improved early outcomes with T-cell replete (TCR) compared with T-cell depleted (TCD) haploidentical stem cell transplantation (haploSCT) ASH Annu Meet Abstr. 2011;118:320. [Google Scholar]
- 137.Solomon SR, et al. Haploidentical transplantation using T cell replete peripheral blood stem cells and myeloablative conditioning in patients with high-risk hematologic malignancies who lack conventional donors is well tolerated and produces excellent relapse-free survival: results of a prospective phase II trial. Biol Blood Marrow Transplant. 2012;18:1859–1866. doi: 10.1016/j.bbmt.2012.06.019. [DOI] [PubMed] [Google Scholar]
- 138.Raiola AM, et al. Unmanipulated haploidentical bone marrow transplantation and posttransplantation cyclophosphamide for hematologic malignancies after myeloablative conditioning. Biol Blood Marrow Transplant. 2013;19:117–122. doi: 10.1016/j.bbmt.2012.08.014. [DOI] [PubMed] [Google Scholar]