

Significance
Understanding how proteins are regulated through the binding of an effector molecule at a nonoverlapping site is a central problem in protein allostery. However, connecting the changes in structural dynamics to changes in the binding affinity can be challenging. Here, we investigate the allosteric mechanism involving a promiscuous protein that serves as a hub for a variety of intrinsically disordered proteins (IDPs). By using a coarse-grained model to simulate the interactions between the protein and two IDPs, we were able to recapitulate experimental binding affinities, capture relevant structural dynamics, and provide a thermodynamic and kinetic description of the allosteric mechanism. It is presumable that the features identified in this allosteric mechanism are conserved between the central protein and other IDPs.
Keywords: allostery, thermodynamics, kinetics, coupled folding and binding, intrinsically disordered proteins
Abstract
The kinase-inducible domain interacting (KIX) domain of the CREB binding protein (CBP) is capable of simultaneously binding two intrinsically disordered transcription factors, such as the mixed-lineage leukemia (MLL) and c-Myb peptides, at isolated interaction sites. In vitro, the affinity for binding c-Myb is approximately doubled when KIX is in complex with MLL, which suggests a positive cooperative binding mechanism, and the affinity for MLL is also slightly increased when KIX is first bound by c-Myb. Expanding the scope of recent NMR and computational studies, we explore the allosteric mechanism at a detailed molecular level that directly connects the microscopic structural dynamics to the macroscopic shift in binding affinities. To this end, we have performed molecular dynamics simulations of free KIX, KIX-c-Myb, MLL-KIX, and MLL-KIX-c-Myb using a topology-based Gō-like model. Our results capture an increase in affinity for the peptide in the allosteric site when KIX is prebound by a complementary effector and both peptides follow an effector-independent folding-and-binding mechanism. More importantly, we discover that MLL binding lowers the entropic cost for c-Myb binding, and vice versa, by stabilizing the L12-G2 loop and the C-terminal region of the α3 helix on KIX. This work demonstrates the importance of entropy in allosteric signaling between promiscuous molecular recognition sites and can inform the rational design of small molecule stabilizers to target important regions of conformationally dynamic proteins.
The term “allosteric” describes the effects of one ligand (or effector) on the catalysis or binding of another ligand at a nonoverlapping site (1, 2). This classical view can be characterized by three main attributes: (i) only two states exist (e.g., “active” or “inactive”); (ii) the allosteric signal is relayed through a single well-defined pathway; and (iii) a conformational change occurs in the nonoverlapping site. A modern interpretation treats the native state as a conformational ensemble (rather than only two conformational states) whose preexisting population of conformations can be redistributed as a result of some allosteric perturbation that can be transmitted through multiple signaling pathways (3–6). This “unified view” defines allostery in purely structural and thermodynamic terms (i.e., allostery can be controlled by entropy and/or enthalpy) and provides a quantitative framework for classifying allosteric mechanisms.
Allostery is a central pillar in the regulation of gene transcription. The transcriptional coactivator CREB binding protein (CBP), which serves as a keystone in the assembly of the transcriptional machinery, is responsible for regulating transcriptional processes relating to hematopoiesis (7, 8), cell differentiation (9, 10), etc. Contained within CBP is an 87-residue-long domain called kinase-inducible domain interacting (KIX) that is critical for regulating transcriptional activity (for review, see ref. 11) and can be cooperatively targeted by a diverse set of transcription factors (12–17) and small molecules (18–23). The structure of KIX (Fig. 1) can be summarized as a three-helix bundle (α1, residues 597–611; α2, residues 623–640; α3, residues 646–669) containing a mobile L12-G2 loop (residues 614–621) and an N-terminal 310 helix (residues 591–594). More importantly, KIX consists of at least two binding sites that are located on opposite surfaces of the protein, which can simultaneously bind, for example, the activation domain of the mixed lineage leukemia (MLL) protein and the activation domain of the protooncogene protein c-Myb (Fig. 1), both of which are expressed concurrently during hematopoietic precursor cell development and therefore play an important role in blood cell proliferation (24–27).
Fig. 1.

Different representations of the MLL-KIX-c-Myb ternary complex (PDB ID code 2AGH).
The binding of either MLL or c-Myb to one of these promiscuous sites can stabilize the C terminus of the α3 helix of KIX and/or decrease the mobility of the L12-G2 loop (4, 20, 28–32), but the functional role of these changes remains to be delineated. Furthermore, in vitro, KIX alone demonstrates a ∼2-fold lower affinity for c-Myb and ∼1.6-fold lower affinity for MLL compared with KIX bound to MLL or c-Myb, respectively (13). Although numerous efforts have been put forth to clarify the molecular mechanism for cooperative binding in KIX, the current allosteric picture detailing the thermodynamic and structural effects of MLL and/or c-Myb binding remains incomplete. Although all-atom molecular dynamics (MD) simulations complement experiment and have provided valuable insight into the allosteric process (20, 30, 33), they are computationally too expensive to explore timescales where multiple binding events or protein folding/unfolding are necessary to understand the underlying mechanism of action. To address these limitations, we use a topology-based Gō-like model (34, 35) that has shown success in examining coupled protein folding and binding (36–44) as well as modeling ligand-induced structural transitions, and allosteric communication (45–47). We extend its application to the study of allostery and cooperative binding for the first time (to our knowledge) here in the context of the MLL-KIX-c-Myb ternary system. To develop a quantitative picture that connects the microscopic changes in structural dynamics to the macroscopic shift in binding affinities and kinetics, we performed multiple simulations for each of KIX-free, MLL-KIX, KIX-c-Myb, and MLL-KIX-c-Myb, and carried out a comparative analysis of the potential allosteric communication pathways. Through this approach, we detected similarities in protein folding/unfolding, observed multiple c-Myb/MLL binding events, and estimated thermodynamic and kinetic parameters for these processes. Our results show that the binding of either peptide stabilizes the L12-G2 loop as well as increases the helicity of the α3 helix, both of which play a role in controlling the size of the hydrophobic core. Most notably, we find that this reduction in structural dynamics results in an increase in the population of KIX structures that resemble KIX in the ternary complex and the entropic cost for binding a peptide at the allosteric site is lowered when an effector is already bound to KIX. Collectively, our data demonstrate that these observations are sufficient to explain the increased binding affinity for c-Myb and MLL when the other complementary peptide is bound to KIX and can direct the design of small molecules to target important regions of conformationally dynamic proteins.
Results
Binding Affinities and Configurational Entropies.
Using a renormalized Gō-like model that was adjusted to capture experimentally reported helical content and binding affinities (Fig. S1), extensive MD simulations of the free KIX, KIX-c-Myb, MLL-KIX, and MLL-KIX-c-Myb systems were performed. It is important to emphasize that no additional calibration of the intermolecular interactions was used to simulate the MLL-KIX-c-Myb ternary complex. That is, aside from the native contacts, the interaction energies were not explicitly biased toward ternary complex formation (SI Text). The resulting thermodynamic parameters and kinetic rate constants for each simulation system are presented in Table 1 and show that MLL and c-Myb bind cooperatively to KIX rather than compete for KIX binding. The simulations revealed that c-Myb and MLL bind to KIX with a Kd = 30 ± 8 μM and a Kd = 0.8 ± 0.3 μM, respectively. In the presence of MLL, the affinity of KIX for c-Myb is enhanced by ∼10-fold and is accompanied by a ∼4.4-fold decrease in koff and ∼2.4-fold increase in kon. Similarly, the affinity of KIX-c-Myb for MLL is enhanced by ∼8-fold and is accompanied by a ∼3.3-fold decrease in koff and ∼1.9-fold increase in kon. Relative to free KIX, the difference in configurational entropy (−TΔS) at 300 K for forming KIX-c-Myb, MLL-KIX, and MLL-KIX-c-Myb was ∼0.8, ∼1.2, and ∼1.4 kcal/mol, respectively (Table 1).
Table 1.
Thermodynamic parameters and kinetic rate constants
| Ligand | Binding to | Expt. Kd*, μM | Simulated Kd†, μM | Simulated koff†, s−1 | Simulated kon†, M−1⋅s−1 | −TΔS‡, kcal/mol |
| c-Myb | KIX | 10 ± 2 | 30 ± 8 | 5.92E+06 ± 0.94E+06 | 0.20E+12 ± 0.02E+12 | 0.8 |
| c-Myb | KIX-MLL | 4 ± 1 | 3 ± 1 | 1.36E+06 ± 0.24E+06 | 0.48E+12 ± 0.17E+12 | 1.4 |
| MLL | KIX | 2.8 ± 0.4 | 0.8 ± 0.3 | 2.46E+06 ± 0.54E+06 | 3.19E+12 ± 0.60E+12 | 1.2 |
| MLL | KIX-c-Myb | 1.7 ± 0.1 | 0.1 ± 0.1 | 0.75E+06 ± 0.10E+06 | 6.02E+12 ± 0.57E+12 | 1.4 |
Experimental Kd published in ref. 13.
Averaged over three groups of 20 simulations.
Calculated at 300 K relative to KIX-free.
Intrinsically Disordered Protein Binding Mechanism.
We examined the relationship between c-Myb binding and c-Myb folding by constructing free-energy profiles along the fraction of intramolecular (Qintra) and intermolecular (Qinter) native contacts (Fig. 2). In both the presence and absence of MLL, the free-energy minimum shows c-Myb as being ∼40–80% folded and bound to KIX with ∼70–95% of its intermolecular native contacts intact. However, when c-Myb loses all of its intermolecular native contacts, it is ∼10–50% helical. Free c-Myb was not found to exist in a preformed helical state and, instead, followed a folding-and-binding mechanism that appears to be independent of MLL. Similar results that support a folding-and-binding mechanism were also observed for MLL binding to free KIX (Fig. S2).
Fig. 2.

Two-dimensional free-energy surfaces as a function of c-Myb folding and binding in (A) the absence of MLL and (B) the presence of MLL. Qintra is the fraction of native intramolecular contacts in c-Myb, whereas Qinter is the fraction of native intermolecular contacts formed between KIX-c-Myb.
To further probe the effects of MLL on c-Myb binding, we compared the order of contact formation between KIX and c-Myb by monitoring the contact appearance order. Leu302 of c-Myb is predominantly first to form its intermolecular native contacts (Fig. S3). Then, other intermolecular native contacts are formed sequentially outward toward both termini of c-Myb. The order in which the intermolecular native contacts were formed between KIX-c-Myb was also found to be independent of MLL binding. Similarly, the order of contact formation between MLL and KIX was also found to be independent of c-Myb binding (Fig. S4).
KIX Dynamics.
Fig. 3 shows the root-mean-square fluctuation of the highest ranked mode computed from principal component analysis (PCA) and projected onto the average simulation structure for each system. In free KIX, the C terminus of the α3 helix and the L12-G2 loop were the most mobile. In fact, the average structure shows a completely unfolded α3 C terminus beginning at around residue 657. Upon binding c-Myb, the mobility of the α3 helix is slightly reduced, whereas the L12-G2 loop remains as flexible as in free KIX. Aside from the C terminus of the α3 helix, the average structures of KIX-free and KIX-c-Myb are very similar. In contrast, MLL binding stabilizes the C-terminal residues of α3 and regains the secondary structure of this helix. Also, the L12-G2 loop appears to be less mobile than in free KIX or KIX-c-Myb. Finally, the ternary complex shows a rigid α3 helix and only some flexibility in the L12-G2 loop, similar to the MLL-KIX binary system. Consistent with these findings, Fig. S5 shows that the binding of MLL and/or c-Myb results in a significant increase in α3 helicity.
Fig. 3.
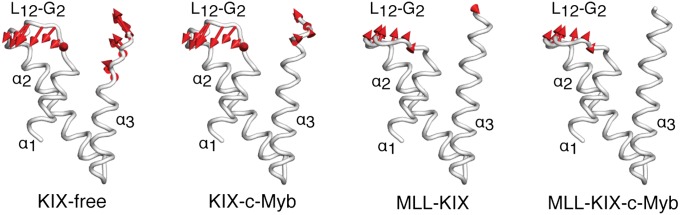
Root-mean-square fluctuations from PCA. The average KIX structure from each simulation is shown in white, and the ligands were omitted for clarity. The red arrows point along the direction of the lowest frequency mode (highest ranked eigenvector), and the amplitude is directly proportional to the length of the arrow. Only fluctuations greater than 1.0 Å are displayed.
Representative free-energy surfaces along the L12-G2 loop root-mean-square deviation (rmsd) (with respect to the NMR structure) and the hydrophobic core size, which effectively describes the distance between helices α1 and α3, were computed and are displayed in Fig. 4. Overall, the conformational sampling of KIX along the two collective variables is the most restricted when both peptides are bound. Relative to KIX-free, the binding of MLL and/or c-Myb decreases the extent of sampling of the loop with the ternary complex being affected the most. The maximum opening of the hydrophobic core is also reduced from ∼5.5 to ∼4 Å when the loop deviates significantly from the reference NMR structure. Again, this trend is most pronounced in the ternary complex. In all cases, the lowest free-energy minimum has a hydrophobic core that is open by ∼3.5–4 Å, but the loop rmsd within each minimum is restricted to ∼4–7 Å when MLL is present, and expands to a wider range (∼4–10 Å) when MLL is absent. Additional free-energy profiles calculated with respect to the α3 helix rmsd, which is believed to contribute to the allosteric binding of c-Myb, are reported in Fig. S6 and show similar results to Fig. 4. Overall, all of the free-energy surfaces show a reduction in the conformational space being sampled when one or both peptides are bound to KIX (i.e., the colored area became smaller upon binding MLL and/or c-Myb and narrowed toward a ternary-like KIX).
Fig. 4.

Representative 2D free-energy surfaces as a function of the hydrophobic core size and the L12-G2 loop rmsd. All rmsd values were calculated relative to the NMR structure, which had a hydrophobic core size of ∼3.6 Å. All ΔS were calculated with respect to KIX-free at 300 K (Methods and Table 1).
Discussion
It has been hypothesized that protein intrinsic disorder offers numerous benefits such as fast protein turnover (48), high specificity for a diverse set of targets (48–52), and high specificity with low affinity binding (49, 50). In addition, it has been predicted that allosteric coupling between independent sites is maximized in the presence of intrinsic disorder (53). In intrinsically disordered protein systems, like those in the present study, there is an intricate relationship between binding and folding (36–39, 54–60). We observed that both c-Myb and MLL first bind to KIX before forming a folded peptide, and the free-energy surfaces between binary and ternary KIX complexes were virtually indistinguishable for both MLL and c-Myb (Fig. 2 and Fig. S2). Additionally, MLL and c-Myb, which were tuned to have a peak helicity of ∼5% and ∼25%, respectively, to match experiment (Fig. S1), achieved over 80% helicity upon binding to KIX. This binding-induced-folding mechanism is consistent with kinetic experiments of KIX-c-Myb (i.e., rapid mixing of KIX with c-Myb) (55, 61, 62), and our results suggest that effector binding allosterically regulates peptide binding at the second site without altering the binding mechanism.
We explored this further by examining the intermolecular contact appearance order for KIX and c-Myb to see whether MLL binding alters the sequence in which contacts between c-Myb and KIX are formed with a low probability of breaking. Fig. S3A shows that Leu302, which is positioned in the middle of the helix and inserts its side chain deep into the hydrophobic core in the native NMR structure (28), is first to form most of its interactions with the α1 and α3 helices of KIX. According to Fig. 2, c-Myb is mostly unstructured at this stage, with only ∼20–40% helicity. Then, binding proceeds outward toward both termini of c-Myb and is concomitant with c-Myb folding (Fig. S3A). The N terminus of c-Myb, which interacts exclusively with the C terminus of the α3 helix, has a slightly higher probability than the C terminus for forming last, which may imply a potential role for α3 in regulating c-Myb binding. These results were similar for c-Myb binding to the binary complex (Fig. S3B) and demonstrate that MLL binding does not have a significant impact on the order in which c-Myb residues contact KIX.
Previous studies have observed changes in the KIX structure resulting from transcription factor binding, and both MLL and c-Myb have been shown to allosterically affect the affinity of KIX for the complementary peptide (4, 20, 28–32). However, a connection between the changes in structural dynamics in KIX and how this directly leads to shifts in transcription factor binding affinities remain tenuous. To bridge this gap, we used a coarse-grained simulation model that is capable of capturing both the microscopic structural changes and macroscopic ligand binding events. Consistent with cooperative binding, we found that the binding affinity for c-Myb is increased by an order of magnitude in the presence of MLL while the affinity for MLL is also increased by a factor of 8 when c-Myb is present (Table 1). These observations are in agreement with experiment, albeit with an approximately fivefold difference in both cases (13), but given the simplicity of our simulation model, it is remarkable that we are able to capture the same allosteric trends as seen in experiment. We also observed relative decreases in koff (∼4.4-fold and ∼3.3-fold for c-Myb and MLL, respectively) and small increases in kon (∼2.4-fold and ∼1.9-fold for c-Myb and MLL, respectively) when an effector was bound first (Table 1). These changes in kinetics combined with our observations of an effector-independent binding-induced-folding mechanism suggest that prebinding of the first peptide likely stabilizes the flexible/unfolded parts of the KIX structure so that the second peptide can (i) form a native intermolecular contact more quickly (i.e., small increase in kon) and (ii) stay bound for much longer (i.e., decrease koff).
To further understand the allosteric mechanism, we investigated how MLL and/or c-Myb binding can affect the dynamics of KIX (Figs. 3 and 4, and Fig. S6). Overall, we found that binding of MLL and/or c-Myb resulted in an increase in stability (or a reduction in dynamics) of the α3 C terminus and the L12-G2 loop. This is consistent with observations made in previous NMR and computational studies, which have led to the suggestion that the increased structural stability of KIX may play a role in allosteric signaling (4, 20, 28–32). For example, NMR backbone order parameter measurements of KIX have demonstrated that the L12-G2 loop and the C-terminal residues of the α3 helix are more rigid when MLL is bound to KIX (29). Thus, our findings are not only supported by experiment but they also provide a direct causative link between changes in the local structural dynamics and its effects on the global binding affinities. From the observations made in Fig. 4 and Fig. S6, we calculated the configurational entropy for each system (Table 1 and Eq. 1) by examining the distribution of configurations along all three collective variables (i.e., L12-G2 loop rmsd, α3 rmsd, and hydrophobic core size). The energetic cost for binding either MLL or c-Myb to KIX-free was –TΔΔS = 0.6 kcal/mol higher than when binding to its complementary binary complex (Table 1, Fig. 4, and Fig. S6). Thus, our analyses show that effector-bound KIX lowers the entropic cost for binding the complementary peptide at the second site, and this is achieved by stabilizing the KIX structure. The observed reduction in entropy due to effector binding explains the decrease in koff and the small increase in kon and this notion of “prepaying the entropic cost” in allostery has been found to play a role in the tryptophan RNA binding attenuation protein (TRAP) (63, 64) and the catabolite activator protein (CAP) (5, 65, 66). Describing allostery within a thermodynamic framework (3–6) allows us to organize the information in a quantitative manner, and it enables comparisons between entropically driven and/or enthalpically driven allosteric mechanisms.
Previous NMR studies have shown that a minor excited state of KIX, which is believed to be a higher affinity conformer for c-Myb and pKID, becomes 7% populated when bound by MLL and has been characterized by a rearrangement of the KIX hydrophobic core (29, 32). In a recent metadynamics-based MD simulation study (31) of the MLL-KIX binary structure (29), it was demonstrated that when the L12-G2 loop was in the “up” state then this resulted in opening of the KIX hydrophobic core, which was said to allow KIX to spontaneously interconvert between a major lower energy state and a minor higher energy state. Essentially, the authors proffered that KIX mediates cooperativity between transcription factors through conformational selection of a higher affinity conformer. This means that, if conformational selection for a 7% binding-competent state were at play, we should expect to see an observable population increase of a minor state in our free-energy surfaces. However, we find no evidence to support this. Instead, upon binding an effector, we observe a narrowing of the preexisting KIX ensemble toward the highly populated minimum, which corresponds to a ternary-like KIX structure (Fig. 4 and Fig. S6). We also point out that, although the enhanced sampling MD simulations of the MLL-KIX complex were informative, simulations of the binary complex alone are insufficient to show, unequivocally, that the observed changes in structural dynamics due to the presence of an effector would have any direct effect on binding affinities of a second transactivation domain. However, our simulations of the free KIX, MLL-KIX, KIX-c-Myb, and MLL-KIX-c-Myb systems provide a clear and systematic way to directly relate the microscopic allosteric effects and the macroscopic changes in the binding affinity that result from MLL and/or c-Myb binding to KIX.
In the accompanying paper, Shammas and coworkers (67) have performed fluorescence stopped-flow measurements to investigate the binding kinetics of peptides from five different transcription factors that can all bind to KIX (c-Myb and pKID bind at the c-Myb site, whereas MLL, HBZ, and E2A bind at the MLL site). Consistent with a coupled folding-and-binding mechanism, they found that the helicity for c-Myb (as well as all of the other peptides) increased upon mixing with KIX, which is consistent with our results. They also observed a positive allosteric effect and identified relative decreases in koff of 3.9 ± 0.1 and 4.1 ± 0.1 for c-Myb and MLL, respectively, that are similar to those reported above. They also demonstrated that the reduction in the dissociation rate constant was generally true for four different pairs of KIX binding partners, and, based upon their data, they proposed a common allosteric mechanism where the entropic cost of binding the second ligand is paid on binding the first ligand rather than involving the formation of a binding-competent low-populated conformer of KIX (29, 31, 32). Our simulation results show good agreement with their kinetics experiments and confirm the hypothesis that MLL binding to KIX lowers the entropic cost for c-Myb binding, and vice versa, by stabilizing the L12-G2 loop and α3 helix. However, they found a relative decrease, rather than a small relative increase, in kon of 2.9 ± 0.1 and 2.8 ± 0.1 for c-Myb and MLL, respectively, and, when the favorable long-range electrostatics was screened with salt, only negligible differences in kon were observed. This can be attributed to differences in temperature or in how a bound state is detected in experiment versus computation. Overall, both bodies of work are in good complementary agreement and support an allosteric mechanism whereby the binding of an effector prepays the entropic cost for binding a complementary peptide in the secondary site.
If a change in the structural stability (or dynamics) is what drives the allosteric mechanism, one could explore alternative ways to modulate the stability of KIX to affect binding affinity. It was previously proposed that stabilizing the C terminus of α3 through MLL binding would result in favorable side-chain electrostatic interactions between residues Glu665 and Glu666 of α3 and Arg294 and Lys291 of c-Myb, (28). Not surprisingly, these electrostatically complementary residues are essentially conserved in all known c-Myb and CBP/p300 sequences (28). Table S1 reports the top six point mutant candidates that were predicted from AGADIR (68–72) to increase the helical propensity of the α3 helix and therefore can potentially affect cooperative binding (SI Text). Out of all of the C-terminal residues located in the α3 helix that make no intermolecular native contacts with either c-Myb or MLL, only six point mutations at either Lys667 or Ser670 caused a significant increase in the predicted helicity of α3 (∼3.5–12.7%) relative to the wild-type sequence. Thus, we predict that, even in the absence of MLL, one or more of these strategically selected point mutations could mimic the effects of MLL binding to KIX and result in an increased c-Myb binding affinity. These predictions can be verified experimentally and could inform the design of small molecules that could stabilize KIX and allow for further structural characterization. For instance, Mapp and coworkers recently solved the first crystal structure of KIX complexed to a small molecule referred to as fragment 1–10 (20). It was found that the L12-G2 loop had large temperature factors (or B factors), the C-terminal residues of the helix α3 were unresolved, and the size of the hydrophobic core was reduced in comparison with the NMR ternary structure. This suggests that the entropy of KIX would not be reduced upon binding fragment 1–10, and, in light of the observations made in the current study, we would predict that this would not lead to an increase in affinity of KIX for c-Myb. This strategy of complementing experiment with computation can be applied generally to other conformationally dynamic proteins and potentially lead to a faster discovery of effective small-molecule modulators.
In conclusion, we have carried out a set of simulations of KIX-free, MLL-KIX, KIX-c-Myb, and MLL-KIX-c-Myb to better understand how cooperative binding is regulated in this particular transcriptional activation system. Remarkably, our simplified model was able to capture relevant changes in thermodynamics, binding kinetics, and structural dynamics, all of which were in good agreement with experiment. Specifically, we discovered that the intermolecular native contact formation between c-Myb and KIX is unaffected by MLL binding, and vice versa, and that the folding-and-binding mechanism is well preserved. Furthermore, our results confirm that KIX occupation by a single transactivation domain was found to stabilize the L12-G2 loop and increase the helicity of the α3 helix. This lowered the entropic cost for binding a complementary peptide at the allosteric site and was accompanied by a narrowing of the preexisting KIX ensemble toward a ternary-like KIX conformation. We also proposed that a well-chosen mutation(s) or small molecule(s) targeted toward stabilizing the α3 helix and/or the L12-G2 loop would be effective in controlling cooperative binding. As a whole, our study directly links the effects of the microscopic changes in structural dynamics to the macroscopic binding affinities and provides a thermodynamic description of the allosteric mechanism involved in KIX cooperative binding.
Methods
Simulation Model and Setup.
An initial Cα-based Gō-like model (SI Text) constructed from the NMR coordinates of the MLL-KIX-c-Myb ternary complex [Protein Data Bank (PDB) ID code 2AGH] (28) was generated using the Multiscale Modeling Tools for Structural Biology (MMTSB) Gō-Model Builder (www.mmtsb.org) (73). MLL (19 residues ranging from 839 to 857), KIX (87 residues ranging 586–672), and c-Myb (25 residues ranging from 291 to 315) contained 22, 198, and 40 intramolecular native contacts, respectively, whereas the total number of intermolecular native contacts between MLL-KIX and KIX-c-Myb were 28 and 29, respectively. Structural models of the individual monomers and binary complexes were derived directly from the ternary complex model. The coarse-grained Gō-like model was then calibrated to match the helical content and binary dissociation constants reported in previous experiments (SI Text and Fig. S1). Each of the four simulation models (i.e., free KIX, MLL-KIX, KIX-c-Myb, and MLL-KIX-c-Myb) was simulated for 15 μs using CHARMM (74, 75) and repeated 60 times to sample multiple c-Myb/MLL binding events in the presence and absence of a complementary effector peptide (SI Text).
Analysis.
Two-dimensional free-energy surfaces were computed at 300 K by constructing 2D histograms along two reaction coordinates of interest. Qintra and Qinter correspond to the fraction of native intramolecular and intermolecular contacts, respectively. The rmsd of the L12-G2 loop (residues 614–621) was calculated after fitting parts of helices α1, α2, and α3 (residues 600–613, 622–662) of KIX from each simulation snapshot to the native NMR structure (31). Similarly, the rmsd of helix α3 (residues 646–669) was calculated by prefitting the rigid N-terminal portion of helix α3 (residues 646–657) to the native NMR structure. The size of the hydrophobic core (residues 607–612, 650–661) (29), whose shape can be approximated by an ellipsoid (31), was calculated from the square root of the second eigenvalue of the gyration tensor (76).
We estimated the configurational entropy, S, using the following:
| [1] |
where kB is the Boltzmann constant and p(L,A,H) is the probability of occupying a particular state along the three collective variables L, A, and H (corresponding to the L12-G2 loop rmsd, helix α3 rmsd, and KIX hydrophobic core size, respectively) in a given simulation. All ΔS were calculated with respect to free KIX at 300 K.
PCA was carried out to study the dynamics of KIX for each system (SI Text). Each simulation snapshot was superimposed onto the three helices of KIX (residues 600–613, 622–662) in the native NMR structure, and the eigenvalues and eigenvectors were obtained by diagonalizing the covariance matrix constructed from the fluctuations of residues 600–669.
Supplementary Material
Acknowledgments
We acknowledge valuable scientific discussions with Drs. Logan Ahlstrom, Bin Zhang, Karunesh Arora, Afra Panahi, and Alex Dickson, as well as Dr. Ningkun Wang and Jean Lodge from the Mapp group. We also thank Garrett Goh and Dr. Sarah Shammas for critical reading of the manuscript. Funding for this work was provided by National Institutes of Health Grant GM037554.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1405831111/-/DCSupplemental.
References
- 1.Monod J, Jacob F. General conclusions: Teleonomic mechanisms in cellular metabolism, growth, and differentiation. Cold Spring Harbor Symp Quant Biol. 1961;26:389–401. doi: 10.1101/sqb.1961.026.01.048. [DOI] [PubMed] [Google Scholar]
- 2.Monod J, Wyman J, Changeux J-P. On the nature of allosteric transitions: A plausible model. J Mol Biol. 1965;12(1):88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- 3.Tsai C-J, Del Sol A, Nussinov R. Protein allostery, signal transmission and dynamics: A classification scheme of allosteric mechanisms. Mol Biosyst. 2009;5(3):207–216. doi: 10.1039/b819720b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.del Sol A, Tsai CJ, Ma B, Nussinov R. The origin of allosteric functional modulation: Multiple pre-existing pathways. Structure. 2009;17(8):1042–1050. doi: 10.1016/j.str.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tsai C-J, del Sol A, Nussinov R. Allostery: Absence of a change in shape does not imply that allostery is not at play. J Mol Biol. 2008;378(1):1–11. doi: 10.1016/j.jmb.2008.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsai C-J, Nussinov R. A unified view of “how allostery works.”. PLoS Comput Biol. 2014;10(2):e1003394. doi: 10.1371/journal.pcbi.1003394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iyer NG, Özdag H, Caldas C. p300/CBP and cancer. Oncogene. 2004;23(24):4225–4231. doi: 10.1038/sj.onc.1207118. [DOI] [PubMed] [Google Scholar]
- 8.Kimbrel EA, et al. Systematic in vivo structure-function analysis of p300 in hematopoiesis. Blood. 2009;114(23):4804–4812. doi: 10.1182/blood-2009-04-217794. [DOI] [PubMed] [Google Scholar]
- 9.Kasper LH, et al. Conditional knockout mice reveal distinct functions for the global transcriptional coactivators CBP and p300 in T-cell development. Mol Cell Biol. 2006;26(3):789–809. doi: 10.1128/MCB.26.3.789-809.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iyer NG, et al. p300 is required for orderly G1/S transition in human cancer cells. Oncogene. 2007;26(1):21–29. doi: 10.1038/sj.onc.1209771. [DOI] [PubMed] [Google Scholar]
- 11.Thakur JK, Yadav A, Yadav G. Molecular recognition by the KIX domain and its role in gene regulation. Nucleic Acids Res. 2014;42(4):2112–2125. doi: 10.1093/nar/gkt1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Radhakrishnan I, et al. Structural analyses of CREB-CBP transcriptional activator-coactivator complexes by NMR spectroscopy: Implications for mapping the boundaries of structural domains. J Mol Biol. 1999;287(5):859–865. doi: 10.1006/jmbi.1999.2658. [DOI] [PubMed] [Google Scholar]
- 13.Goto NK, Zor T, Martinez-Yamout M, Dyson HJ, Wright PE. Cooperativity in transcription factor binding to the coactivator CREB-binding protein (CBP). The mixed lineage leukemia protein (MLL) activation domain binds to an allosteric site on the KIX domain. J Biol Chem. 2002;277(45):43168–43174. doi: 10.1074/jbc.M207660200. [DOI] [PubMed] [Google Scholar]
- 14.Campbell KM, Lumb KJ. Structurally distinct modes of recognition of the KIX domain of CBP by Jun and CREB. Biochemistry. 2002;41(47):13956–13964. doi: 10.1021/bi026222m. [DOI] [PubMed] [Google Scholar]
- 15.Vendel AC, McBryant SJ, Lumb KJ. KIX-mediated assembly of the CBP-CREB-HTLV-1 tax coactivator-activator complex. Biochemistry. 2003;42(43):12481–12487. doi: 10.1021/bi0353023. [DOI] [PubMed] [Google Scholar]
- 16.Zor T, De Guzman RN, Dyson HJ, Wright PE. Solution structure of the KIX domain of CBP bound to the transactivation domain of c-Myb. J Mol Biol. 2004;337(3):521–534. doi: 10.1016/j.jmb.2004.01.038. [DOI] [PubMed] [Google Scholar]
- 17.Vendel AC, Lumb KJ. NMR mapping of the HIV-1 Tat interaction surface of the KIX domain of the human coactivator CBP. Biochemistry. 2004;43(4):904–908. doi: 10.1021/bi035612l. [DOI] [PubMed] [Google Scholar]
- 18.Lee LW, Mapp AK. Transcriptional switches: Chemical approaches to gene regulation. J Biol Chem. 2010;285(15):11033–11038. doi: 10.1074/jbc.R109.075044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Majmudar CY, et al. Sekikaic acid and lobaric acid target a dynamic interface of the coactivator CBP/p300. Angew Chem Int Ed Engl. 2012;51(45):11258–11262. doi: 10.1002/anie.201206815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang N, et al. Ordering a dynamic protein via a small-molecule stabilizer. J Am Chem Soc. 2013;135(9):3363–3366. doi: 10.1021/ja3122334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bates CA, Pomerantz WC, Mapp AK. Transcriptional tools: Small molecules for modulating CBP KIX-dependent transcriptional activators. Biopolymers. 2011;95(1):17–23. doi: 10.1002/bip.21548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pomerantz WC, et al. Profiling the dynamic interfaces of fluorinated transcription complexes for ligand discovery and characterization. ACS Chem Biol. 2012;7(8):1345–1350. doi: 10.1021/cb3002733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lodge JM, Rettenmaier TJ, Wells JA, Pomerantz WC, Mapp AK. Fp tethering: A screening technique to rapidly identify compounds that disrupt protein–protein interactions. Medchemcomm. 2014;5:370–375. doi: 10.1039/C3MD00356F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jin S, et al. c-Myb binds MLL through menin in human leukemia cells and is an important driver of MLL-associated leukemogenesis. J Clin Invest. 2010;120(2):593–606. doi: 10.1172/JCI38030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hess JL, et al. c-Myb is an essential downstream target for homeobox-mediated transformation of hematopoietic cells. Blood. 2006;108(1):297–304. doi: 10.1182/blood-2005-12-5014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Graf T. Myb: A transcriptional activator linking proliferation and differentiation in hematopoietic cells. Curr Opin Genet Dev. 1992;2(2):249–255. doi: 10.1016/s0959-437x(05)80281-3. [DOI] [PubMed] [Google Scholar]
- 27.Kasper LH, et al. Genetic interaction between mutations in c-Myb and the KIX domains of CBP and p300 affects multiple blood cell lineages and influences both gene activation and repression. PLoS One. 2013;8(12):e82684. doi: 10.1371/journal.pone.0082684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.De Guzman RN, Goto NK, Dyson HJ, Wright PE. Structural basis for cooperative transcription factor binding to the CBP coactivator. J Mol Biol. 2006;355(5):1005–1013. doi: 10.1016/j.jmb.2005.09.059. [DOI] [PubMed] [Google Scholar]
- 29.Brüschweiler S, Konrat R, Tollinger M. Allosteric communication in the KIX domain proceeds through dynamic repacking of the hydrophobic core. ACS Chem Biol. 2013;8(7):1600–1610. doi: 10.1021/cb4002188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Korkmaz EN, Nussinov R, Haliloğlu T. Conformational control of the binding of the transactivation domain of the MLL protein and c-Myb to the KIX domain of CREB. PLoS Comput Biol. 2012;8(3):e1002420. doi: 10.1371/journal.pcbi.1002420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Palazzesi F, Barducci A, Tollinger M, Parrinello M. The allosteric communication pathways in KIX domain of CBP. Proc Natl Acad Sci USA. 2013;110(35):14237–14242. doi: 10.1073/pnas.1313548110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brüschweiler S, et al. Direct observation of the dynamic process underlying allosteric signal transmission. J Am Chem Soc. 2009;131(8):3063–3068. doi: 10.1021/ja809947w. [DOI] [PubMed] [Google Scholar]
- 33.Umezawa K, Ikebe J, Takano M, Nakamura H, Higo J. Conformational ensembles of an intrinsically disordered protein Pkid with and without a KIX domain in explicit solvent investigated by all-atom multicanonical molecular dynamics. Biomolecules. 2012;2(1):104–121. doi: 10.3390/biom2010104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Karanicolas J, Brooks CL., 3rd Improved Gō-like models demonstrate the robustness of protein folding mechanisms towards non-native interactions. J Mol Biol. 2003;334(2):309–325. doi: 10.1016/j.jmb.2003.09.047. [DOI] [PubMed] [Google Scholar]
- 35.Karanicolas J, Brooks CL., 3rd The origins of asymmetry in the folding transition states of protein L and protein G. Protein Sci. 2002;11(10):2351–2361. doi: 10.1110/ps.0205402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Turjanski AG, Best R, Hummer G, Gutkind JS. The folding and binding pathway of the transcription factor Creb to CBP. FASEB J. 2007;21(5):A270–A270. [Google Scholar]
- 37.Ganguly D, Chen JH. Topology-based modeling of intrinsically disordered proteins: Balancing intrinsic folding and intermolecular interactions. Proteins. 2011;79(4):1251–1266. doi: 10.1002/prot.22960. [DOI] [PubMed] [Google Scholar]
- 38.De Sancho D, Best RB. Modulation of an IDP binding mechanism and rates by helix propensity and non-native interactions: Association of HIF1α with CBP. Mol Biosyst. 2012;8(1):256–267. doi: 10.1039/c1mb05252g. [DOI] [PubMed] [Google Scholar]
- 39.Ahlstrom LS, Dickson A, Brooks CL., 3rd Binding and folding of the small bacterial chaperone HdeA. J Phys Chem B. 2013;117(42):13219–13225. doi: 10.1021/jp403264s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Clementi C, Jennings PA, Onuchic JN. How native-state topology affects the folding of dihydrofolate reductase and interleukin-1β. Proc Natl Acad Sci USA. 2000;97(11):5871–5876. doi: 10.1073/pnas.100547897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Levy Y, Cho SS, Onuchic JN, Wolynes PG. A survey of flexible protein binding mechanisms and their transition states using native topology based energy landscapes. J Mol Biol. 2005;346(4):1121–1145. doi: 10.1016/j.jmb.2004.12.021. [DOI] [PubMed] [Google Scholar]
- 42.Levy Y, Onuchic JN. Mechanisms of protein assembly: Lessons from minimalist models. Acc Chem Res. 2006;39(2):135–142. doi: 10.1021/ar040204a. [DOI] [PubMed] [Google Scholar]
- 43.Levy Y, Wolynes PG, Onuchic JN. Protein topology determines binding mechanism. Proc Natl Acad Sci USA. 2004;101(2):511–516. doi: 10.1073/pnas.2534828100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shea JE, Onuchic JN, Brooks CL., 3rd Exploring the origins of topological frustration: Design of a minimally frustrated model of fragment B of protein A. Proc Natl Acad Sci USA. 1999;96(22):12512–12517. doi: 10.1073/pnas.96.22.12512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jana B, Morcos F, Onuchic JN. From structure to function: The convergence of structure based models and co-evolutionary information. Phys Chem Chem Phys. 2014;16(14):6496–6507. doi: 10.1039/c3cp55275f. [DOI] [PubMed] [Google Scholar]
- 46.Whitford PC, Gosavi S, Onuchic JN. Conformational transitions in adenylate kinase. Allosteric communication reduces misligation. J Biol Chem. 2008;283(4):2042–2048. doi: 10.1074/jbc.M707632200. [DOI] [PubMed] [Google Scholar]
- 47.Hyeon C, Jennings PA, Adams JA, Onuchic JN. Ligand-induced global transitions in the catalytic domain of protein kinase A. Proc Natl Acad Sci USA. 2009;106(9):3023–3028. doi: 10.1073/pnas.0813266106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wright PE, Dyson HJ. Intrinsically unstructured proteins: Re-assessing the protein structure-function paradigm. J Mol Biol. 1999;293(2):321–331. doi: 10.1006/jmbi.1999.3110. [DOI] [PubMed] [Google Scholar]
- 49.Uversky VN, Oldfield CJ, Dunker AK. Showing your ID: Intrinsic disorder as an ID for recognition, regulation and cell signaling. J Mol Recognit. 2005;18(5):343–384. doi: 10.1002/jmr.747. [DOI] [PubMed] [Google Scholar]
- 50.Liu J, et al. Intrinsic disorder in transcription factors. Biochemistry. 2006;45(22):6873–6888. doi: 10.1021/bi0602718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Uversky VN. Natively unfolded proteins: A point where biology waits for physics. Protein Sci. 2002;11(4):739–756. doi: 10.1110/ps.4210102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dunker AK, et al. Intrinsically disordered protein. J Mol Graph Model. 2001;19(1):26–59. doi: 10.1016/s1093-3263(00)00138-8. [DOI] [PubMed] [Google Scholar]
- 53.Hilser VJ, Thompson EB. Intrinsic disorder as a mechanism to optimize allosteric coupling in proteins. Proc Natl Acad Sci USA. 2007;104(20):8311–8315. doi: 10.1073/pnas.0700329104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sugase K, Dyson HJ, Wright PE. Mechanism of coupled folding and binding of an intrinsically disordered protein. Nature. 2007;447(7147):1021–1025. doi: 10.1038/nature05858. [DOI] [PubMed] [Google Scholar]
- 55.Gianni S, Morrone A, Giri R, Brunori M. A folding-after-binding mechanism describes the recognition between the transactivation domain of c-Myb and the KIX domain of the CREB-binding protein. Biochem Biophys Res Commun. 2012;428(2):205–209. doi: 10.1016/j.bbrc.2012.09.112. [DOI] [PubMed] [Google Scholar]
- 56.Ganguly D, Chen JH. Concerted involvement of long-range electrostatic interactions and fly-casting in recognition of intrinsically disordered proteins. Biophys J. 2010;98(3):257A. [Google Scholar]
- 57.Dyson HJ, Wright PE. Coupling of folding and binding for unstructured proteins. Curr Opin Struct Biol. 2002;12(1):54–60. doi: 10.1016/s0959-440x(02)00289-0. [DOI] [PubMed] [Google Scholar]
- 58.Wright PE, Dyson HJ. Linking folding and binding. Curr Opin Struct Biol. 2009;19(1):31–38. doi: 10.1016/j.sbi.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ganguly D, Zhang W, Chen J. Electrostatically accelerated encounter and folding for facile recognition of intrinsically disordered proteins. PLoS Comput Biol. 2013;9(11):e1003363. doi: 10.1371/journal.pcbi.1003363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rogers JM, Steward A, Clarke J. Folding and binding of an intrinsically disordered protein: Fast, but not “diffusion-limited.”. J Am Chem Soc. 2013;135(4):1415–1422. doi: 10.1021/ja309527h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Giri R, Morrone A, Toto A, Brunori M, Gianni S. Structure of the transition state for the binding of c-Myb and KIX highlights an unexpected order for a disordered system. Proc Natl Acad Sci USA. 2013;110(37):14942–14947. doi: 10.1073/pnas.1307337110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shammas SL, Travis AJ, Clarke J. Remarkably fast coupled folding and binding of the intrinsically disordered transactivation domain of cMyb to CBP KIX. J Phys Chem B. 2013;117(42):13346–13356. doi: 10.1021/jp404267e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McElroy C, Manfredo A, Wendt A, Gollnick P, Foster M. TROSY-NMR studies of the 91kDa TRAP protein reveal allosteric control of a gene regulatory protein by ligand-altered flexibility. J Mol Biol. 2002;323(3):463–473. doi: 10.1016/s0022-2836(02)00940-3. [DOI] [PubMed] [Google Scholar]
- 64.Heddle JG, et al. Dynamic allostery in the ring protein TRAP. J Mol Biol. 2007;371(1):154–167. doi: 10.1016/j.jmb.2007.05.013. [DOI] [PubMed] [Google Scholar]
- 65.Boyer JA, Lee AL. Monitoring aromatic picosecond to nanosecond dynamics in proteins via 13C relaxation: Expanding perturbation mapping of the rigidifying core mutation, V54A, in eglin c. Biochemistry. 2008;47(17):4876–4886. doi: 10.1021/bi702330t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Popovych N, Sun S, Ebright RH, Kalodimos CG. Dynamically driven protein allostery. Nat Struct Mol Biol. 2006;13(9):831–838. doi: 10.1038/nsmb1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shammas SL, Travis AJ, Clarke J. Allostery within a transcription coactivator is predominantly mediated through dissociation rate constants. Proc Natl Acad Sci USA. 2014;111:12055–12060. doi: 10.1073/pnas.1405815111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Muñoz V, Serrano L. Elucidating the folding problem of helical peptides using empirical parameters. II. Helix macrodipole effects and rational modification of the helical content of natural peptides. J Mol Biol. 1995;245(3):275–296. doi: 10.1006/jmbi.1994.0023. [DOI] [PubMed] [Google Scholar]
- 69.Muñoz V, Serrano L. Elucidating the folding problem of helical peptides using empirical parameters. Nat Struct Biol. 1994;1(6):399–409. doi: 10.1038/nsb0694-399. [DOI] [PubMed] [Google Scholar]
- 70.Muñoz V, Serrano L. Elucidating the folding problem of helical peptides using empirical parameters. III. Temperature and pH dependence. J Mol Biol. 1995;245(3):297–308. doi: 10.1006/jmbi.1994.0024. [DOI] [PubMed] [Google Scholar]
- 71.Muñoz V, Serrano L. Development of the multiple sequence approximation within the AGADIR model of alpha-helix formation: Comparison with Zimm-Bragg and Lifson-Roig formalisms. Biopolymers. 1997;41(5):495–509. doi: 10.1002/(SICI)1097-0282(19970415)41:5<495::AID-BIP2>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 72.Lacroix E, Viguera AR, Serrano L. Elucidating the folding problem of alpha-helices: Local motifs, long-range electrostatics, ionic-strength dependence and prediction of NMR parameters. J Mol Biol. 1998;284(1):173–191. doi: 10.1006/jmbi.1998.2145. [DOI] [PubMed] [Google Scholar]
- 73.Feig M, Karanicolas J, Brooks CL., 3rd MMTSB Tool Set: Enhanced sampling and multiscale modeling methods for applications in structural biology. J Mol Graph Model. 2004;22(5):377–395. doi: 10.1016/j.jmgm.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 74.Brooks BR, et al. CHARMM: The biomolecular simulation program. J Comput Chem. 2009;30(10):1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Brooks BR, et al. CHARMM—a program for macromolecular energy, minimization, and dynamics calculations. J Comput Chem. 1983;4(2):187–217. [Google Scholar]
- 76.Vymětal J, Vondrášek J. Gyration- and inertia-tensor-based collective coordinates for metadynamics. Application on the conformational behavior of polyalanine peptides and Trp-cage folding. J Phys Chem A. 2011;115(41):11455–11465. doi: 10.1021/jp2065612. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.