

Significance
The Red Queen hypothesis proposes that host defense genes evolve to counter the adverse effects of rapidly evolving invasive viruses. Although 3D structures of host–viral protein complexes have provided great insights into the molecular conflicts between them, a single structure represents only an evolutionary snapshot. Here we present the atomic details of the step-by-step arms race between tomato mosaic virus replication protein and the host inhibitor protein Tm-1, in which host recognition of a viral molecule, viral adaptive evasion of the recognition, host counteradaptation, and viral counter-counteradaptation are depicted by determination of the complex structures of Tm-1 variants and the viral protein and by biochemical analyses and molecular dynamics simulations of the interactions between these proteins.
Keywords: protein complex, virus resistance
Abstract
The tomato mosaic virus (ToMV) resistance gene Tm-1 encodes a protein that shows no sequence homology to functionally characterized proteins. Tm-1 binds ToMV replication proteins and thereby inhibits replication complex formation. ToMV mutants that overcome this resistance have amino acid substitutions in the helicase domain of the replication proteins (ToMV-Hel). A small region of Tm-1 in the genome of the wild tomato Solanum habrochaites has been under positive selection during its antagonistic coevolution with ToMV. Here we report crystal structures for the N-terminal inhibitory domains of Tm-1 and a natural Tm-1 variant with an I91-to-T substitution that has a greater ability to inhibit ToMV RNA replication and their complexes with ToMV-Hel. Each complex contains a Tm-1 dimer and two ToMV-Hel monomers with the interfaces between Tm-1 and ToMV-Hel bridged by ATP. Residues in ToMV-Hel and Tm-1 involved in antagonistic coevolution are found at the interface. The structural differences between ToMV-Hel in its free form and in complex with Tm-1 suggest that Tm-1 affects nucleoside triphosphatase activity of ToMV-Hel, and this effect was confirmed experimentally. Molecular dynamics simulations of complexes formed by Tm-1 with ToMV-Hel variants showed how the amino acid changes in ToMV-Hel impair the interaction with Tm-1 to overcome the resistance. With these findings, together with the biochemical properties of the interactions between ToMV-Hel and Tm-1 variants and effects of the mutations in the polymorphic residues of Tm-1, an atomic view of a step-by-step coevolutionary arms race between a plant resistance protein and a viral protein emerges.
Viruses can affect the fitness of their hosts and thus often impose a selection pressure. Cellular organisms have evolved a variety of defense systems against invading viruses. Virus-specific molecular patterns, such as dsRNA or the RNA 5′-triphosphate, are targets of host innate immune systems that have broad antiviral specificity; whereas individual viral proteins may be targeted by a specific host resistance system. In mammals, restriction factors prevent the propagation of specific groups of viruses (1). In plants, resistance proteins directly or indirectly recognize the targeted viral protein (gene-for-gene resistance systems) and then trigger a defense reaction or inhibit the viral protein’s function (2–4).
Viruses are able to evolve rapidly and acquire mutations that can escape or antagonize the host defense systems. To counter rapidly evolving viruses, the sequences of many host restriction factor genes are subject to positive selection and, consequently, mutate rapidly (5, 6). Molecular evolutionary approaches have identified residues that are important for resistance in host defense protein sequences. Such information, in conjunction with the tertiary structures of related proteins, greatly facilitates our understanding of virus–host evolutionary arms races (7).
The tomato mosaic virus (ToMV) resistance gene Tm-1 was bred from the wild tomato Solanum habrochaites S. Knapp & D.M. Spooner into the cultivated tomato Solanum lycopersicum L. to protect the latter from ToMV infection (8). Tm-1 is a 754-amino acid protein that binds ToMV replication proteins and contains at least two domains according to an RPS-BLAST search of the Conserved Domain Database (9): an uncharacterized N-terminal region (residues M1–K431) and a TIM-barrel–like C-terminal domain (residues T484–E754) (Fig. 1A) (10). An Escherichia coli-expressed N-terminal fragment of Tm-1 [residues 1–431, hereafter Tm-1(431)] inhibits ToMV RNA replication in vitro (11). In S. habrochaites, the Tm-1 gene sequence encoding residues T79–D112 has been shown to be under positive selection, suggesting that these residues have evolved to counter ToMV infection and are important for inhibition of ToMV RNA replication (12). We also found that a single naturally occurring amino acid change (I91 to T) in Tm-1 makes it a stronger inhibitor of ToMV RNA replication, enabling it to inhibit the replication of a Tm-1 resistance-breaking ToMV mutant, LT1, that contains Q979-to-E and H984-to-Y substitutions in the replication proteins (12). However, in LT1-inoculated S. habrochaites plants that have a Tm-1 allele with a T residue at position 91 [Tm-1(I91T)], mutant viruses that escaped from the inhibition by Tm-1(I91T) emerged spontaneously. These viruses have an additional mutation, E979-to-K [LT1(E979K)] or D1097-to-Y [LT1(D1097Y)], in the replication proteins. Replication of another resistance-breaking ToMV mutant T21 containing D1097-to-V and R1100-to-Q substitutions in the replication proteins is not inhibited by Tm-1(I91T). These findings indicate that the ToMV–Tm-1 system should serve as an appropriate model for a virus–host coevolutionary arms race, because, as described above, several ToMV mutants with different sensitivities to Tm-1 and Tm-1(I91T) exist.
Fig. 1.

Structure of the N-terminal inhibitory domain of Tm-1. (A) The domain organization of Tm-1 with the NN and NC domains colored blue. The C-terminal domain is colored green. The residue positions that define the domain boundaries are identified. The red line highlights the sequence under positive selection. (B) The 3D structure of Tm-1(431) is shown as a ribbon diagram. To differentiate between the two polypeptide chains in the Tm-1 homodimer, one is colored blue and the other cyan. Residues T79–D112, which are under positive selection, are colored red. Sequences with no interpretable electron density (S39–K45, L80–A89, and S202–G210) are shown as dashed lines. All structures in this and other figures were generated by PyMol (32). (C) Topology of Tm-1(431). α-Helices and β-sheets are shown as magenta cylinders and cyan arrows, respectively. The diagram was created using TopDraw (33). (D) Inhibitory activities of Tm-1(431), Tm-1(201), and their I91T mutants on ToMV RNA replication in vitro. Genomic RNAs of the WT ToMV, LT1, and LT1(E979K) strains were translated and replicated in vitro in the presence of the indicated Tm-1 derivatives. Incorporation of [α-32P]CTP into the synthesized genomic RNA (G) and replicative form RNA (RF) is shown.
ToMV is a positive-strand RNA virus and its replication proteins—a 126-kDa protein and its translational read-through product, a 183-kDa protein—are encoded in its RNA genome (13). The 126-kDa protein contains the methyltransferase domain, involved in RNA 5′ capping, and the helicase domain. In addition to the aforementioned domains, the 183-kDa protein also contains the RNA-dependent RNA polymerase domain in the read-through region. Tm-1 inhibits the formation of ToMV replication complexes on host intracellular membranes and consequently inhibits negative-strand viral RNA synthesis (14). The amino acid changes found in Tm-1 resistance-breaking mutants locate at the helicase domain of ToMV replication proteins (ToMV-Hel) and decrease the affinity of Tm-1 for the replication proteins (12). We recently determined a crystal structure of ToMV-Hel (residues S666–Q1116 of the replication proteins) (15). ToMV-Hel contains two canonical RecA-like α/β domains (1A and 2A), which form the helicase core, and a structurally unique N-terminal domain. The positions of mutations in Tm-1 resistance-breaking mutants have been mapped to the surface of the molecule (15).
In this study we present an atomic view that describes the coevolution of ToMV and the resistance gene Tm-1 as deduced from the crystal structures of complexes between fragments of ToMV-Hel and Tm-1.
Results
Structure of the Inhibitory Domain of Tm-1.
We determined a 2.7-Å resolution crystal structure of selenomethionine (SeMet)-labeled Tm-1(431) by multiwavelength anomalous diffraction phasing (Table S1). The asymmetric unit of the crystal contains three Tm-1(431) homodimers with missing electron density for residues S39–K45, L80–A89, and S202–G210. The three Tm-1(431) dimers have an rmsd value of 1.04 Å, suggesting that the dimer structures are basically identical.
Tm-1(431) contains two discrete structural domains, residues M1–S201 (referred to herein as the “NN domain”) and residues K211–K431 (the “NC domain”) (Fig. 1 A–C). The two domains are connected by residues S202–G210, for which electron density was not found and thus suggesting the presence of a disordered interdomain loop. The NN domain contains a parallel five-stranded β-sheet surrounded by four helices on one side and two helices on the other; the tertiary structure is most similar to that of the nonhydrolyzing bacterial UDP-GlcNAc 2-epimerase [Protein Data Bank (PDB) ID code 3BEO] (Fig. S1A), according to the distance alignment matrix method (DALI) server (16). The NC domain contains a parallel six-stranded β-sheet surrounded by five helices on one side and two helices on the other, and an additional antiparallel three-stranded β-sheet with a short α-helix was found. The NC domain structure is most similar to the N-terminal domain of the Thermotoga maritima ribose-binding protein (PDB ID code 2FN8) (Fig. S1B), according to the DALI server (16). The region under positive selection (T79–D112; shown in red in Fig. 1B) forms part of the Tm-1 surface and contains α3 and residues L80–A89, which, because this sequence has no interpretable electron density, may be a disordered loop.
In the crystal structure, Tm-1(431) forms the homodimer via interactions between the NN domains (Fig. 1B and Tables S2 and S3). Because the NN domain contains the positively selected region, we next expressed and purified the NN domain as the construct Tm-1(201) containing residues M1–S201 and its variant, Tm-1(201/I91T). These fragments inhibited ToMV RNA replication in vitro (Fig. 1D). Although determination of the crystal structure of Tm-1(201) was unsuccessful, we were able to determine that of Tm-1(201/I91T) (Fig. S1C) and found that deletion of the NC domain did not affect the structure of the NN domain [the mean Cα rmsd value for superpositioned Tm-1(201/I91T) and the NN domain of Tm-1(431) is 0.46 Å].
Biochemical Characterization of ToMV-Hel–Tm-1 Interactions.
Although Tm-1 resistance is overcome by mutations in ToMV-Hel, it has not been determined if Tm-1 could bind purified ToMV-Hel. Therefore we characterized the interaction between ToMV-Hel and Tm-1(431) by size-exclusion chromatography (SEC) and found that neither Tm-1(431) nor Tm-1(431/I91T) bound ToMV-Hel in the absence of ATP (Fig. 2A). In the presence of ATP the individual Tm-1(431) or Tm-1(431/I91T) and ToMV-Hel peaks almost disappeared, and new peaks of greater molecular mass appeared in which both Tm-1(431) or Tm-1(431/I91T) and ToMV-Hel were present as shown by SDS/PAGE (Fig. 2A). Thus, complexes between Tm-1(431) or Tm-1(431/I91T) and ToMV-Hel were formed with the aid of ATP.
Fig. 2.

Tm-1(431) binds ToMV-Hel only when ATP is present. (A) Size-exclusion gel chromatographs of mixtures of ToMV-Hel and Tm-1(431) or Tm-1(431/I91T) in the presence or absence of ATP. The elution positions for the molecular-mass markers myoglobin (17 kDa), ovalbumin (44 kDa), γ-globulin (158 kDa), and thyroglobulin (670 kDa) are identified by arrows above the top chromatogram. Insets show Coomassie-stained SDS/PAGE gels for chromatographic fractions. (B) (Upper) ITC assessments of the interaction between ToMV-Hel and Tm-1(431) or Tm-1(431/I91T). (Lower) The integrated heat is plotted against the molar ratio of Tm-1(431) or Tm-1(431/I91T) to ToMV-Hel in the cell after subtracting the heat dilution. The binding parameters are shown at the bottom of the figure.
To obtain further insight into the binding mechanism of Tm-1(431) and Tm-1(431/I91T) to ToMV-Hel, we used isothermal titration calorimetry (ITC) to determine the thermodynamic binding parameters and included adenosine 5′-O-(3-thio) triphosphate (ATPγS), a slowly hydrolysable ATP analog, instead of ATP to circumvent the need to correct for the heat of ATP hydrolysis by ToMV-Hel. The results suggest that Tm-1(431) and Tm-1(431/I91T) bind ToMV-Hel with a 1:1 stoichiometry (Fig. 2B). Consistent with previous results suggesting that the I91-to-T substitution strengthens Tm-1 inhibitory activity (12), the ITC results indicate that Tm-1(431/I91T) binds ToMV-Hel more strongly than does Tm-1(431) [an approximately sevenfold difference in the Kd values: 3.1 × 10−7 M and 2.1 × 10−6 M for Tm-1(431/I91T) and Tm-1(431), respectively]. Furthermore, the association of ToMV-Hel and Tm-1(431/I91T) is more enthalpic than that of ToMV-Hel and Tm-1(431) (ΔH = −16.4 kcal/mol and −12.8 kcal/mol, respectively).
Overall Structure of the ToMV-Hel–Tm-1(431) Complex.
Because ATP is required for the ToMV-Hel–Tm-1(431) complex formation but also is hydrolyzed by ToMV-Hel (17), we cocrystallized the ToMV-Hel–Tm-1(431) complex in the presence of ATPγS and solved its structure at 2.5 Å (Fig. 3A and Table S1). The asymmetric unit of the crystal contains a tetrameric complex with a 2:2 stoichiometry consisting of a Tm-1(431) homodimer and two monomeric ToMV-Hel molecules. The complex has dimensions of ∼135 × 110 × 90 Å and resembles a “lobster,” with the NN and NC domains of Tm-1(431) forming the body and the head, respectively, and ToMV-Hel forming a pair of claws (Fig. 3A). Each half of the complex is roughly related by a rotation along the long axis of the tetramer; however, the two heterodimeric structures are not identical. The average Cα rmsd values for the two Tm-1(431) monomers and the two ToMV-Hel monomers in the complex are 2.37 Å (for 411 Cα atoms) and 1.82 Å (for 426 Cα atoms), respectively, whereas the average Cα rmsd value for the ToMV-Hel–Tm-1(431) heterodimers is 4.49 Å (for 829 Cα atoms), suggesting that the relative positions and orientations of the two ToMV-Hel–Tm-1(431) heterodimers differ and that the differences may be caused by crystal-packing interactions.
Fig. 3.

Crystal structure of the ToMV-Hel–Tm-1(431) complex. (A) Ribbon diagram of the ToMV-Hel–Tm-1(431) complex before (Left) and after (Right) a 90° rotation around the vertical axis. The Tm-1 molecules are colored blue and cyan, and the ToMV-Hel molecules are colored violet and light pink. Residues T79–D112 of Tm-1(431) are colored red. ATPγS molecules are represented as orange stick models. (B) Interaction interface of Tm-1(431) and ToMV-Hel. The residues of Tm-1(431) (blue) and ToMV-Hel (magenta) that make direct contact with each other at the interface are identified by name. The ATPγS molecule is represented as an orange stick model. Oxygen atoms are colored red, nitrogen atoms are blue, and sulfur atoms are yellow. The dashed lines enclose the four sets of interacting residues (see text). (C) A close-up view of the ATPγS-binding site. An ATPγS Fo-Fc difference density in the ToMV-Hel–Tm-1(431) complex is contoured at 3σ and is shown as an orange grid. ATPγS is represented as a stick model, with oxygen atoms in red, nitrogen atoms in blue, sulfur atoms in yellow, and carbon and phosphorus atoms in orange. Hydrogen bonds are shown as dashed blue lines.
The Binding Interface of ToMV-Hel–Tm-1(431).
The two interfaces of ToMV-Hel and Tm-1(431) in the tetramer are nearly equivalent. Each interface consists of the N-terminal region of the Tm-1 NN domain that contains T79–D112, which is under positive selection (Fig. 3B, Left), and two regions of ToMV-Hel, I1094–Y1109 in the C-terminal region and H975–M986 in the region connecting the 1A and 2A domains (Fig. 3B, Right). At least 21 residues in Tm-1(431) and 23 residues in ToMV-Hel contact each other directly (Fig. 3B, Fig. S2 A and B, and Tables S4 and S5). Notably, an ATPγS molecule is found in each ToMV-Hel–Tm-1(431) interface, in addition to those found in the two ToMV-Hel nucleoside triphosphatase (NTPase) active sites.
The interaction between ToMV-Hel and Tm-1(431) is mediated by a combination of hydrophobic interactions and hydrogen bonds and can be parsed into four sets of residues (Fig. 3B and Fig. S2B): (i) T55–W57 and E59 in the loop connecting β2 and α2 of Tm-1(431) and two α-helices and an intervening loop in the C-terminal region of ToMV-Hel (E1099–Y1104), in which two hydrogen bonds [T55(O)–S1102(N) and W57(N)–R1100(O)] are formed; (ii) K72 and L75 in α2 and H78 and L80 in the loop between α2 and α3 of Tm-1(431) and S1102, Y1104, L1105, M1108, and Y1109 in the ToMV-Hel C-terminal α-helix, which interact via hydrophobic interactions; (iii) the flexible loop between α2 and α3 (G81 and T84–D90) of Tm-1(431) and the α-helix formed by N978–R980 and the loop formed by Y981–H984 between the ToMV-Hel 1A and 2A domains, for which five pairs of hydrogen bonds [T84(OG1)–H984(NE2), M85(O)–G983(N), M85(O)–H984(N), F88(O)–Q979(NE), and F88(N)–Q979(O)] are formed; additionally, the carbonyl oxygen atoms of Tm-1(431) A89 and D90 hydrogen bond with R1065(NH1) of ToMV-Hel; (iv) the N-terminal portion of Tm-1(431) α3 (I91, L94, A95, and I98) interacts with certain residues in the sequence that connects domain 1A and 2A (H975, F976, N978, Q979, and H984) and the two C-terminal α-helices of ToMV-Hel (I1094, D1097, L1098, V1101, L1105, M1108, and Y1109) mainly through hydrophobic interactions, with a hydrogen bond between I91(O) and H975(NE2). All known ToMV-Hel residues that are changed in the resistance-breaking mutants, namely Q979, H984, D1097, and R1100, make direct contact with Tm-1(431) (Fig. S2 C–F).
A Role for ATP in the Complex Formation.
Because ATP is required for ToMV-Hel–Tm-1(431) association and because ATPγS is found in the ToMV-Hel NTPase catalytic sites and interfaces of the complex structure, we investigated whether the presence of ATP in either or both sites is important for complex formation. ToMV-Hel hydrolyzes GTP, UTP, and CTP, as well as ATP (17), indicating that these nucleoside triphosphates bind in the ToMV-Hel active site. However, when examined by the SEC and ITC, ToMV-Hel and Tm-1(431/I91T) could form a complex only in the presence of ATP, ADP, or ATPγS but not in the presence of GTP or guanosine 5′-O-(3-thio) triphosphate (GTPγS) (Fig. S3 A and B). These results suggest the dispensability of an NTP at the ToMV-Hel active site for complex formation and emphasize the importance of bound ATP at the ToMV-Hel–Tm-1(431) interface. In the crystal structure, ATPγS is positioned in the groove of Tm-1 that is surrounded by T16, D18, and K20 in α1, R92 in α3, G124–G127 in α4, and T55 and S56 in the loop between β2 and β3 (Fig. 3C and Table S6). The adenine ring of ATPγS directly contacts Tm-1(431) S56 and R92 and ToMV-Hel D1097 and R1100 (Table S6). Interestingly, the side chains of Tm-1(431) R92 and ToMV-Hel R1100 are in van der Waals distances with the purine base of ATPγS (Fig. S2F). Thus, ATP may act as a bridge connecting ToMV-Hel and Tm-1(431).
The presence of an ATPγS-binding cavity in Tm-1(431) suggests that Tm-1 has an intrinsic ability to bind ATP. Indeed, according to the results of an ITC experiment, Tm-1(431) bound ATP with a Kd of 1.4 × 10−4 M without hydrolyzing it (Fig. S3C). Conversely, the binding affinity of Tm-1(431) for GTP was less than 70-fold lower (Kd > 1.0 × 10−2 M).
Conformational Differences Between Free and Tm-1(431)–Bound ToMV-Hel.
To investigate whether the binding of Tm-1(431) influences the conformation of ToMV-Hel, and vice versa, we compared the structures of their free forms and their complexed forms. No dramatic structural differences were found for free and ToMV-Hel–bound Tm-1(431) (Cα rmsd, 1.004 Å), although a disorder-to-order transition involving residues L80–A89 and changes in the orientations of the side chains involved in ATPγS binding were observed (Fig. 4A). Conversely, the structures of free (PDB ID code 3VKW) (15) and complexed ToMV-Hel are strikingly different. When the 2A domains of the two ToMV-Hel molecules are superpositioned, the relative orientations of the 1A and N-terminal domains are each rotated by ∼25° [calculated by DymDom (18)] (Fig. 4B). Furthermore, in the complex, the N terminus of ToMV-Hel is oriented in the opposite direction of that found for free ToMV-Hel (Fig. 4B). The following changes also are observed in ToMV-Hel after binding to Tm-1(431). (i) F88(N) of Tm-1 forms a hydrogen bond with Q979(O) of ToMV-Hel. (ii) The side chain conformation of R980 is altered so that its NH2 can hydrogen bond with Q935(OE1). (iii) Q935(N) forms a hydrogen bond with D933(OD1). (iv) D933(OD2) hydrogen bonds with K839(NZ), and a conformational change in the K839 side chain at the NTPase active site also is observed (Fig. 4C). Therefore, Tm-1 may induce a conformational change in ToMV-Hel by promoting a new hydrogen bond network that could influence the position of the side chain of K839, which is important for NTPase activity (19, 20).
Fig. 4.

Conformational changes in Tm-1(431) and ToMV-Hel upon complex formation. (A, Left) Superposition of the Tm-1(431) NN-domain in its free (yellow) and ToMV-Hel–bound (blue) forms. ATPγS is represented by orange stick models. (Right) An enlargement of the ATPγS binding site. The side chains of residues that interact with ATPγS are shown as stick models; the color coding is the same as in the left panel. (B) Superposition of ToMV-Hel in its free (PDB ID code 3VKW; yellow) and Tm-1(431)–bound (magenta) forms. A close-up view of the N-terminal region is shown on the right. (C) A conformational ripple effect induced by Tm-1 binding to ToMV-Hel. Upon Tm-1 binding, the side chains of the identified residues change their positions. (D) Inhibition of ToMV-Hel ATPase activity by Tm-1(431/I91T). Hydrolysis of ATP by ToMV-Hel in the absence (black trace) or presence (red trace) of equal molar amounts of Tm-1(431/I91T) at 15 °C was measured using ITC.
Next, using ITC, we examined the ATPase activity of ToMV-Hel in the presence or absence of Tm-1(431/I91T). As deduced from the structure, ToMV-Hel complexed with Tm-1(431/I91T) showed less ATPase activity (a 32% reduction in kcat) than did free ToMV-Hel (Fig. 4D). The reduction in the ToMV-Hel ATPase activity by Tm-1 may account, in part, for Tm-1’s inhibition of ToMV RNA replication.
Comparisons of the ToMV-Resistant and -Susceptible Tm-1 Alleles.
In S. habrochaites, both ToMV-resistant and -susceptible Tm-1 alleles have been maintained by balancing selection (12). In full-length Tm-1 (754 residues), 30 residues differ in the products of the Tm-1 alleles of the ToMV-resistant (US Department of Agriculture National Plant Germplasm System accession no. PI126445, identical to Tm-1GCR237 used for structural determination) and ToMV-susceptible (accession no. PI390515) S. habrochaites plants. Twenty-two of those residues are in the NN domain, and 16 of those 22 residues are between T79–D112, the positive-selection sequence. As described above, at least 21 residues in Tm-1(431) make direct contact with ToMV-Hel in the crystal structure, and, notably, 11 of them differ in Tm-1PI126445 and Tm-1PI390515 (Fig. 5A). To determine the importance of these residue changes for resistance, we constructed single-site Tm-1PI126445 mutants in which one of the residues was replaced with the residue in the corresponding position in Tm-1PI390515. These mutant proteins were synthesized by in vitro translation and assessed for their ability to inhibit ToMV RNA replication. Surprisingly, many of the mutations decreased the inhibitory activity of Tm-1PI126445 (Fig. 5B). Therefore, the Tm-1 variable region cannot change readily while maintaining Tm-1 inhibitory activity; this limitation is in keeping with the limited sequence diversity within S. habrochaites ToMV-resistant Tm-1 alleles (12). In addition, although the cDNA nucleotide sequences of Tm-1PI126445 and Tm-1PI390515 are 98% identical, two or three nucleotides in the codons encoding the residues at position 57, 87, 91, and 100 are altered (Fig. 5B). The Tm-1PI126445 residues at these positions are important for the inhibitory activity (Fig. 5B), and W57, T87, and I91 directly contact ToMV-Hel (Tables S4 and S5).
Fig. 5.
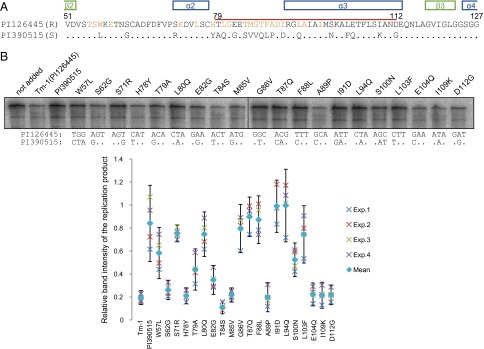
Comparisons of Tm-1 alleles from ToMV-resistant and ToMV-susceptible S. habrochaites. (A) Alignment of the amino acid sequences of ToMV-resistant Tm-1PI126445 and ToMV-susceptible Tm-1PI390515 between positions 51 and 127. Residues that are identical in the two sequences are shown as dots in the sequence of Tm-1PI390515. Residues that interact directly with ToMV-Hel in the ToMV-Hel–Tm-1(431) crystal structure are colored orange. The positively selected region (residues T79–D112) is marked by a red line. Secondary structures are shown above the alignment. (B) Effects of single-site mutations in Tm-1PI126445 polymorphic residues on the inhibitory activity. The indicated amino acid changes were introduced individually into the gene encoding Tm-1PI126445. Each mutant protein was synthesized by in vitro translation and then was tested for its ability to inhibit ToMV RNA replication in vitro. Experiments were performed four times using individually prepared evacuolated BY-2 protoplast extracts. (Lower) Band intensities of the replication products were plotted relative to their intensities in the absence of Tm-1 in each experiment (crosses). The mean values of the four experiments were also plotted (diamonds). Error bars indicate 95% confidence intervals. (Upper) The gel image is a result of Exp. 3. The relevant nucleotide sequences in Tm-1PI126445 and Tm-1PI390515 are shown below the gel.
Effect of the Tm-1 I91-to-T Substitution on ToMV-Hel–Tm-1 Binding.
To understand how the I91-to-T substitution in Tm-1 increases its inhibitory activity, we solved the structure of the ToMV-Hel–Tm-1(431/I91T) complex crystallized in the presence of ATPγS (Table S1). The overall structure of this complex is very similar to that of the ToMV-Hel–Tm-1(431) complex (Cα rmsd, 0.228 Å) (Fig. 6A). Tm-1(431) I91 makes hydrophobic interactions with seven ToMV-Hel residues, and its backbone carbonyl oxygen makes a hydrogen bond with H975(NE2) (Fig. 6B and Tables S4 and S5). In the ToMV-Hel–Tm-1(431/I91T) structure, T91(OG1) and T91(O) hydrogen bond directly with Q979(OE1) and H975(NE2), respectively, and T91(OG1) and T91(CG2) are involved in a hydrogen bond network containing water molecules and I1094(O) and D1097(OD2) (Fig. 6C and Tables S4 and S5). These observations are consistent with the ITC result (Fig. 2B) showing that Tm-1(431/I91T) binds to ToMV-Hel more tightly than does Tm-1(431).
Fig. 6.

Structure of the ToMV-Hel–Tm-1(431/I91T) complex. (A) Structural comparison of the ToMV-Hel–Tm-1(431) (blue) and ToMV-Hel–Tm-1(431/I91T) (cyan) complexes. (B) The interaction of Tm-1(431) I91 with ToMV-Hel. The atoms of I91 are shown as blue stick models. The ToMV-Hel residues that interact with I91 are partially colored light pink, and their nitrogen and oxygen atoms are colored blue and red, respectively. The hydrogen bond is shown as a dashed cyan line. (C) The interaction of Tm-1(431/I91T) T91 with ToMV-Hel. The atoms of T91 are shown as blue stick models except for the oxygen atoms, which are shown as red stick models. The ToMV-Hel residues that interact with T91 are partially colored light pink; their nitrogen and oxygen atoms are colored blue and red, respectively. Hydrogen bonds are shown as dashed cyan lines. Water molecules are shown as red spheres.
Molecular Dynamics Simulations to Assess How Amino Acid Substitutions in ToMV-Hel Allow It to Circumvent Inhibition by Tm-1.
To understand how the amino acid changes in ToMV-Hel affect its interaction with Tm-1, we simulated stable structures of the complexes formed by Tm-1 and each of the ToMV-Hel mutants using molecular dynamics (MD), having built each complex from the crystal structure of the ToMV-Hel–Tm-1(431) complex (Fig. 7A). The hydrogen bond between the Q979(NE2) of WT ToMV-Hel and Tm-1 F88(O) is lost when Q979 is replaced by E as found in LT1-Hel (Fig. 7B). In addition, although H984 in ToMV-Hel forms hydrogen bonds with Tm-1 T84(OG1) and M85(O), such hydrogen bonds are not possible for Y984 in LT1-Hel (Fig. 7B). Disruption of these hydrogen bonds also causes separation of ToMV-Hel N978–Y984 and Tm-1 G81–F88, indicating that the hydrophobic interactions are compromised (Fig. 7B). In T21-Hel, V1097 and Q1100 (replacing D and R, respectively, in WT ToMV-Hel) do not interact with ATPγS, although these mutations do not appear to affect the direct interaction with Tm-1 residues (Fig. 7B). The inability of V1097 and Q1100 to interact with ATPγS allows the separation of T21-Hel and Tm-1 and the disruption of certain hydrophobic interactions, for example, that between ToMV-Hel L1098 and Tm-1 I91. These results again highlight the importance of the sandwiched ATP at the complex interface.
Fig. 7.

Structural view of the arms race between ToMV-Hel and Tm-1. (A) Close-up view of the crystal structure of the interface of the ToMV-Hel–Tm-1(431) complex. The ToMV-Hel residues that interact with Tm-1(431) are partially colored light pink; their nitrogen and oxygen atoms are colored blue and red, respectively. The Tm-1 residues that interact with ToMV-Hel are partially colored cyan; their nitrogen and oxygen atoms are colored blue and red, respectively. ATPγS is shown as an orange stick model. Hydrogen bonds are shown as dashed cyan lines. Water molecules are shown as red spheres. (B) MD-simulated complexes of Tm-1(431) and LT1-Hel (containing the Q979-to-E and H984-to-Y substitutions) (Left) or T21-Hel (containing the D1097-to-V and R1100-to-Q substitutions) (Right) using the crystal structure of ToMV-Hel–Tm-1(431) as the template. (C) MD structures of LT1-Hel–Tm-1(I91T) using the crystal structure of the ToMV-Hel–Tm-1(431/I91T) as the template. (D) MD structures of LT1(E979K)-Hel and LT1(D1097Y)-Hel–Tm-1(I91T) using the crystal structure of the ToMV-Hel–Tm-1(431/I91T) as the template.
Tm-1(I91T) and its truncated variants inhibit LT1 RNA replication (Fig. 1D); however, LT1 escapes inhibition when E979 or D1097 is replaced by a K or Y, respectively (12). In the crystal structure of ToMV-Hel–Tm-1(431/I91T), both key residues of ToMV-Hel [Q979(NE2) and D1097(OD2)] hydrogen bond with T91 side-chain atoms of Tm-1(431/I91T) directly and through water molecules, respectively (Fig. 6C). The LT1-Hel–Tm-1(431/I91T) complex was simulated using the crystal structure of the ToMV-Hel–Tm-1(431/I91T) complex as the template. In this modeled complex, Tm-1(I91T) T91(OG1) hydrogen bonds directly with LT1-Hel E979(OE2) (Fig. 7C). In addition, Tm-1(I91T) T91(CG2) participates in a hydrogen bond network involving a water molecule, LT1-Hel E979(OE1), and D1097(OD2) (Fig. 7C). Consequently, the interacting loops of LT1-Hel and Tm-1(I91T) remain adjacent to each other even though Y984 is not involved in hydrogen bonds that were formed by WT ToMV-Hel H984. The E979-to-K substitution in LT1-Hel prevents the formation of hydrogen bonds between LT1(E979K)-Hel K979 and Tm-1(I91T) T91 and a water molecule (Fig. 7D). The D1097Y substitution in LT1-Hel also disrupts the hydrogen bonds formed with Tm-1 (I91T) T91 via a water molecule and the hydrogen bond formed with ATPγS (Fig. 7D).
Discussion
Structural Basis for the Recognition of ToMV-Hel by Tm-1(431).
We present here the crystal structures of ToMV-Hel complexed with Tm-1(431) and Tm-1(431/I91T); we used these structures to identify their protein–protein interaction (PPI) interface. Two characteristic stabilizing mechanisms appear to be present for the interaction of ToMV-Hel and Tm-1(431): an ATP bridge (represented by ATPγS in the crystal structures) and a disorder-to-order transition involving Tm-1 residues L80–A89.
ATPγS is positioned in the cavity of Tm-1 so that many residues interact with its adenine ring, ribose moiety, and phosphates (Fig. 3C and Table S6). Because Tm-1 homologs also are found in fungi, bacteria, and archaea, we proposed that Tm-1 has a primary function other than virus resistance in plants and that its ability to bind ToMV replication proteins and inhibit viral replication is an accidental evolutionary event (10). The ATP-binding site of Tm-1 resembles the substrate-binding site of bacterial UDP-GlcNAc 2-epimerase (Fig. S1A). As documented in our ITC experiment, Tm-1(431) binds ATP (Fig. S3C); therefore, the ability of Tm-1 to bind ATP (and other related molecules) may be part of its primary function. Tm-1 probably binds ATP before forming a complex with ToMV-Hel because in the complex crystal structure ToMV-Hel D1097 and R1100 contact the adenine base of ATPγS and thereby serve as a lid (Fig. 3C). The inability of free Tm-1(431/I91T) to bind GTP (Fig. S3C) and to bind ToMV-Hel in the presence of GTP or GTPγS (Fig. S3 A and B) is consistent with this proposal. Allosteric and direct interactions involving small molecules have been proposed as means for stabilizing protein complexes (21). ATP and related molecules possibly act to glue ToMV-Hel and Tm-1 together and are direct stabilizers. Small molecule-stabilized PPIs can act as sensors for the concentrations of the stabilizing small molecules and thereby regulate downstream functions (21). However, because ATP is abundant in the cytoplasm, it is unlikely that it regulates the formation of the ToMV replication protein–Tm-1 complex; rather, Tm-1 would be present mainly in an ATP-bound form that can bind ToMV-Hel.
Regarding the disorder-to-order transition, intrinsically disordered regions of proteins often have been found at PPI sites (22, 23). Residues L80–A89 appear to be disordered in free Tm-1(431) (Fig. 1) but ordered when complexed with ToMV-Hel (Fig. 3). ToMV-Hel Q979 and H984, which are mutated in LT1, interact with these residues of Tm-1 via hydrogen bonds and hydrophobic interactions. Notably, this region and α3 of Tm-1 comprise the positively selected region (T79–D112) involved in antagonistic coevolution with ToMV. Probably, mutations in the disordered region do not affect the overall structure of Tm-1 and thus could have undergone rapid mutation over time without constraints regarding the primary function of Tm-1.
The I91-to-T substitution increased Tm-1 inhibitory activity (Fig. 2B), whereas the I91-to-D substitution abolished it (Fig. 5B), indicating that the residue at position 91 in Tm-1 is key for the interaction between Tm-1 and ToMV-Hel. In the complexes, the I91 or T91 side chain is located geometrically at the center of the interaction interface and is surrounded by seven or six interacting ToMV-Hel residues, respectively (Figs. 3B and 6 B and C, Fig. S2B, and Tables S4 and S5). Therefore, the residue at position 91 in Tm-1 possibly serves as a hot spot for ToMV-Hel–Tm-1 association. Such hot spots have been found in many PPI sites (24, 25).
Possible Mechanisms for Inhibiting ToMV RNA Replication by Tm-1.
The structures of ToMV-Hel in the free form and in the ToMV-Hel–Tm-1(431) complex are strikingly different (Fig. 4B), and the structural changes may be induced by Tm-1 binding. Although Tm-1 binds residues distal to the ToMV-Hel NTPase active site, the binding results in conformational changes at the active site (Fig. 4C) so that Tm-1 partially inhibits the ATPase activity of ToMV-Hel (Fig. 4D). Tm-1 F88, which interacts with ToMV-Hel Q979 to trigger the conformational change, is important for the inhibitory activity for ToMV RNA replication (Fig. 5B). Because helicases undergo multiple conformational changes upon binding to ATP or nucleic acids during catalysis (26), the inhibition may be viewed as an allosteric effect because the conformational change in ToMV-Hel is constrained by Tm-1. In tobacco mosaic virus, closely related to ToMV, mutations in the NTPase active site have a deleterious effect on viral infectivity (20). If ToMV-Hel NTPase activity is required multiple times per replication, a partial reduction of the NTPase activity may have a big effect on the inhibition of RNA replication. We proposed that ToMV-Hel must undergo a conformational change to interact with host factors during the assembly of the replication complex (27). In a cell-free replication system, Tm-1 inhibits the binding of replication proteins to the host membrane proteins TOM1 and ARL8 and the sequestration of replication templates, which are tightly linked to and precede the formation of the replication complex, in the membranous compartment (14). How partial inhibition of helicase NTPase activity affects overall replication, or if other mechanisms (e.g., inhibition of a conformational change required for replication complex assembly) also contribute to the inhibition by Tm-1 will be addressed in future studies.
The Recognition–Evasion Arms Race Between ToMV and Tm-1.
In combination with molecular evolution studies, structural determination of antiviral proteins provides insights into how hosts recognize viruses, how they have evolved against viruses, and vice versa. For example, in vertebrates, protein kinase R, an important component of the antiviral innate immunity system, has evolved so that mutations arise at positions distal from its interface with the antagonizing poxvirus protein K3L as well as directly at the interface (28, 29). As noted above, we previously found that the sequence T79–D112 in Tm-1 is under positive selection (12); now we show that these residues are directly involved in binding Tm-1 to ToMV-Hel. Residues in flexible loops usually are not subject to structural/functional constraints and may be mutated without resulting in deleterious effects; this property might have accelerated the evolution of Tm-1 alleles until resistant alleles against ToMV emerged. Our mutagenesis study suggests that once Tm-1 gained the ability to bind ToMV-Hel, the identities of residues between positions 79 and 112 have been maintained under a functional constraint (Fig. 5). ToMV has escaped Tm-1 inhibition via changes in the residues targeted by Tm-1. Two possible sets of residue changes have been reported in spontaneously emerged ToMV mutants (LT1- and T21-type) (30, 31). Our MD simulations show that these mutations can (partially) disrupt the ToMV-Hel–Tm-1 interaction (Fig. 7). Note that T21 carries a higher fitness cost to overcome the resistance than LT1 (12). Possibly, to counteract the effect of the LT1-type resistance-breaking ToMV mutant, host plants have evolved the alleles encoding Tm-1(I91T) that can form a hydrogen-bond network involving water molecules and that has a greater affinity for ToMV-Hel (Figs. 2 and 6). Our MD simulation confirmed that the LT1-Hel–Tm-1(431/I91T) complex is stable (Fig. 7). To evade inhibition by Tm-1(I91T), LT1 incorporates an E979-to-K or D1097-to-Y substitution. The MD simulations suggest that each of these substitutions can disrupt Tm-1(I91T) binding (Fig. 7). Taken together, the crystal structures of the ToMV-Hel–Tm-1 complexes and the MD simulations reveal the atomic details of host recognition of a viral molecule, viral evasion of that recognition for adaptation, host counteradaptation, and viral counter-counteradaptation during a virus–host arms race.
Materials and Methods
Tm-1(431), Tm-1(201), and their derivatives containing the I91-to-T substitution were expressed as maltose-binding protein fusion proteins in E. coli and were purified as described previously (11). The ToMV-L strain and proteins encoded by its genome were used as the WT system. WT ToMV-Hel and its mutant forms were expressed as thioredoxin-hexahistidine fusion proteins in E. coli and were purified as described previously (16). ToMV-Hel–Tm-1(431)-type complexes were purified by a Superdex-200 SEC after loading a 1:1 molar mixture of the appropriate two proteins in the presence of an excess amount of ATP. Protein crystals were grown in hanging drops containing equal volumes of protein and reservoir solutions. Cryoprotected crystals were flash-frozen in liquid nitrogen before diffraction data were collected at the SPring-8 beamline BL38B1 or the Photon Factory beamlines BL-5A and NWE-12A. Diffraction data and refinement statistics are given in Table S1. The Tm-1(431) structure was determined by the multiwavelength anomalous diffraction phasing method using a SeMet-labeled protein crystal. Other structures were determined by molecular replacement with the appropriate structures as the template. Full details are provided in SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank Drs. Toshio Furuya and Takane Yokotagawa for assistance with MD simulations; the staff at the SPring-8 beamline BL38B1 for help, as approved by the Japan Synchrotron Radiation Research Institute Priority Program for Disaster Affected Quantum Beam Facilities through Grants 2011A2031 (to E.K.) and 2013A6848 (to H.M.); and the staffs at the BL-5A and NWE-12A beamlines at Photon Factory of Japan through Grants 2011P005 (to E.K.) and 2013G148 (to H.M.). This work was supported in part by a grant from the Program for Promotion of Basic Research Activities for Innovative Biosciences, Japan (to E.K. and M.I.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: Crystallography, atomic coordinates, and structure factors have been deposited in the Protein Data Bank, www.pdb.org (ID codes 3WRV [free Tm-1(431)], 3WRW [Tm-1(201/I91T)], 3WRX [ToMV-Hel-Tm-1(431) complex], and 3WRY [ToMV-Hel-Tm-1(431) complex]).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1407888111/-/DCSupplemental.
References
- 1.Yan N, Chen ZJ. Intrinsic antiviral immunity. Nat Immunol. 2012;13(3):214–222. doi: 10.1038/ni.2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gómez P, Rodríguez-Hernández AM, Moury B, Aranda MA. Genetic resistance for the sustainable control of plant virus diseases: Breeding, mechanisms and durability. Eur J Plant Pathol. 2009;125(1):1–22. [Google Scholar]
- 3.Gururani MA, et al. Plant disease resistance genes: Current status and future directions. Physiol Mol Plant Pathol. 2012;78:51–65. [Google Scholar]
- 4.Mandadi KK, Scholthof K-BG. Plant immune responses against viruses: How does a virus cause disease? Plant Cell. 2013;25(5):1489–1505. doi: 10.1105/tpc.113.111658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johnson W. Rapid adversarial co-evolution of viruses and cellular restriction factors. In: Cullen BR, editor. Intrinsic Immunity, Current Topics in Microbiology and Immunology. Vol 371. Berlin: Springer; 2013. pp. 123–151. [DOI] [PubMed] [Google Scholar]
- 6.Duggal NK, Emerman M. Evolutionary conflicts between viruses and restriction factors shape immunity. Nat Rev Immunol. 2012;12(10):687–695. doi: 10.1038/nri3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Daugherty MD, Malik HS. Rules of engagement: Molecular insights from host-virus arms races. Annu Rev Genet. 2012;46(1):677–700. doi: 10.1146/annurev-genet-110711-155522. [DOI] [PubMed] [Google Scholar]
- 8.Pelham J. Resistance in tomato to tobacco mosaic virus. Euphytica. 1966;15(2):258–267. [Google Scholar]
- 9.Marchler-Bauer A, et al. CDD: A conserved domain database for the functional annotation of proteins. Nucleic Acids Res. 2011;39(Database issue):D225–D229. doi: 10.1093/nar/gkq1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ishibashi K, Masuda K, Naito S, Meshi T, Ishikawa M. An inhibitor of viral RNA replication is encoded by a plant resistance gene. Proc Natl Acad Sci USA. 2007;104(34):13833–13838. doi: 10.1073/pnas.0703203104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kato M, Ishibashi K, Kobayashi C, Ishikawa M, Katoh E. Expression, purification, and functional characterization of an N-terminal fragment of the tomato mosaic virus resistance protein Tm-1. Protein Expr Purif. 2013;89(1):1–6. doi: 10.1016/j.pep.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 12.Ishibashi K, et al. Coevolution and hierarchical interactions of Tomato mosaic virus and the resistance gene Tm-1. PLoS Pathog. 2012;8(10):e1002975. doi: 10.1371/journal.ppat.1002975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ishikawa M, Okada Y. Replication of tobamovirus RNA. Proceedings of the Japan Academy, Series B. 2004;80(5):215–224. [Google Scholar]
- 14.Ishibashi K, Ishikawa M. The resistance protein Tm-1 inhibits formation of a Tomato mosaic virus replication protein-host membrane protein complex. J Virol. 2013;87(14):7933–7939. doi: 10.1128/JVI.00743-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nishikiori M, et al. Crystal structure of the superfamily 1 helicase from Tomato mosaic virus. J Virol. 2012;86(14):7565–7576. doi: 10.1128/JVI.00118-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Holm L, Rosenström P. Dali server: Conservation mapping in 3D. Nucleic Acids Res. 2010;38(Web Server issue):W545-9. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xiang H, et al. Expression, purification, and functional characterization of a stable helicase domain from a tomato mosaic virus replication protein. Protein Expr Purif. 2012;81(1):89–95. doi: 10.1016/j.pep.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 18.Hayward S, Berendsen HJ. Systematic analysis of domain motions in proteins from conformational change: New results on citrate synthase and T4 lysozyme. Proteins. 1998;30(2):144–154. [PubMed] [Google Scholar]
- 19.Hall MC, Matson SW. Helicase motifs: The engine that powers DNA unwinding. Mol Microbiol. 1999;34(5):867–877. doi: 10.1046/j.1365-2958.1999.01659.x. [DOI] [PubMed] [Google Scholar]
- 20.Wang X, Kelman Z, Culver JN. Helicase ATPase activity of the Tobacco mosaic virus 126-kDa protein modulates replicase complex assembly. Virology. 2010;402(2):292–302. doi: 10.1016/j.virol.2010.03.019. [DOI] [PubMed] [Google Scholar]
- 21.Thiel P, Kaiser M, Ottmann C. Small-molecule stabilization of protein-protein interactions: An underestimated concept in drug discovery? Angew Chem Int Ed Engl. 2012;51(9):2012–2018. doi: 10.1002/anie.201107616. [DOI] [PubMed] [Google Scholar]
- 22.Dunker AK, Brown CJ, Lawson JD, Iakoucheva LM, Obradović Z. Intrinsic disorder and protein function. Biochemistry. 2002;41(21):6573–6582. doi: 10.1021/bi012159+. [DOI] [PubMed] [Google Scholar]
- 23.Fong JH, et al. Intrinsic disorder in protein interactions: Insights from a comprehensive structural analysis. PLOS Comput Biol. 2009;5(3):e1000316. doi: 10.1371/journal.pcbi.1000316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clackson T, Wells JA. A hot spot of binding energy in a hormone-receptor interface. Science. 1995;267(5196):383–386. doi: 10.1126/science.7529940. [DOI] [PubMed] [Google Scholar]
- 25.Bogan AA, Thorn KS. Anatomy of hot spots in protein interfaces. J Mol Biol. 1998;280(1):1–9. doi: 10.1006/jmbi.1998.1843. [DOI] [PubMed] [Google Scholar]
- 26.Ding SC, Pyle AM. Molecular mechanics of RNA rranslocases. In: Eckhard J, editor. Methods Enzymology. Vol 511. San Diego: Academic; 2012. pp. 131–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ishibashi K, Miyashita S, Katoh E, Ishikawa M. Host membrane proteins involved in the replication of tobamovirus RNA. Curr Opin Virol. 2012;2(6):699–704. doi: 10.1016/j.coviro.2012.09.011. [DOI] [PubMed] [Google Scholar]
- 28.Elde NC, Child SJ, Geballe AP, Malik HS. Protein kinase R reveals an evolutionary model for defeating viral mimicry. Nature. 2009;457(7228):485–489. doi: 10.1038/nature07529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rothenburg S, Seo EJ, Gibbs JS, Dever TE, Dittmar K. Rapid evolution of protein kinase PKR alters sensitivity to viral inhibitors. Nat Struct Mol Biol. 2009;16(1):63–70. doi: 10.1038/nsmb.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meshi T, et al. Two concomitant base substitutions in the putative replicase genes of tobacco mosaic virus confer the ability to overcome the effects of a tomato resistance gene, Tm-1. EMBO J. 1988;7(6):1575–1581. doi: 10.1002/j.1460-2075.1988.tb02982.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Strasser M, Pfitzner AJP. The double-resistance-breaking Tomato mosaic virus strain ToMV1-2 contains two independent single resistance-breaking domains. Arch Virol. 2007;152(5):903–914. doi: 10.1007/s00705-006-0915-8. [DOI] [PubMed] [Google Scholar]
- 32.DeLano WL. 2002. The PyMOL molecular graphics system. Available at www.pymol.org. Accessed July 24, 2014.
- 33.Bond CS. TopDraw: A sketchpad for protein structure topology cartoons. Bioinformatics. 2003;19(2):311–312. doi: 10.1093/bioinformatics/19.2.311. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.