

Abstract
Aims: Peripheral artery disease is caused by the restriction or occlusion of arteries supplying the leg. Better understanding of the molecular mechanisms underpinning tissue response to ischemia is urgently needed to improve therapeutic options. The aim of this study is to investigate hypoxia-induced miR-210 regulation and its role in a mouse model of hindlimb ischemia. Results: miR-210 expression was induced by femoral artery dissection. To study the role of miR-210, its function was inhibited by the systemic administration of a miR-210 complementary locked nucleic acid (LNA)-oligonucleotide (anti-miR-210). In the ischemic skeletal muscle, anti-miR-210 caused a marked decrease of miR-210 compared with LNA-scramble control, while miR-210 target expression increased accordingly. Histological evaluation of acute tissue damage showed that miR-210 inhibition increased both apoptosis at 1 day and necrosis at 3 days. Capillary density decrease caused by ischemia was significantly more pronounced in anti-miR-210-treated mice; residual limb perfusion decreased accordingly. To investigate the molecular mechanisms underpinning the increased damage triggered by miR-210 blockade, we tested the impact of anti-miR-210 treatment on the transcriptome. Gene expression analysis highlighted the deregulation of mitochondrial function and redox balance. Accordingly, oxidative damage was more severe in the ischemic limb of anti-miR-210-treated mice and miR-210 inhibition increased oxidative metabolism. Further, oxidative-stress resistant p66Shc-null mice displayed decreased tissue damage following ischemia. Innovation: This study identifies miR-210 as a crucial element in the adaptive mechanisms to acute peripheral ischemia. Conclusions: The physiopathological significance of miR-210 is context dependent. In the ischemic skeletal muscle it seems to be cytoprotective, regulating oxidative metabolism and oxidative stress. Antioxid. Redox Signal. 21, 1177–1188.
Introduction
Peripheral artery disease is a frequent condition, affecting almost 6% of the U.S. population aged ≥40 (36), which is mostly caused by stenosis, embolism, or thrombosis involving the arteries supplying the leg (37). Abrupt arterial occlusion leads to acute ischemia, while restriction of blood flow due to arterial stenosis most commonly causes chronic ischemia. According to several parameters such as the affected vessel, the degree of occlusion, and the presence of collaterals, peripheral artery disease can cause mild claudication to major tissue loss and may require revascularization surgery (37, 43). Unfortunately, delayed treatment of acute patients can result in morbidity, amputation, and/or death (27). Moreover, chronic patients who benefit from successful revascularization often suffer from high rate of recurrent symptoms or revision surgery and many still require progressive amputations (42). Patients affected by chronic critical limb ischemia, the most severe form of the disease, are particularly at risk. Within 1 year of diagnosis, 40%–50% will experience an amputation and 20%–25% will die (42).
Innovation.
We investigated the role of hypoxia-induced miR-210 in ischemia response. To this aim, we used a mouse model of acute hindlimb ischemia and the systemic administration of a miR-210 complementary locked nucleic acid-oligonucleotide as inhibitory agent. We found that miR-210 has cytoprotective effects in the skeletal muscle, regulating oxidative metabolism and oxidative stress. Thus, miR-210 is a crucial element of the adaptive mechanisms to acute peripheral ischemia. miR-210 is considered a master miRNA of hypoxic response, since it was found upregulated by hypoxia in virtually all the cells and tissues tested to date. Thus, with all due limitations, our findings may not be restricted to peripheral ischemic disease.
Recent evidence shows that ischemia induces profound changes in the expression of microRNAs (miRNAs), small non-protein-coding RNAs that act as negative regulators of gene expression (14). Hypoxia is a crucial component of both acute and chronic ischemia and one specific miRNA, miR-210, is robustly upregulated by both hypoxia and ischemia (5, 7, 12, 26). Between the articulate program of cellular adaptive mechanisms aimed at relieving tissue hypoxia and removing irreversibly damaged cells, miR-210 can be considered a master miRNA of hypoxic response, since it was found upregulated by hypoxia in virtually all the cells and tissues tested to date (7, 12). Mechanistically, miR-210 is a target of the hypoxia-inducible factor 1-alpha (HIF1alpha), which binds to its promoter and activates transcription, upon low oxygen exposure (12). The instrumental role of miR-210 in the regulation of cell response to hypoxia is confirmed by preclinical and clinical evidences (21). Indeed, miR-210 has been found to be upregulated upon brain transient focal ischemia in rats, in mouse ischemic wounds, after human myocardial infarction and was proposed as a blood biomarker in acute cerebral ischemia (3, 4, 28, 49). miR-210 was also found upregulated in most solid tumors and its expression correlates with an adverse clinical outcome and with metastatic potential (12, 26). In keeping with these data, miR-210 has been proposed as a novel tumor hypoxia marker (18).
Evidence accumulated in numerous culture systems shows reduced survival of cells devoid of miR-210 in hypoxia, but also in normoxia, in certain cell types (13, 17, 22, 39, 41, 45). Further, miR-210 has a crucial cytoprotective role in mesenchymal stem cells exposed to anoxia after ischemia/reoxygenation preconditioning, supporting their survival after transplantation in the infarcted heart (30). Although the molecular mechanisms supporting these events are complex, miR-210 directly represses the apoptotic component CASP8AP2 (30) and other apoptosis-related genes such as DAPK1 and, at least in humans, AIFM3 (24, 35, 45). However, miR-210 function is complex and context-dependent, since miR-210 overexpression in normoxia induces apoptosis in certain cancer cell cultures (17, 39).
miR-210 also influences mitochondrial metabolism: targeting the iron–sulfur cluster scaffold protein ISCU1/2, miR-210 inhibits the mitochondrial electron transport chain, prompting the shift from mitochondrial respiration to glycolysis observed in hypoxia (6, 8, 15, 17, 46). Accordingly, miR-210 can also modulate the mitochondrial generation of reactive oxygen species (ROS) (6, 8, 17).
In this study, we investigated the regulation and the role of miR-210 in a rodent model of acute hindlimb ischemia. We found that miR-210 blockade increased apoptosis and necrosis in addition to oxidative damage associated to ischemia.
Results
Hindlimb ischemia induces miR-210 expression
To evaluate whether miR-210 was modulated following acute ischemia, the femoral artery of 2-month-old CD1 male mice was removed to induce unilateral hindlimb ischemia. Gastrocnemius muscles were harvested at 0 (nonischemic) 1, 3, 7, and 14 days after surgery, total RNA was extracted, and miR-210 levels were measured by quantitative real-time PCR (qPCR). Following hindlimb ischemia, miR-210 expression progressively increased up to day 7 and then declined at day 14 (Fig. 1).
FIG. 1.

Hindlimb ischemia induces miR-210 expression. Gastrocnemius muscles were harvested from non ischemic or ischemic mice 1, 3, 7, and 14 days after femoral artery dissection. The bar graph shows miR-210 level measured by qPCR and expressed as fold induction versus non ischemic muscles (n=3; *p<0.05; **p<0.01). qPCR, quantitative real-time PCR.
Functional miR-210 blockade by anti-miR-210
To investigate the role played by miR-210 in the tissue damage induced by ischemia, we focused our attention to the first 3 days after femoral artery dissection, when the peak of tissue degeneration is reached and vascular and muscular regeneration had not started yet (Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/ars) (47). To this aim, we blocked miR-210 function by systemic administration of a 15mer complementary locked nucleic acid (LNA) oligonucleotide (anti-miR-210) in ischemic mice. Twelve mg/kg of anti-miR-210 or scrambled (SCR) LNA sequences were injected in the tail vein 2 days before surgery and mice were sacrificed 3 days after femoral artery removal (i.e., 5 days after LNA-oligonucleotide injection). As shown in Figure 2A, anti-miR-210 treatment efficiently inhibited miR-210 expression, both in ischemic and in nonischemic controlateral muscles. Indeed, upon anti-miR-210 treatment, miR-210 levels in the ischemic muscles were lower than those observed in untreated nonischemic muscles. Conversely, SCR treatment did not affect miR-210 induction upon femoral artery removal, confirming the specificity of anti-miR-210 action. In keeping with these observations, miR-210 levels were similarly decreased in the liver of anti-miR-210-treated mice (not shown).
FIG. 2.

miR-210 blockade by anti-miR-210 treatment. Anti-miR-210 or SCR oligonucleotides were injected in the tail vein 2 days before surgery. Mice were sacrificed 3 days after femoral artery dissection and total RNA was extracted from ischemic and controlateral gastrocnemius. (A) The bar graph shows miR-210 levels measured by qPCR and expressed as fold change versus untreated nonischemic muscles. Anti-miR-210 treatment efficiently inhibited miR-210 expression induced by ischemia and the SCR sequence did not affect this induction (n=6–8; ***p<0.001). (B) The level of the indicated miR-210 targets was measured in the gastrocnemius muscles by qPCR, 3 days after ischemia induction (n=6–8). The heat map shows average expression levels where green and red colors indicate down- or upregulation, respectively. Most miR-210 targets were de-repressed in anti-miR-210 compared to SCR-treated ischemic muscles. (C) Representative western blotting analysis of P4hb, Rod1, and α-tubulin (loading control) protein levels in ischemic gastrocnemius muscles (n=3). The protein expression of both miR-210 targets increased in anti-miR-210-treated mice compared with SCR. SCR, scrambled.
To evaluate whether the observed miR-210 decrease in anti-miR-210-treated mice was functionally effective, a subset of well-established miR-210 targets was measured in the ischemic gastrocnemius muscle. We found that, to a different extent, most miR-210 targets were de-repressed in anti-miR-210 compared with SCR-treated ischemic muscles, with the only exception of Mnt, that was decreased, albeit nonsignificantly (Fig. 2B, C). It has been reported that miR-210 and miR-147b have similar functional activities (2). When miR-147b levels were measured, we found that miR-147b was undetectable by qPCR in non ischemic muscles. Following ischemia, miR-147b levels passed detection threshold (Supplementary Fig. S2), indicating that miR-147b and miR-210 are both induced by ischemia. However, miR-210 blockade by anti-miR-210 in ischemic muscles did not affect miR-147b levels compared to ischemic SCR control (1.1±0.3-fold change, not significant). These data further confirm the specificity of the treatment and indicate no compensatory super-induction of miR-147b in the adopted experimental conditions.
Anti-miR-210 increases apoptosis induced by ischemia
Hindlimb ischemia has been shown to induce cell death by both apoptosis and necrosis (47). Given the anti-apoptotic role of miR-210 (12), we investigated whether miR-210 inhibition affected skeletal muscle apoptotic response upon ischemia. To this aim, femoral artery was removed and ischemic mice were analyzed 8 h after surgery, when necrosis was not present yet. As expected, terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick-endlabeling (TUNEL)-positive nuclei were easily detectable in gastrocnemius muscle sections of SCR-treated mice and greatly increased when miR-210 was blocked (Fig. 3A). As shown in the bar graph (Fig. 3B), the percentage of apoptotic nuclei was more than threefold higher in anti-miR-210 than in SCR-treated mice, indicating that miR-210 inhibition enhanced apoptosis following acute hindlimb ischemia.
FIG. 3.

Ischemia-induced apoptosis is increased by miR-210 inhibition.(A) Representative sections of gastrocnemius muscles obtained from both SCR and anti-miR-210-treated mice, at 8 h of ischemia. TUNEL-positive nuclei are stained in green and nuclei are counter-stained by Hoechst 33342 (blue fluorescence). Magnification 400×; calibration bar 25 μm. (B) The bar graph shows the percentage of TUNEL-positive nuclei (n=7; #p<0.02).
Anti-miR-210 increases ischemic damage
Ischemia leads to myofiber degeneration, necrosis, and decreased capillary density (47). Thus, we tested whether the higher apoptosis levels induced by miR-210 inhibition were followed by more tissue damage. To this aim, mice were injected with anti-miR-210 or SCR and gastrocnemius muscles were analyzed 1 and 3 days after femoral artery dissection.
Morphometric analysis of non ischemic muscles did not show any overt difference between SCR and anti-miR-210-treated mice (Fig. 4A). As expected, upon femoral artery dissection, areas of necrotic myofibers were present at both timepoints (Fig. 4A). Necrotic areas were evaluated by morphological criteria, differential eosin staining, and presence of infiltrating cells both into and near the degenerating myofibers. Quantification showed that the percentage of necrotic areas was significantly higher in anti-miR-210-treated muscles both at 1 and 3 days of ischemia (Fig. 4B). These results were confirmed by in vivo systemic administration of Evans Blue Dye (EBD), which can permeate and stain only damaged myofibers (23). Indeed, myofibers devoid of an intact cell membrane allow EBD penetration and emit red fluorescence in fluorescent microscopy while healthy myofibers remain dark (Fig. 5A). Figure 5B shows that the percentage of EBD-positive areas was significantly higher in anti-miR-210-treated muscles compared with SCR, confirming more extensive damage. To further strengthen this notion, we measured capillary density in ischemic muscles. As expected, capillary density decreased in ischemic respect to non ischemic gastrocnemius muscles, in both groups. However, this decrease was significantly more pronounced in anti-miR-210-treated mice (Fig. 6A, B). This difference in capillary density is likely functionally relevant, since it was mirrored by a small but significant decrease of residual perfusion (Fig. 6C). Conversely, differences in arteriolar length density did not reach statistical significance (not shown).
FIG. 4.

Ischemic muscle damage is increased by miR-210 inhibition. (A) Representative hematoxilin/eosin-stained sections of gastrocnemius muscles obtained from both SCR and anti-miR-210-treated mice, 1 and 3 days after femoral artery dissection. Magnification 200×. Calibration bar 100 μm. (B) More necrosis in anti-miR-210 ischemic gastrocnemius muscles. The bar graph shows the percentage of necrotic areas respect to whole section area (**p<0.01; ***p<0.0001, n=5–6). To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
FIG. 5.

Anti-miR-210 treatment increases ischemic muscle damage measured by EBD. (A) Representative EBD stained sections of gastrocnemius muscles 3 days after ischemia. EBD was administered intraperitoneally, 16 h before sacrifice. Red fluorescence indicates damaged myofibers, permeable to EBD; conversely healthy myofibers are dark. Nuclei were highlighted by Hoechst 33342 staining (blue fluorescence). Magnification 200×, Calibration bar 100 μm. (B) Bar graph showing the percentage of EBD-positive areas with respect to whole area of the section (°p<0.04; n=7).
FIG. 6.

miR-210 blockade increases capillaries damage. (A) Hematoxilin/eosin representative stainings of both non ischemic (upper panels) and ischemic (lower panels) gastrocnemius muscles 3 days after ischemia. Magnification 1000×. Calibration bar 50 μm. Black arrows indicate capillaries. Yellow arrows indicate capillaries with a trapped erythrocyte. (B) Quantification of capillaries/mm2 (#p<0.04; n=8). (C) Bar graph showing laser Doppler perfusion imaging measurement 3 days after femoral artery dissection. Percentage of residual perfusion is expressed as ischemic/non ischemic ratio (*p<0.02, n=18).
No difference in Hif pathway activation upon miR-210 inhibition in ischemic mice
It has been shown in certain cell culture systems that miR-210 inhibition leads to decreased HIF1alpha (official symbol for mouse: Hif1a) stabilization following ischemia, decreasing HIF1alpha-dependent response (29, 39). To test whether the increased sensitivity to ischemic damage upon miR-210 inactivation was due to decreased HIF1alpha pathway activation, the expression of several HIF1alpha target genes was measured. qPCR analysis shows that HIF1alpha pathway was similarly activated by ischemia in both SCR and anti-miR-210 mice (Supplementary Fig. S3A). Accordingly, similar HIF1alpha and HIF2alpha protein levels were observed after miR-210 blocking in ischemic muscles compared with controls (Supplementary Fig. S3B), indicating that miR-210 acted with different mechanisms in the context of peripheral acute ischemia.
miR-210 blockade increases oxidative stress
To investigate the molecular mechanisms underpinning the increased apoptosis and tissue damage triggered by miR-210 blockade, we tested the impact of miR-210 inhibition on the transcriptome. To this aim, we measured gene expression profiles of gastrocnemius muscles derived from SCR and anti-miR-210-treated mice, in the presence or absence of hindlimb ischemia for 3 days. Unsupervised hierarchical clustering analysis segregated the profiles of SCR and anti-miR-210 samples, confirming the biological relevance of miR-210 activity (Supplementary Fig. S4A). Class comparison analysis of SCR versus anti-miR-210 muscles was performed and qPCR validated (Supplementary Fig. S4B, C), identifying 338 transcripts differentially expressed in nonischemic tissues and 420 transcripts differentially expressed in the ischemic ones. As expected, the comparison between ischemic versus nonischemic SCR-treated muscles identified a much higher number (2340) of differentially expressed transcripts.
To facilitate the interpretation of the complex gene expression changes observed in SCR and anti-miR-210-treated groups, we used Ingenuity Pathway Analysis software (IPA), exploring for enriched biological functions and pathways. We applied a low-stringency approach, prioritizing sensitivity to maximize the identification of the potentially involved categories. Among the top scoring functions, we identified mitochondrial dysfunction, glutathione depletion, oxidative stress, and NRF2-mediated oxidative stress response and apoptosis, all indicating that miR-210 inhibition may lead to increased oxidative tissue damage. To experimentally validate these findings, we assayed protein nitrosylation, an oxidative damage marker. Figure 7A shows that nitrotyrosine staining was more pronounced and diffuse in anti-miR-210-treated mice after 1 day of ischemia. A similar pattern was observed at 3 days, but higher background was present, possibly due to the dominant presence of necrotic tissue (not shown).
FIG. 7.

Role of oxidative stress in increased tissue damage induced by miR-210 inhibition. (A) Increased protein nitrosylation following miR-210 blockade in ischemic muscles. Representative nitrotyrosine immunostainings of anti-miR-210 and SCR-treated ischemic muscles, 1 day after femoral artery dissection (n=6). Brown nitrotyrosine staining was more intense and diffuse in anti-miR-210-treated mice. Nuclei are stained in light blue by hematoxilin. Magnification 400×; calibration bar 100 μm. (B) The bar graph shows the percentage of necrotic areas in 3 days ischemic mice (***p<0.001; n=6–7). Ischemic p66Shc−/− mice were resistant to increased tissue damage induced by miR-210 inhibition. (C) The bar graph represents the number of capillaries/mm2 in ischemic and non ischemic mice (*p<0.03; ***p<0.00003; n=6–7). Ischemic p66Shc−/− mice were resistant to capillary density decrease upon miR-210 blockade. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
To validate this finding, we tested whether it was also true in isolated cells. In particular, given the implication of miR-210 in oxidative metabolism (5, 12), mitochondrial ROS were measured. To this aim, miR-210 was inhibited in hypoxic cultured myotubes and mitochondrial oxidants were assayed by MitoSOX fluorogenic dye staining. C2C12 murine myoblasts were transfected with anti-miR-210 or SCR LNA. After 48 h of differentiation, myotubes were cultured in hypoxic conditions for 24 h and then stained with MitoSOX fluorescent dye. In keeping with in vivo data, MitoSOX-associated fluorescence was increased when miR-210 was inhibited (Supplementary Fig. S5). We conclude that miR-210 inhibition is associated to increased oxidative stress that may, in turn, increase ischemia-induced muscle damage.
p66Shc null mice are resistant to increased tissue damage upon miR-210 blockade
p66Shc is a lifespan-regulating protein contributing to mitochondrial ROS metabolism and regulating the mitochondrial apoptosis pathway (19). Indeed, p66Shc null mice are resistant to several ROS-mediated injuries (19). In particular, we previously demonstrated that 129 SvEv p66Shc null mice (p66Shc−/−) are resistant to ischemia-induced oxidative damage in skeletal muscles and vascular structures (47). To clarify whether mitochondrial ROS played a causal role in the increased tissue damage induced by miR-210 blockade, we tested the effect of miR-210 blocking in the presence or absence of p66Shc. To this aim, both p66Shc−/− and p66Shc wt mice were injected with SCR or anti-miR-210 LNA, 2 days before femoral artery dissection. Next, mice were sacrificed 3 days after surgery for histological evaluation. When p66Shc wt gastrocnemius muscles were analyzed, a dramatic damage increase was observed in anti-miR-210 muscles compared to SCR (Fig. 7B). These data are in agreement with those obtained from CD1 wt mice (Fig. 4), albeit the difference between anti-miR-210 and SCR-treated mice was even more dramatic, possibly due to mouse strain specificities. Conversely, p66Shc−/− muscles showed minimal tissue damage not only in SCR-treated mice, but also upon miR-210 blockade (Fig. 7B). Similar results were obtained when capillary density was evaluated. As previously published (47), capillary density of normoperfused gastrocnemius muscles was significantly lower in p66Shc−/− than in p66Shc wt mice (Fig. 7C). In ischemic p66Shc wt mice, capillary density was further decreased by anti-miR-210 treatment. Conversely, p66Shc−/− mice were resistant to the capillary density decrease induced by ischemia, and only minimal capillary rarefaction was observed following miR-210 blocking (Fig. 7C). In conclusion, these data indicate that miR-210 blockade is virtually ineffective in p66Shc−/− mice, indicating that oxidative stress of mitochondrial origin is a crucial player in the increased muscle damage observed upon miR-210 blockade in ischemic mice.
Anti-miR-210 treatment induces a shift from glycolytic to oxidative metabolism in normoxic muscles
As previously noticed, histological analysis of nonischemic skeletal muscles derived from SCR and anti-miR-210-treated mice did not display any overt alteration (Figs. 4 and 5) and no decrease in capillary density was observed (Fig. 6). However previous observations in cell culture showed that normoxic levels of miR-210 were functionally relevant (9, 13, 25). In keeping with these observations, we found that upon miR-210 inhibition, miR-210 targets were de-repressed in normoperfused nonischemic muscles as well (Supplementary Fig. S6). Thus, we investigated whether more subtle metabolic alterations were occurring. To this aim, the extensor digitorum longus (EDL) muscle, a fast-twitch muscle composed mainly by type IIB (glycolitic) and type IIA (intermediate glycolitic/oxidative) fibers (31) was analyzed using the NADH-TR-diaphorase staining (Fig. 8A). The intensity of the staining, that identifies mitochondria and sarcoplasmic reticulum, is related to the metabolism: a light staining indicates type IIB fibers with glycolytic metabolism, while a darker staining indicates a more oxidative metabolism (type IIA fibers) (50). As shown in Figure 8B, miR-210 down modulation increased NADH-TR-diaphorase staining both at 2 and 5 days of anti-miR-210 treatment. Analysis of the individual fibers showed that anti-miR-210 increased the fibers with a type IIA-pattern, while more glycolytic type IIB-like fibers remained prevalent in the SCR-treated muscles (Fig. 8C). However, anti-miR-210 treatment, at least in the adopted conditions, did not alter myofiber specification. Indeed, the abundance of fiber-type specific isoforms of myosin (MyH1, 4 and 7) and troponin (Tnn1 and 2) was unchanged (not shown). In keeping with these observation, Aconitase activity was significantly increased in anti-miR-210 EDL muscle extracts (Fig. 9A). In addition, Complex I activity, measured in crude mitochondrial preparation of gastrocnemius muscles, showed a significant increase when miR-210 was blocked (Fig. 9B).
FIG. 8.

Anti-miR-210 shifts metabolism of EDL from glycolytic to oxidative. Frozen sections of EDL muscle were stained with NADH-TR-diaphorase and analyzed to evaluate metabolic differences between mice treated with anti-miR-210 and SCR. (A) representative pictures of day 2 muscles are shown. White arrows indicate type IIB fibers, while black arrows indicate type IIA fibers. Calibration bar 50 μm. (B) The bar graph shows increased intensity of NADH-TR-diaphorase staining expressed in arbitrary units, at the indicated timepoints (n=8; ***p<0.001). (C) The bar graph shows the percentage of type IIA fibers, indicating an increase of this fiber type in mice treated with anti-miR-210, for 2 or 5 days (n=8; ***p<0.001). EDL, estensor digitorum longus.
FIG. 9.

Metabolic activity assays. (A) The bar graph represents Aconitase activity fold change in EDL muscle extracts (*p<0.01; n=9). (B) The bar graph shows Complex I activity measured in crude mitochondrial preparations of gastrocnemius muscles (*p<0.05; n=5).
In conclusion, these data indicated a shift toward a more oxidative metabolism, confirming the relevance of miR-210 also in normoperfused skeletal muscles.
Discussion
In this study, we identified an anti-apoptotic, pro-survival role of miR-210 in skeletal muscles exposed acute ischemia. Specifically, acute pharmacological inhibition of miR-210 increased the levels of apoptosis and necrosis in a mouse model of hindlimb ischemia. Our findings are in keeping with numerous observations indicating that miR-210 inhibition increases apoptosis and cell death in a variety of cell culture systems (7, 12, 26). We and others found that miR-210 blockade induces endothelial cell apoptosis and increases cell death in hypoxia (6, 13). Moreover, when miR-210 is blocked, cell death is significantly more pronounced in differentiated myoblast cultures upon mitochondrial damage and oxidative stress (9). Our observations are also in agreement with a previous in vivo study that used a reciprocal gain of function approach: It was found that miR-210 overexpression can inhibit apoptosis, increase angiogenesis, and improve cardiac function in a murine model of myocardial infarction (24).
Here, miR-210 was expressed at physiological levels and its function was inhibited by a specific LNA-antisense sequence. The effectiveness of this approach was demonstrated not only by the decreased miR-210 levels but, more importantly, by the de-repression of many experimentally validated miR-210 target mRNAs. It is however worth noting that not all targets were de-repressed and de-repression levels were highly variable. While different explanations are possible, one should keep into consideration that certain targets may be mostly affected at the translational level and that tissue specificity in miR-210 targeting efficiency may be present (1).
It is worth noting that, while miR-210 was robustly induced by ischemia 3 days after femoral artery dissection, its levels further increased at 7 days and remained high at 14 days. Later, high levels of miR-210 may be due to the compound effect of the still unresolved ischemia with myogenic regeneration, which in the adopted experimental setting, is absent at day 3, starts at day 7, and peaks at day 14 (G.Z. and F.M., unpublished). Indeed, we have previously shown that miR-210 is induced during myogenic differentiation and its levels were elevated in regenerating skeletal muscles following cardiotoxin damage (9).
In a recent article, Bertero et al. showed that miR-210 and miR-147b share a “minimal” 6-nucleotides seed sequence and have similar functional activities in A549 adenocarcinoma cell line (2). We found that miR-147b is induced by ischemia, increasing the number of overlapping features between miR-147b and miR-210. However, we did not find evidence of any compensatory mechanism of miR-147b overexpression triggered by miR-210 acute inhibition. Further investigation is needed to ascertain whether the same is true upon congenital or chronic miR-210 loss of function.
We also investigated the molecular mechanisms underpinning miR-210 action in peripheral acute ischemia. A hypoxia-induced positive feedback loop promoting HIF1alpha stability through miR-210 has been observed, providing a possible explanation for the increased apoptosis and tissue damage observed following miR-210 inhibition in ischemic muscles (29, 39). However, we did not find any significant difference in the activation of HIF1alpha-pathway, possibly due to differences in the experimental systems. Indeed, the studies of Puissegur et al. (39) and of Kelly et al. (29) were performed in cancer cell cultures exposed to hypoxia, while, in our case, nontransformed tissues were exposed to ischemia.
A possible mechanism involved in the regulation of cell survival by miR-210 is the direct targeting of miR-210 of apoptotic genes such as CASP8AP2 (30) and DAPK1 (24). Moreover, genome wide analysis of gene expression indicated that miR-210 inhibition affected, directly or indirectly, several genes modulating mitochondrial function and oxidative stress. Indeed, increased levels of oxidative damage were found in ischemic muscles when miR-210 was blocked by anti-miR-210. These findings are in keeping with numerous observations in culture systems. We and others found that anti-miR-210 increased ROS levels in normoxic (9) and hypoxic-differentiated myotubes and in endothelial cells (6), and in other cell systems (17, 35). Moreover, experiments performed in both primary endothelial and cancer cells show that miR-210 is a crucial regulator of mitochondrial metabolism: by downregulating the expression of ISCU1/2, NDUFA2, COX10, and SDH, miR-210 disrupts the mitochondrial electron transport activity, repressing mitochondrial oxidative phosphorylation (6, 8, 15, 17, 46). In support of a prominent role of miR-210 in the regulation of metabolism, we found that, even in non ischemic muscles, miR-210 blockade induced a shift toward a more oxidative metabolism.
The p66Shc gene encodes an adaptor protein for cell signaling and its ablation in mice causes life-span prolongation without overt pathological consequence, confers resistance to oxidative stress, and correlates with reduced levels of apoptosis (19, 34). A fraction of p66Shc localizes to mitochondria where it binds to cytochrome c and acts as oxidoreductase, generating ROS and leading to organelle dysfunction and cell death (20). p66Shc−/− mice are resistant to several ROS-mediated injuries; in particular, we previously demonstrated that p66Shc−/− mice are resistant to ischemia-induced oxidative damage on skeletal muscles and vascular structures (47). We now show that p66Shc−/− mice were also largely resistant to the additional damage induced by miR-210 inhibition upon hindlimb ischemia, further implicating mitochondrial dysfunction as an underpinning mechanism.
Finally miR-210 may also influence the function of inflammatory cells that, in turn, have a great influence in tissue response to ischemia. Indeed, miRNA-210 negatively regulates the production of proinflammatory cytokines by targeting NF-κB1 in murine macrophages (40).
It has been reported that miR-210 inhibits DNA damage in hypoxic cells (11). Thus, it is likely that DNA damage repair may be increased in ischemic limbs upon miR-210 blocking. However, if present, this potentially positive effect seems outweighed by all the other detrimental effects. One should also keep in mind that miR-210 function appears to be largely context dependent. For instance, Biswas et al. showed that miR-210 attenuates keratinocyte proliferation and impairs closure in a murine model of ischemic wounds (3). Thus, while miR-210 seems to have a cytoprotective role, it might also have a detrimental role in the ensuing regeneration. A specific experimental setting will be necessary to further explore this issue. Another cautionary note is represented by the fact that, in our experimental setting, we acutely blocked miR-210 function. Different results might be obtained upon congenital deletion of this miRNAs, where compensatory mechanisms may be established.
Proposed mechanisms of miR-210 action in the specific context of acute peripheral ischemia are summarized in Figure 10. In hypoxic cells, mitochondrial formation of ROS is increased and metabolism switches from oxidative phosphorylation to glycolysis, minimizing oxidative damage (38). Thus, inhibition of mitochondrial respiration can be deleterious in normoxia and positive in hypoxia. miR-210 is an integral part of oxidative phosphorylation inhibition: we found that miR-210 blockade stimulates oxidative metabolism boosting ROS levels and this has a negative impact on ischemic skeletal muscles.
FIG. 10.
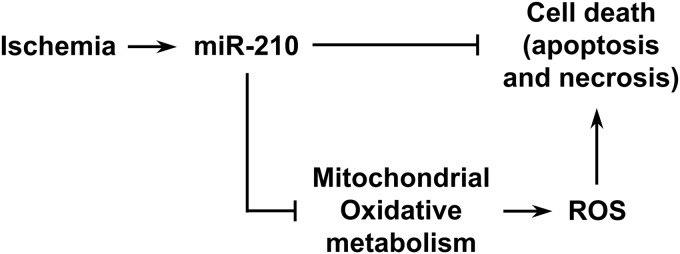
Model of the mechanisms underpinning miR-210 antiapoptotic, cytoprotective function. Upon miR-210 blocking, mitochondrial oxidative phosphorylation is de-repressed, increasing ROS levels and triggering cell death. Other sources of ROS, such as NADPH oxidase (not indicated), may also contribute to determine ROS levels. ROS, reactive oxygen species.
In conclusion, we identified a crucial role played by miR-210 in the adaptive mechanisms to acute peripheral ischemia. Together, our findings implicate miR-210 as a miRNA with cytoprotective effects in the skeletal muscle, regulating oxidative metabolism and oxidative stress.
Materials and Methods
Animal model and surgical procedures
Unless differently specified, 2-month-old CD1 male mice were used. For certain experiments, 2 to 3-month-old 129 Sv-Ev p66Shc wt and p66Shc−/− mice were used (34). Experimental procedures complied with the Guidelines of the Italian National Institutes of Health and with the Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources, National Academy of Sciences, Bethesda, MD) and were approved by the institutional Animal Care and Use Committee. Before all surgical and perfusion procedures, mice were anesthetized with an intraperitoneal injection of 1 mg/kg Medetomidine (Domitor; VETEM) and 75 mg/kg Ketamine (Ketavet 100; Intervet Farmaceutici). Dissection of the left femoral artery and blood flow measurement by Laser Doppler Perfusion Imaging (Lisca) were previously described (10). In vivo down modulation of miR-210 was carried out by tail vein injection of LNA oligonucleotides against miR-210 (anti-miR-210) or SCR control sequence (In vivo LNAmicroRNA Inhibitors; Exiqon). Specifically, the following 15mers LNA-enhanced sequences with complete phosphothioate backbone were used: anti-miR-210, GCTGTCACACGCACA; SCR, CGTCTAGCCACCTAG. Twelve mg/kg of anti-miR-210 or SCR LNA were diluted in 200 μl of saline and injected in the tail vein of two different groups of mice. Next, mice were divided into two subgroups, one group was sacrificed 2 days after treatment (nonischemic mice) and the second underwent femoral artery dissection (ischemic mice). For RNA extraction, muscles were snap frozen in liquid nitrogen. For histological analysis, mice were perfused via left ventricle with phosphate-buffered saline (PBS), followed by 10% buffered formalin, at 100 mmHg for 10 min. Therefore, gastrocnemius muscles were harvested, fixed, and paraffin embedded. For the evaluation of myofibers permeability, 1% EBD (Sigma) in PBS (pH 7.5) was used. EBD solution 1% volume relative to body mass was intraperitoneally injected 16 h before sacrifice. Thereafter, gastrocnemius muscles were harvested and frozen in OCT embedding medium.
Histological, morphometric, and NADH-TR-diaphorase analyses
Apoptosis mediated by DNAse I and II was identified by TUNEL assay (ApoAlert DNA fragmentation assay kit; Clontech) according to the manufacturer's instructions. Hematoxylin/eosine sections of paraffin-embedded gastrocnemius muscles were prepared as previously described (44). Necrotic muscle fibers were identified by morphology, differential eosin staining, and presence of infiltrating cells into and near the degenerating fibers on the whole section at 400×magnification. Capillary density was measured counting the number of capillary profiles in 30–40 random fields/section, at 1000×magnification (48). Immunohistochemistry for Nitrotyrosine (rabbit polyclonal antibody; Calbiochem) was carried out according to standard procedures on 3 μm gastrocnemius sections.
Frozen sections (10 μm) of EBD muscles were fixed in cold acetone (−20°C) for 10 min, washed in PBS, and mounted with fluorescence mounting medium.
For NADH-TR-diaphorase staining, EDL muscles were dissected, harvested, and frozen in OCT embedding medium. Eight micrometer frozen sections were fixed using 4% paraphormaldehyde for 10 min, incubated for 30 min at 37°C with 10 mg/5 ml NBT (N6876; Sigma) and 8 mg/5 ml NADH (N8129; Sigma) in 1:1 ratio, and then washed thrice with water. Unbound NBT was removed by washes with 30%, 60%, and 90% acetone solutions in increasing and then decreasing concentrations. Samples were rinsed several times with water and then mounted with aqueous mounting medium onto a labeled glass slide. Whole sections were analyzed.
A Zeiss Axioplan 2 fluorescence microscope with image analyzer KS300 software was used to acquire images and to measure areas. All histological and morphometric analyses were carried out by two blinded readers with comparable results.
Metabolic activity assays
Aconitase activity was measured in EDL muscle extracts using the Aconitase Assay Kit (ab83459; Abcam) according to manufacturer's instructions. Gastrocnemius muscles crude mitochondrial preparations were assayed for Complex I activity using the Complex I Enzyme Activity Microplate Assay Kit (ab109721; Abcam) according to manufacturer's instructions.
Cell culture and MitoSOX assay
Mouse C2C12 myoblast cell line (ATCC) was cultured in high-glucose Dulbecco's modified Eagle's medium (DMEM), supplemented with 20% fetal calf serum (growing medium) as described previously (33). For miR-210 down modulation, the same LNA oligonucleotides against miR-210 or a SCR sequence used in vivo (40 nM; both from Exiqon) were transfected using siRNA transfection reagent (SC29528; Santa Cruz Biotechnology). After 16 h, cells were washed and differentiation medium (DMEM supplemented with 2% horse serum) was added. Forty-eight hours later, differentiated myotubes cells were exposed to 1% oxygen tension in a hypoxic incubator and maintained in hypoxia for 24 h. Mitochondrial oxidants were assayed as previously reported (9). Briefly, cells were incubated with 5 μM MitoSOX (Molecular Probes; Invitrogen) for 10 min at 37°C in the hypoxic incubator. Then cells were fixed with 4% paraformaldehyde, nuclei were stained with Hoechst 33342 and fluorescence was revealed by fluorescence microscopy (Olympus IX51, with image analyzer Soft Imaging System Cell F) using the same exposure conditions for each sample and quantified using Adobe Photoshop CS2. Intensity of MitoSOX fluorescence was normalized for the number of Hoechst 33342 positive nuclei.
Gene expression profiles
Total RNA was extracted using TRIzol (Invitrogen) and the TissueLyser system (Qiagen). RNA was further purified using the RNAeasy mini kit (Qiagen) following the RNA cleanup protocol as indicated by the manufacturer. RNA purity and integrity were assessed by spectophotometric analysis and agarose gel electrophoresis. Illumina MouseWG-6 v2.0 Expression BeadChips were used for this study. Total RNA (300 ng) was analyzed according to the manufacturer's instructions. Gene expression profiles were analyzed using the class comparison between-groups function of BRB-ArrayTools (Richard Simon and BRBArrayTools Development Team). False discovery rate was computed per gene using the Benjamini and Hochberg method. The complete dataset of our study is available from the National Center for Biotechnology Information Gene Expression Omnibus database (GEO entry GSE43340). Data validation was performed by qPCR.
Pathway analysis was performed using Ingenuity Pathways Knowledge Base-v8.8 (Ingenuity Systems) as reference set and assuming direct and indirect relationships. Fisher's exact test p-value<0.05 was deemed as statistically significant.
miRNA and mRNA quantification
miRNA levels were analyzed using TaqMan qPCR assay (1 ng per assay) and quantified with the 7900 HT Fast Real Time PCR System (Applied Biosystems) as previously described (32). Primers for miR-210, miR-147b, and miR-16 and the reagents for reverse transcriptase and qPCRs were all purchased from Applied Biosystems. miR-210 expression level in each sample was normalized to miR-16 expression as, under the experimental conditions of the present study, miR-16 was not modulated by ischemia or miR-210. mRNAs levels were analyzed using the SYBR-GREEN qPCR method (5 ng per assay; Qiagen) and quantified with 7900 HT Fast Real Time PCR System (Applied Biosystems) as previously described (16). mRNA expression was normalized to RPL13 levels, not modulated by ischemia or miR-210. For both miRNAs and mRNAs, relative expression was calculated using the comparative Ct method (2–Delta Delta Ct). Heat maps were generated using Genesis software (version 1.7.5; Graz University of Technology, Institute for Genomics and Bioinformatics).
Western blotting
Frozen tissues were homogenized in ice-cold RIPA buffer containing 1 mM PMSF (Sigma) and Protease Inhibitor Cocktail (Thermo Scientific) (300 μl buffer/5 mg tissue) using TissueLyser system (Qiagen). The homogenate was centrifuged for 20 min at 12,000 rpm at 4°C and then the supernatant was used for SDS-PAGE. Fifty micrograms of total proteins after 5 min boiling in 4×Laemmli buffer were separated in SDS-PAGE and transferred to a nitrocellulose membrane (Bio-Rad) by standard procedures. The membranes were incubated with the following antibodies: anti-P4hb (C-2; Santa Cruz Biotechnologies), anti-α-tubulin (B-5-1-2; Sigma), anti-Rod1 (F-30; Santa Cruz Biotechnologies), anti-Hif-1a (h1alpha67; Novus Biologicals), and anti-Hif-2a (Novus Biologicals). Horseradish peroxidase-linked secondary antibodies were diluted 1/2000 and SuperSignal West Dura Extended Duration Substrate was used for chemiluminescence development. Expression levels were scanned by Molecular Imager ChemiDoc XRS System using the Quantity One software (Bio-Rad).
Statistical analysis
Unless differently stated, variables were analyzed by Student's t-test and ANOVA. A value of p<0.05 was deemed statistically significant. Results are reported as mean±SEM values.
Supplementary Material
Abbreviations Used
- DMEM
Dulbecco's modified Eagle's medium
- EBD
Evans Blue Dye
- EDL
estensor digitorum longus
- HIF1alpha
hypoxia-inducible factor 1alpha
- LNA
locked nucleic acid
- miRNA
microRNA
- PBS
phosphate-buffered saline
- qPCR
quantitative real-time PCR
- ROS
reactive oxygen species
- SCR
scrambled
Acknowledgments
Dr. Giovanni Pani (Università Cattolica School of Medicine, Rome, Italy) is acknowledged for his invaluable advice on oxidative stress mouse models. This work was supported by Ministero della Salute and Associazione Italiana per la Ricerca sul Cancro (Grant AIRC IG-11436).
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell 136: 215–233, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bertero T, Grosso S, Robbe-Sermesant K, Lebrigand K, Henaoui IS, Puissegur MP, Fourre S, Zaragosi LE, Mazure NM, Ponzio G, Cardinaud B, Barbry P, Rezzonico R, and Mari B. “Seed-Milarity” confers to hsa-miR-210 and hsa-miR-147b similar functional activity. PLoS One 7: e44919, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Biswas S, Roy S, Banerjee J, Hussain SR, Khanna S, Meenakshisundaram G, Kuppusamy P, Friedman A, and Sen CK. Hypoxia inducible microRNA 210 attenuates keratinocyte proliferation and impairs closure in a murine model of ischemic wounds. Proc Natl Acad Sci U S A 107: 6976–6981, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bostjancic E, Zidar N, and Glavac D. MicroRNA microarray expression profiling in human myocardial infarction. Dis Markers 27: 255–268, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chan SY. and Loscalzo J. MicroRNA-210: a unique and pleiotropic hypoxamir. Cell Cycle 9: 1072–1083, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chan SY, Zhang YY, Hemann C, Mahoney CE, Zweier JL, and Loscalzo J. MicroRNA-210 controls mitochondrial metabolism during hypoxia by repressing the iron-sulfur cluster assembly proteins ISCU1/2. Cell Metab 10: 273–284, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chan YC, Banerjee J, Choi SY, and Sen CK. miR-210: the master hypoxamir. Microcirculation 19: 215–223, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen Z, Li Y, Zhang H, Huang P, and Luthra R. Hypoxia-regulated microRNA-210 modulates mitochondrial function and decreases ISCU and COX10 expression. Oncogene 29: 4362–4368, 2010 [DOI] [PubMed] [Google Scholar]
- 9.Cicchillitti L, Di Stefano V, Isaia E, Crimaldi L, Fasanaro P, Ambrosino V, Antonini A, Capogrossi MC, Gaetano C, Piaggio G, and Martelli F. Hypoxia-inducible Factor 1-alpha Induces miR-210 in normoxic differentiating myoblasts. J Biol Chem 287: 44761–44771, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Couffinhal T, Silver M, Zheng LP, Kearney M, Witzenbichler B, and Isner JM. Mouse model of angiogenesis. Am J Pathol 152: 1667–1679, 1998 [PMC free article] [PubMed] [Google Scholar]
- 11.Crosby ME, Kulshreshtha R, Ivan M, and Glazer PM. MicroRNA regulation of DNA repair gene expression in hypoxic stress. Cancer Res 69: 1221–1229, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Devlin C, Greco S, Martelli F, and Ivan M. miR-210: more than a silent player in hypoxia. IUBMB Life 63: 94–100, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fasanaro P, D'Alessandra Y, Di Stefano V, Melchionna R, Romani S, Pompilio G, Capogrossi MC, and Martelli F. MicroRNA-210 modulates endothelial cell response to hypoxia and inhibits the receptor tyrosine kinase ligand Ephrin-A3. J Biol Chem 283: 15878–15883, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fasanaro P, Greco S, Ivan M, Capogrossi MC, and Martelli F. microRNA: emerging therapeutic targets in acute ischemic diseases. Pharmacol Ther 125: 92–104, 2010 [DOI] [PubMed] [Google Scholar]
- 15.Fasanaro P, Greco S, Lorenzi M, Pescatori M, Brioschi M, Kulshreshtha R, Banfi C, Stubbs A, Calin GA, Ivan M, Capogrossi MC, and Martelli F. An integrated approach for experimental target identification of hypoxia-induced miR-210. J Biol Chem 284: 35134–35143, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fasanaro P, Romani S, Voellenkle C, Maimone B, Capogrossi MC, and Martelli F. ROD1 is a seedless target gene of hypoxia-induced miR-210. PLoS One 7: e44651, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Favaro E, Ramachandran A, McCormick R, Gee H, Blancher C, Crosby M, Devlin C, Blick C, Buffa F, Li JL, Vojnovic B, Pires das Neves R, Glazer P, Iborra F, Ivan M, Ragoussis J, and Harris AL. MicroRNA-210 regulates mitochondrial free radical response to hypoxia and krebs cycle in cancer cells by targeting iron sulfur cluster protein ISCU. PLoS One 5: e10345, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gee HE, Camps C, Buffa FM, Patiar S, Winter SC, Betts G, Homer J, Corbridge R, Cox G, West CM, Ragoussis J, and Harris AL. hsa-mir-210 is a marker of tumor hypoxia and a prognostic factor in head and neck cancer. Cancer 116: 2148–2158, 2010 [DOI] [PubMed] [Google Scholar]
- 19.Gertz M. and Steegborn C. The Lifespan-regulator p66Shc in mitochondria: redox enzyme or redox sensor? Antioxid Redox Signal 13: 1417–1428, 2010 [DOI] [PubMed] [Google Scholar]
- 20.Giorgio M, Migliaccio E, Orsini F, Paolucci D, Moroni M, Contursi C, Pelliccia G, Luzi L, Minucci S, Marcaccio M, Pinton P, Rizzuto R, Bernardi P, Paolucci F, and Pelicci PG. Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell 122: 221–233, 2005 [DOI] [PubMed] [Google Scholar]
- 21.Gorospe M, Tominaga K, Wu X, Fahling M, and Ivan M. Post-transcriptional control of the hypoxic response by RNA-binding proteins and MicroRNAs. Front Mol Neurosci 4: 7, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gou D, Ramchandran R, Peng X, Yao L, Kang K, Sarkar J, Wang Z, Zhou G, and Raj JU. miR-210 has an antiapoptotic effect in pulmonary artery smooth muscle cells during hypoxia. Am J Physiol Lung Cell Mol Physiol 303: L682–L691, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hamer PW, McGeachie JM, Davies MJ, and Grounds MD. Evans Blue dye as an in vivo marker of myofibre damage: optimising parameters for detecting initial myofibre membrane permeability. J Anat 200: 69–79, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hu S, Huang M, Li Z, Jia F, Ghosh Z, Lijkwan MA, Fasanaro P, Sun N, Wang X, Martelli F, Robbins RC, and Wu JC. MicroRNA-210 as a novel therapy for treatment of ischemic heart disease. Circulation 122: S124–S131, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang X, Ding L, Bennewith KL, Tong RT, Welford SM, Ang KK, Story M, Le QT, and Giaccia AJ. Hypoxia-inducible mir-210 regulates normoxic gene expression involved in tumor initiation. Mol Cell 35: 856–867, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang X, Le QT, and Giaccia AJ. MiR-210—micromanager of the hypoxia pathway. Trends Mol Med 16: 230–237, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jaffery Z, Thornton SN, and White CJ. Acute limb ischemia. Am J Med Sci 342: 226–234, 2011 [DOI] [PubMed] [Google Scholar]
- 28.Jeyaseelan K, Lim KY, and Armugam A. MicroRNA expression in the blood and brain of rats subjected to transient focal ischemia by middle cerebral artery occlusion. Stroke 39: 959–966, 2008 [DOI] [PubMed] [Google Scholar]
- 29.Kelly TJ, Souza AL, Clish CB, and Puigserver P. A hypoxia-induced positive feedback loop promotes hypoxia-inducible factor 1alpha stability through miR-210 suppression of glycerol-3-phosphate dehydrogenase 1-like. Mol Cell Biol 31: 2696–2706, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim HW, Haider HK, Jiang S, and Ashraf M. Ischemic preconditioning augments survival of stem cells via miR-210 expression by targeting caspase-8-associated protein 2. J Biol Chem 284: 33161–33168, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klont RE, Brocks L, and Eikelenboom G. Muscle fibre type and meat quality. Meat Sci 49S1: S219–S229, 1998 [PubMed] [Google Scholar]
- 32.Magenta A, Cencioni C, Fasanaro P, Zaccagnini G, Greco S, Sarra-Ferraris G, Antonini A, Martelli F, and Capogrossi MC. miR-200c is upregulated by oxidative stress and induces endothelial cell apoptosis and senescence via ZEB1 inhibition. Cell Death Differ 18: 1628–1639, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martelli F, Cenciarelli C, Santarelli G, Polikar B, Felsani A, and Caruso M. MyoD induces retinoblastoma gene expression during myogenic differentiation. Oncogene 9: 3579–3590, 1994 [PubMed] [Google Scholar]
- 34.Migliaccio E, Giorgio M, Mele S, Pelicci G, Reboldi P, Pandolfi PP, Lanfrancone L, and Pelicci PG. The p66shc adaptor protein controls oxidative stress response and life span in mammals. Nature 402: 309–313, 1999 [DOI] [PubMed] [Google Scholar]
- 35.Mutharasan RK, Nagpal V, Ichikawa Y, and Ardehali H. microRNA-210 is upregulated in hypoxic cardiomyocytes through Akt- and p53-dependent pathways and exerts cytoprotective effects. Am J Physiol Heart Circ Physiol 301: H1519–H1530, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pande RL, Perlstein TS, Beckman JA, and Creager MA. Secondary prevention and mortality in peripheral artery disease: National Health and Nutrition Examination Study, 1999 to 2004. Circulation 124: 17–23, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peach G, Griffin M, Jones KG, Thompson MM, and Hinchliffe RJ. Diagnosis and management of peripheral arterial disease. BMJ 345: e5208, 2012 [DOI] [PubMed] [Google Scholar]
- 38.Prabhakar NR. and Semenza GL. Adaptive and maladaptive cardiorespiratory responses to continuous and intermittent hypoxia mediated by hypoxia-inducible factors 1 and 2. Physiol Rev 92: 967–1003, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Puissegur MP, Mazure NM, Bertero T, Pradelli L, Grosso S, Robbe-Sermesant K, Maurin T, Lebrigand K, Cardinaud B, Hofman V, Fourre S, Magnone V, Ricci JE, Pouyssegur J, Gounon P, Hofman P, Barbry P, and Mari B. miR-210 is overexpressed in late stages of lung cancer and mediates mitochondrial alterations associated with modulation of HIF-1 activity. Cell Death Differ 18: 465–478, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Qi J, Qiao Y, Wang P, Li S, Zhao W, and Gao C. microRNA-210 negatively regulates LPS-induced production of proinflammatory cytokines by targeting NF-kappaB1 in murine macrophages. FEBS Lett 586: 1201–1207, 2012 [DOI] [PubMed] [Google Scholar]
- 41.Quero L, Dubois L, Lieuwes NG, Hennequin C, and Lambin P. miR-210 as a marker of chronic hypoxia, but not a therapeutic target in prostate cancer. Radiother Oncol 101: 203–208, 2011 [DOI] [PubMed] [Google Scholar]
- 42.Setacci C, de Donato G, Teraa M, Moll FL, Ricco JB, Becker F, Robert-Ebadi H, Cao P, Eckstein HH, De Rango P, Diehm N, Schmidli J, Dick F, Davies AH, Lepantalo M, and Apelqvist J. Chapter IV: treatment of critical limb ischaemia. Eur J Vasc Endovasc Surg 42Suppl 2: S43–S59, 2011 [DOI] [PubMed] [Google Scholar]
- 43.Shanmugasundaram M, Ram VK, Luft UC, Szerlip M, and Alpert JS. Peripheral arterial disease—what do we need to know? Clin Cardiol 34: 478–482, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Turrini P, Gaetano C, Antonelli A, Capogrossi MC, and Aloe L. Nerve growth factor induces angiogenic activity in a mouse model of hindlimb ischemia. Neurosci Lett 323: 109–112, 2002 [DOI] [PubMed] [Google Scholar]
- 45.Yang W, Sun T, Cao J, Liu F, Tian Y, and Zhu W. Downregulation of miR-210 expression inhibits proliferation, induces apoptosis and enhances radiosensitivity in hypoxic human hepatoma cells in vitro. Exp Cell Res 318: 944–954, 2012 [DOI] [PubMed] [Google Scholar]
- 46.Yoshioka Y, Kosaka N, Ochiya T, and Kato T. Micromanaging iron homeostasis: hypoxia-inducible micro-RNA-210 suppresses iron homeostasis-related proteins. J Biol Chem 287: 34110–34119, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zaccagnini G, Martelli F, Fasanaro P, Magenta A, Gaetano C, Di Carlo A, Biglioli P, Giorgio M, Martin-Padura I, Pelicci PG, and Capogrossi MC. p66ShcA modulates tissue response to hindlimb ischemia. Circulation 109: 2917–2923, 2004 [DOI] [PubMed] [Google Scholar]
- 48.Zaccagnini G, Martelli F, Magenta A, Cencioni C, Fasanaro P, Nicoletti C, Biglioli P, Pelicci PG, and Capogrossi MC. p66(ShcA) and oxidative stress modulate myogenic differentiation and skeletal muscle regeneration after hind limb ischemia. J Biol Chem 282: 31453–31459, 2007 [DOI] [PubMed] [Google Scholar]
- 49.Zeng L, Liu J, Wang Y, Wang L, Weng S, Tang Y, Zheng C, Cheng Q, Chen S, and Yang GY. MicroRNA-210 as a novel blood biomarker in acute cerebral ischemia. Front Biosci (Elite Ed) 3: 1265–1272, 2011 [DOI] [PubMed] [Google Scholar]
- 50.Zhang MY, Zhang WJ, and Medler S. The continuum of hybrid IIX/IIB fibers in normal mouse muscles: MHC isoform proportions and spatial distribution within single fibers. Am J Physiol Regul Integr Comp Physiol 299: R1582–R1591, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.