

Significance
Spliceostatins are bacterial natural products that show promising anticancer activity. Understanding how the bacterium makes spliceostatins will aid efforts toward a sustainable route for their production. Moreover, altering the chemical structure of a natural product is usually necessary to improve its pharmaceutical properties. For example, the parent spliceostatin molecule contains an unstable hemiketal chemical group. Contrary to previous hypotheses, we report on the identification of a dioxygenase enzyme responsible for hemiketal biosynthesis. Deletion of the corresponding dioxygenase gene led to a strain that produces exclusively spliceostatin congeners that are more stable than, and as active as, the parent compound, when derivatized to increase cell permeability. The strain generated in this study will be the basis for future development.
Keywords: thailanstatin, natural product, bacteria, secondary metabolite
Abstract
Spliceostatins are potent spliceosome inhibitors biosynthesized by a hybrid nonribosomal peptide synthetase−polyketide synthase (NRPS−PKS) system of the trans-acyl transferase (AT) type. Burkholderia sp. FERM BP-3421 produces hemiketal spliceostatins, such as FR901464, as well as analogs containing a terminal carboxylic acid. We provide genetic and biochemical evidence for hemiketal biosynthesis by oxidative decarboxylation rather than the previously hypothesized Baeyer–Villiger oxidative release postulated to be catalyzed by a flavin-dependent monooxygenase (FMO) activity internal to the last module of the PKS. Inactivation of Fe(II)/α-ketoglutarate–dependent dioxygenase gene fr9P led to loss of hemiketal congeners, whereas the mutant was still able to produce all major carboxylic acid-type compounds. FMO mutants, on the other hand, produced both hemiketal and carboxylic acid analogs containing an exocyclic methylene instead of an epoxide, indicating that the FMO is involved in epoxidation rather than Baeyer–Villiger oxidation. Moreover, recombinant Fr9P enzyme was shown to catalyze hydroxylation to form β-hydroxy acids, which upon decarboxylation led to hemiketal FR901464. Finally, a third oxygenase activity encoded in the biosynthetic gene cluster, the cytochrome P450 monooxygenase Fr9R, was assigned as a 4-hydroxylase based on gene inactivation results. Identification and deletion of the gene involved in hemiketal formation allowed us to generate a strain—the dioxygenase fr9P− mutant—that accumulates only the carboxylic acid-type spliceostatins, which are as potent as the hemiketal analogs, when derivatized to increase cell permeability, but are chemically more stable.
The spliceosome, a multi-megadalton ribonucleoprotein complex that is involved in mRNA processing in eukaryotic cells, has emerged as a promising target in cancer therapy (1). Misregulation of mRNA splicing and mutations in the splicing machinery has been observed in a variety of cancers. Splicing modulation with small molecules provides a potential avenue for treatment (2, 3). Three classes of bacterial natural products (Fig. 1A)—pladienolide/FD-895 (4–6), spliceostatin/FR901464 (7, 8), and herboxidiene (GEX1A) (9)—have been shown to modulate splicing activity by binding to the SF3b spliceosome subunit (10–12), exhibiting low to subnanomolar half-maximal inhibitory concentration (IC50) values against different cancer cell lines. The mode of action (MoA) and potent cytotoxicity of these agents led to efforts in advancing spliceosome inhibitors as antitumor drugs. Results of a phase I clinical trial of a semisynthetic analog of pladienolide D, E7107, have been recently published (13), providing early clinical evidence for spliceosome inhibition as a potentially viable MoA to treat cancer.
Fig. 1.

Structures of spliceosome inhibitors and hypotheses for spliceostatin biosynthesis. (A) Structural classes of spliceosome inhibitors. Spliceostatin A is a more stable, semisynthetic analog of natural product FR901464 (11, 23). (B) Known spliceostatin analogs that are the subject of this study. (C) Baeyer–Villiger oxidation hypothesis for spliceostatin hemiketal formation according to Zhang et al. (19) (Upper); biosynthesis of acids over hemiketals by FMO domain skipping, adapted from Liu et al. (20) (Lower). ACP, acyl carrier protein; FMO, flavin-dependent monooxygenase; DH, dehydratase; TE, thioesterase. The subscript 9 indicates the PKS module in which the enzyme activities are present. (D) Alternative, oxidative decarboxylation hypothesis. The R group is indicated in C.
The challenging polyketide structures of spliceosome inhibitors coupled with clinical potential have inspired the synthetic chemistry community. As a result, several total synthesis routes to pladienolide, spliceostatin, and herboxidiene classes of natural products have been reported (for the most recent, see refs. 14–16). For spliceostatins, the shortest described synthetic route contains 20 total steps, with the longest linear sequence being 10 steps (14). Harnessing the biosynthetic potential of the producing organisms for compound production and analog generation is an attractive alternative to chemical synthesis. Pladienolide (17) and herboxidiene (18) are biosynthesized by different Streptomyces species via a canonical type I polyketide synthase (PKS) system, whereas biosynthesis of spliceostatins in two Burkholderia strains (FERM BP-3421 and MSMB 43) is encoded by highly homologous, hybrid trans-acyl transferase (AT) PKS–nonribosomal peptide synthetase (NRPS) gene clusters (19, 20).
Hemiketal-bearing compounds such as FR901464 (Fig. 1B, 4) were described from strain FERM BP-3421 in 1997 (8). FERM BP-3421 had been identified as Pseudomonas sp. no. 2663 but was recently reclassified as Burkholderia (21, 22). A more stable, semisynthetic methyl ketal of 4 was later shown to inhibit the spliceosome, and it was thus termed spliceostatin A (Fig. 1A) (11, 23). Subsequently, an analog bearing a terminal carboxylic acid (3) was reported from Burkholderia sp. MSMB 43 (20, 24). Although 3 belongs to the same family of compounds as 4 and 5, the name thailanstatin A was used when reporting compound 3, as it had been isolated from a Burkholderia thailandensis-like strain. It has recently been shown that strain FERM BP-3421 produces carboxylic acids 1, 2, and 3 in addition to hemiketals 4 and 5 (22, 25), whereas only 3 and two chlorohydrin adducts of 3 have been isolated from MSMB 43 (20). We chose to use the term spliceostatin in this manuscript to refer to all congeners 1–5 in an attempt to unify the terminology of this compound class. Spliceostatins bearing a terminal carboxylic acid have been reported to inhibit mRNA splicing in vitro as effectively as the corresponding hemiketal, while having improved stability (20). We have shown that the free acids themselves are less cytotoxic than the hemiketals against cancer cell lines. However, activity is rescued when carboxylic acid compounds are derivatized (e.g., as methyl esters or propyl amides) presumably due to improved cell permeability (22). As such, the carboxyl analog series represents an attractive chemical lead for anticancer drug discovery.
We were particularly intrigued by how hemiketals are biosynthesized, and compelled by the opportunity to leverage biosynthetic knowledge to block hemiketal formation and accumulate stable carboxylic acid analogs in fermentation. The biosynthetic hypothesis proposed by Tang and coworkers (19, 26) embodies an unprecedented Baeyer–Villiger oxidation catalyzed by a flavin-dependent monooxygenase (FMO) domain present in the last module of the trans-AT PKS (Fig. 1C). If this is correct, domain skipping may explain the formation of acids over hemiketals, as proposed by Liu et al. (20). However, we envisioned oxidative decarboxylation as a plausible alternative hypothesis, in which carboxylic acid compounds would be biosynthesized first, followed by hydroxylation and decarboxylation to form the hemiketal congeners (Fig. 1D). We herein assign functions to three oxygenases encoded in the spliceostatin biosynthetic gene cluster and provide genetic and biochemical evidence for the oxidative decarboxylation hypothesis.
Results
Genome Sequencing and Identification of the Spliceostatin Biosynthetic Gene Cluster.
We independently sequenced the genome of Burkholderia sp. FERM BP-3421 using next-generation technologies (454 and Illumina). At the time, biosynthetic studies on spliceostatins had not been published. Based on the structural similarity of the spliceostatins to pederin (27), we hypothesized that the former may likewise be biosynthesized by a trans-AT PKS system. Indeed, through genome mining, we identified a hybrid trans-AT PKS−NRPS biosynthetic gene cluster (Fig. 2) that seemed consistent with spliceostatin biosynthesis, taking into account deviations to the colinearity rule that are common to trans-AT PKSs (28). In general, the DNA sequence we obtained is in good agreement with that since reported by Zhang et al. (19). However, we found four differences that affected the annotation of the PKS genes. Our sequence has a 34-bp insertion in fr9D and a 23-bp insertion in fr9E that join fr9D, fr9E, and fr9F into one large ORF, which we named fr9DEF. Next, our sequence lacks a cytosine in fr9G that eliminates a frameshift present in the sequence deposited by Zhang et al. and joins fr9G and fr9H into another large ORF, which we named fr9GH. Last, the sequence deposited by Zhang et al. contains an extra 2,397 bp located in fr9G corresponding to an exact repeat of the KS-KR domains in fr9G that we did not observe in our sequence. With these changes, our annotation of the fr9 spliceostatin biosynthetic gene cluster from FERM BP-3421 displays the same PKS organization as the homologous tst gene cluster (20) in MSMB 43 (Fig. 2). Proposed revisions to the biosynthetic hypothesis previously published include the role of dehydratase (DH)-like domains, termed pyran synthase (PS), present in modules 4 and 9 (Fig. 2). PS domains have since been shown by the Piel group (29) to catalyze cyclic ether formation during pederin biosynthesis. Sequence alignment confirmed the presence of characteristic, aberrant active-site motifs in PS4 and PS9 (Fig. S1). Thus, PS4 and PS9 are tentatively assigned to catalyze biosynthesis of the two tetrahydropyran rings of spliceostatins. Furthermore, He et al. (30) have recently shown that a thioesterase domain present in module 1 acts as a dehydratase to form the Z-configured double bond.
Fig. 2.
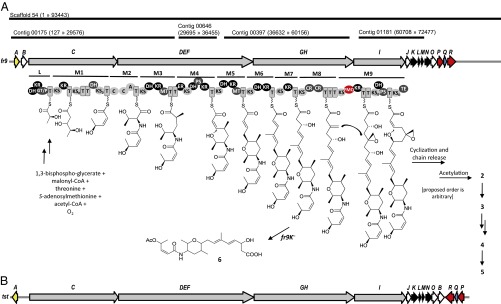
Spliceostatin biosynthetic gene clusters and proposed biosynthesis hypothesis. (A) Gene cluster from Burkholderia sp. FERM BP-3421 (the fr9 nomenclature was first used in ref. 19). Depicted contigs are from 454 sequencing results. Scaffold 54 was obtained by combining 454 and Illumina data. ORFs are represented by arrows (PKS genes in gray, AT in white, regulator in yellow, β-branching cassette in black, protein of unknown function in blue, oxygenases in red). PKS domain key: A, adenylation; C, condensation; CR, crotonase; DH, dehydratase; ER, enoyl reductase; FMO, flavin-dependent monooxygenase; GT/P, glyceryl transferase/phosphatase; KR, ketoreductase; KS, ketosynthase; KS0, KS lacking active-site cysteine; PS, pyran synthase; T, thiolation; TE, thioesterase. L, loading module. M1–M9, extension modules 1–9. (B) Organization of the spliceostatin (thailanstatin, tst) biosynthetic gene cluster in Burkholderia sp. MSMB 43 (20) shown for comparison.
The DNA sequence we initially obtained using 454 pyrosequencing technology was fragmented, containing trans-AT PKS genes on four different contigs (Fig. 2). While we waited for further sequencing data using Illumina technology, we inactivated genes/modules in each contig to test their involvement in spliceostatin biosynthesis. Inactivations of modules 2 and 4 within the PKS-NRPS gene fr9DEF, and an inactivation of modules 5 and 6 that span PKS genes fr9DEF and fr9GH led to mutants that were devoid of detectable spliceostatin production, confirming the involvement of this gene cluster in spliceostatin biosynthesis (Fig. S2). Inactivation of 3-hydroxy-3-methylglutaryl-CoA synthase (HMGS)-like gene fr9K in the fourth contig led to accumulation of a truncated analog with Mr 437.5, which we tentatively assigned as 6 by tandem mass spectrometry (MS/MS) (Fig. 2 and Fig. S3). The conversion of a keto functional group into a carbon branch in polyketides, called β-branching, involves the action of four enzymatic activities—an acyl carrier protein (ACP), a nonelongating and free-standing KS, an HMGS-like enzyme, and a crotonase (CR) or enoyl-CoA dehydratase (28). Inactivation of the HMGS-like gene did not furnish a spliceostatin analog containing a keto group in place of the exocyclic methylene/epoxide, but instead led to apparent interruption of biosynthesis at the time of β-branching (Fig. 2). This result is in accord with the established substrate specificity of trans-AT KSs (31). Canonical cis-AT PKSs evolved by gene duplication, resulting in KS domains that code for a particular metabolite sharing high sequence identity, and allowing the production of structurally modified natural products by domain/module engineering [albeit with possible reduction in reaction rates (32)]. In contrast, trans-AT KS domains are more dissimilar and can be phylogenetically grouped by the type of substrates processed (e.g., β-hydroxylated, α-methylated, α,β-unsaturated, β-branched) (28), indicating that successful PKS engineering toward structure modification of polyketides biosynthesized by trans-AT PKSs may require KS swap or mutagenesis.
Assigning Function to Oxygenase Activities by Gene Inactivation.
Three putative oxygenase activities are encoded in the spliceostatin biosynthetic gene cluster: (i) a FMO domain internal to the last PKS module in fr9GH; (ii) the Fe(II)/α-ketoglutarate–dependent dioxygenase Fr9P; and (iii) the cytochrome P450 Fr9R. Four oxidation reactions are required to obtain the final product 5 from 1′—epoxidation, hydroxylation, formation of the hemiketal, and another hydroxylation (Fig. 3). We began our analysis by determining whether, based on precedence, any of these types of oxygenases would be more likely to catalyze the necessary oxidation steps. Reactions catalyzed by cytochrome P450 monooxygenases are diverse and include hydroxylation of saturated C—H bonds, epoxidation of double bonds, and oxidative decarboxylation (33). Moreover, dual-function P450s have been reported that catalyze sequential epoxidation and hydroxylation of the same substrate (34, 35). Iron/α-ketoglutarate–dependent dioxygenases are also very versatile (36). Well-characterized dioxygenases of this class include taurine hydroxylase (37) and clavulanic acid synthase, which remarkably catalyzes three steps in clavulanic acid biosynthesis, i.e., hydroxylation, oxidative cyclization, and desaturation (38). Reactions described for flavin-dependent monooxygenases, on the other hand, include hydroxylation of aromatic rings, epoxidation of double bonds, and Baeyer–Villiger oxidation (39). Thus, we surmised that the P450 or FMO would be more likely to catalyze the epoxidation step; the needed hydroxylations would be expected to be catalyzed by the P450 or the dioxygenase; and although PKS offloading involving a Baeyer–Villiger oxidation reaction catalyzed by a domain internal to the PKS would be unprecedented, free-standing FMOs have been shown to catalyze Baeyer–Villiger oxidation of natural products (40–43). Therefore, hemiketal biosynthesis could, in principle, be encoded in any of these three oxygenase genes/domain.
Fig. 3.

Proposed function of oxygenase activities within the spliceostatin biosynthetic gene cluster based on gene inactivation results. (A) Phenotype of oxygenase mutants compared with the parent strain. HPLC chromatograms of culture supernatants with detection at 230 nm. (B) Proposed biosynthetic route based on gene inactivation results.
To assign function to the different oxygenases, we inactivated each gene/domain individually (Fig. 3A). The FMO in-frame deletion mutant accumulated compound 1, containing an exocyclic methylene and a free carboxylic acid. None of the other reported spliceostatin analogs could be detected in the FMO mutant. The cytochrome P450 mutant (gene replaced by a tetracycline resistance marker) produced 4-deoxy, carboxyl spliceostatins 1 and 2, along with the previously unidentified hemiketal congener 7, which was determined by liquid chromatography (LC)/MS and NMR analyses to be a 4-deoxy analog of 4. The dioxygenase mutant (gene replaced by a tetracycline resistance marker) produced carboxyl spliceostatins 1, 2, and 3. The combined gene/domain inactivation results are in agreement with the biosynthetic route depicted in Fig. 3B in which the FMO domain catalyzes epoxidation (because deletion of this domain arrests biosynthesis at the exocyclic methylene stage); the cytochrome P450 Fr9R catalyzes hydroxylation of 2 to form 3, and the dioxygenase would be involved in hemiketal formation. The production of compound 7 by the P450 mutant indicates that the dioxygenase may have relaxed substrate specificity and can accept 2 as substrate in the absence of the putative substrate 3. It also rules out the possibility that the P450 is essential for hemiketal formation. It is unclear from these experiments which of the three enzymes, if any, catalyzes the final hydroxylation at C-2 to generate compound 5.
In Vitro Characterization of Fe(II)/α-Ketoglutarate–Dependent Dioxygenase Fr9P.
To further test the involvement of the putative dioxygenase Fr9P in hemiketal formation, we expressed fr9P as a His6-GST–tagged protein in Escherichia coli and used enzyme purified by Ni-affinity chromatography in biochemical assays. Incubation of recombinant Fr9P with 3 led to the formation of 8 (Fig. 4) with +16 mass units, indicating the addition of an oxygen atom to 3. Apparent steady-state kinetic constants of Fr9P were measured in vitro by monitoring the formation of the hydroxylated product by HPLC (Table 1). The 4-deoxy analog 2 is also accepted by Fr9P as substrate to yield 9, albeit with approximately twofold reduced enzyme efficiency (kcat/Km). NMR structure elucidation of derivatized analogs of 8 and 9—designed to increase stability of these β-hydroxy acids—demonstrated that hydroxylation occurred at C-1 to form hemiketals (see below).
Fig. 4.

In vitro activity of Fe(II)/α-ketoglutarate–dependent dioxygenase Fr9P. (A) Reaction catalyzed by Fr9P. See text for comments on the configuration at C-1. (B) HPLC traces with detection at 230 nm. The complete reaction shown contained 3, α-ketoglutarate, sodium ascorbate, Fe(II), and recombinant Fr9P. In the EDTA-containing reaction, a 10-fold molar excess (0.1 mM) compared with Fe(II) was added. All reactions in this panel were carried out under aerobic conditions. (C) Comparison of reactions carried out under aerobic (complete) and anaerobic (nitrogen atmosphere) conditions as evidence for O2 dependance. See Materials and Methods for further details.
Table 1.
Apparent kinetic constants for Fr9P
| Substrate | Km, µM | kcat, min−1 | kcat/Km, µM−1⋅min−1 | Relative enzyme efficiency, % |
| 3 | 60 ± 14 | 86 ± 9 | 1.43 | 100 |
| 2 | 74 ± 7 | 60 ± 6 | 0.81 | 57 |
The kinetic values (at 30 °C, pH 7.5) are average of triplicate measurements ± SD.
The Fr9P-catalyzed reaction is α-ketoglutarate (Km = 4 µM) and Fe(II)-dependent (Fig. 4B). Ascorbate, although not essential, is used to increase activity (effect presumed to be exerted at least in part by avoiding iron oxidation). Molecular oxygen dependence is evidenced by lack of product formation under anaerobic conditions (Fig. 4C). These observations are consistent with reports on other dioxygenases of this class (37). The pH optimum was determined to be 7.5. Phosphate and Bis-Tris buffers were shown to inhibit enzyme activity. MOPS (4-morpholinepropanesulfonic acid), MES (4-morpholineethanesulfonic acid), and Tris⋅HCl buffers yielded similar results and are preferred. The enzyme is inactivated at 37 °C after 20 min under the assay conditions tested. Incubation at 21 °C or 30 °C gives similar results (Fig. S4). It is worth noting that the optimum temperature for Fr9P activity matches the permissive growth temperature of <35 °C for the producing organism FERM BP-3421 (21).
Structure of Products of Fr9P Reaction Confirms Hemiketal Formation.
Product 8 (C28H41NO10, m/z 552.281 predicted and obtained for [M+H+]+) was unstable. Upon lyophilization of aqueous acetonitrile solutions of 8 (HPLC fractions), three major degradation products could be observed (Fig. S5) with masses—as determined by high-resolution MS—corresponding to the loss of CO2 (4, C27H41NO8, m/z 530.273 predicted, 530.274 obtained for [M+Na+]+); CO2 and H2O (10, C27H39NO7, m/z 512.262 predicted and obtained for [M+Na+]+) and corresponding to a truncated aldehyde analog (11, C21H31NO5, m/z 400.210 predicted and obtained for [M+Na+]+). The structures of these degradation products were further elucidated by NMR or by comparison with authentic standards (see below).
We were able to obtain 1D NMR data for 9 (slightly more stable than 8), which suggested that hydroxylation had taken place at C-1 to form a hemiketal, as it was evident by the change in the C-1 chemical shift from δC-1 68.2 to δC-1 95.3. However, instability of 9 (expected for β-hydroxy acids) precluded full structure elucidation. Thus, we aimed to block major routes of decomposition by derivatization. Attempts to generate methyl ketal derivatives of β-hydroxy acids 8 and 9 using various acid catalysts in methanol were unsuccessful. Ultimately, stable n-propyl amide analogs 12 and 13 were prepared using standard amidation conditions. The improved stability of n-propyl amide analogs allowed full structure characterization, including the absolute stereochemistry of the β-hydroxy function. The n-propylamide function in 12 was readily confirmed by a network of heteronuclear multiple-bond correlation spectroscopy (HMBC) correlations from an exchangeable proton signal at δNH 8.01 and methylene resonances at δH 2.39 (δC 45.9) and δH 3.00/3.07 (δC 40.6) to the carbonyl signal at δC 170.4 (Fig. 5). As expected, compound 13 showed a very similar HMBC connectivity profile to that of compound 12. The relative configuration of the amide side chain was determined based on a strong rotating frame nuclear Overhauser effect spectroscopy (ROESY) correlation between H-5 and the hemiketal OH (δOH 6.38 in 12 and δOH 6.35 in 13), which supported an equatorial disposition of the side chain, establishing a 1R, 5R and 1R, 5S orientation for compounds 12 and 13, respectively (Fig. 5). The chair conformation of the hemiketal ring (as shown) and the absolute configuration of the other stereocenters have been well documented in the literature (8). The absolute configuration of the C-1 hydroxy group of 12 and 13 suggests that hydroxylation catalyzed by Fr9P may proceed with inversion of configuration (i.e., conversion of 3→8 and 2→9 giving products with R configuration at C-1; Fig. 4A). However, our experiments do not rule out that the observed R stereochemistry at C-1 of 12 and 13 may be defined posthydroxylation. The spliceostatin hemiketal core exists in equilibrium with its linear open-chain keto-alcohol (44, 45). Conceivably, the isomerization of the hydroxyl group at C-1 of 8 and 9 (or of 12 and 13) could have occurred during this equilibration process.
Fig. 5.

Key ROESY correlations depicting the relative configuration of 12 and 13 along with selected HMBC data and chemical shift assignments for compound 12.
Decarboxylation of β-Hydroxy Acid Intermediate 8 to Yield Hemiketal 4 Is Likely Enzyme Catalyzed.
Given that 4 could be detected upon lyophilization of HPLC fractions containing 8, we considered whether nonenzymatic decarboxylation of 8 is the in vivo route of 4 production. The half-life of 8 at 25 °C (temperature used for compound production) was determined to be 17 h at pH 7.5 and 24 h at pH 6.0 (Fig. S6). Degradation products include the furan 10 and the truncated aldehyde 11 (Fig. 6). Hemiketal 4 is a minor component in both cases, with its formation being favored at lower pH, as it would be expected for nonenzymatic decarboxylation. In contrast, incubation of 8 with a cell-free lysate of module 2 mutant of PKS–NRPS gene fr9DEF (chosen for its lack in spliceostatin production) leads nearly exclusively to 4, suggesting a role for a (as of yet) uncharacterized decarboxylase (Fig. 6). A candidate decarboxylase is the product of fr9Q, the only gene with unassigned, putative function in the spliceostatin gene cluster (Fig. 2). Fr9Q shares sequence similarity to hypothetical proteins/putative cyclases and was proposed by Zhang et al. (19) to play a role in the formation of the first tetrahydropyran ring of spliceostatins. However, because pyran synthase domains in modules 4 and 9 are likely to catalyze the two necessary cyclizations, Fr9Q may well be the missing decarboxylase. A potential mechanism for formation of 4 includes decarboxylation of a β-ketoacid followed by recyclization (Fig. 6C). Alternatively, the decarboxylase may be present outside of the spliceostatin gene cluster. Further experiments will be necessary to test these hypotheses.
Fig. 6.
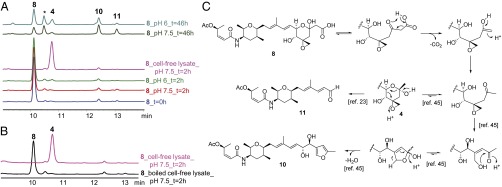
Decarboxylation of β-hydroxy acid 8 to yield 4 is likely enzyme catalyzed in vivo. (A) 8 was obtained using recombinant dioxygenase Fr9P, and then incubated at 25 °C, 200 rpm, under conditions indicated (pH 7.5 or 6.0 and with or without a cell-free lysate of module 2 mutant of PKS–NRPS gene fr9DEF). The mass of the degradation product marked with an asterisk corresponds to 8−H2O; this compound was not isolated. HPLC traces with detection at 230 nm. (B) Negative control using boiled cell-free lysate under the same conditions as in A. HPLC traces with detection at 230 nm. (C) Potential mechanisms for formation of hemiketal 4, furan 10, and aldehyde 11 (23, 45).
The FMO Mutant Is Capable of Producing a Hemiketal Analog.
To test the oxidative decarboxylation hypothesis further, we searched for hemiketal congeners in the FMO mutant. First, we showed that the known exocyclic methylene derivative 1 produced by the FMO mutant is accepted as substrate by dioxygenase Fr9P in vitro (Fig. S7). We were then indeed able to isolate and elucidate the structure of the previously unidentified hemiketal analog 14 (Fig. 7) from the FMO mutant, proving that the FMO is not essential for hemiketal formation in vivo. Although Tang et al. (26) demonstrated that the FMO can catalyze Baeyer–Villiger oxidation in vitro, the truncated, surrogate substrate that was used lacked a double bond. Thus, it is reasonable to speculate that the reaction observed in vitro is not the preferred reaction in vivo. Indeed, the indirect evidence provided here suggests the role for the FMO as an epoxidase.
Fig. 7.
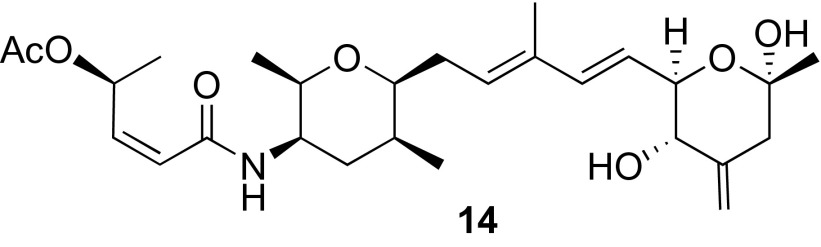
Spliceostatin hemiketal analog isolated from the FMO mutant.
Production of Spliceostatins by Burkholderia sp. MSMB 43 and Deletion of the Dioxygenase Gene tstP.
As mentioned above, the recently described tst gene cluster from Burkholderia sp. MSMB 43 is highly homologous to the fr9 gene cluster. Likewise, the tst cluster encodes three oxygenases, i.e., FMO (OX) domain in TstGH, cytochrome P450 TstR, and dioxygenase TstP (Fig. 2) (20). However, only analogs containing a carboxyl moiety have been described from the MSMB 43 strain, i.e., 3, and two chlorohydrin adducts of 3, one of them containing an isobutyryl group instead of the distal acetyl group (20). In addition to 3, we found that MSMB 43 also produces carboxyl analog 2 and hemiketal congeners 4 and 5 when fermented under the same conditions as FERM BP-3421 (Fig. S8A). The production of both hemiketal- and carboxyl-containing spliceostatins by MSMB 43 suggests that all three oxygenases in the tst cluster are active. To test whether the dioxygenase TstP has a role in hemiketal formation, an in-frame deletion of tstP was performed in MSMB 43. Similar to the inactivation of fr9P in FERM BP-3421, the tstP deletion mutant produced only 2 and 3 in fermentation; no hemiketal-containing spliceostatins were detected in the mutant (Fig. S8B). These results suggest that TstP is involved in hemiketal formation in MSMB 43 and that spliceostatin biosynthesis follows the same logic in the two homologous pathways.
Discussion
Splicing inhibition offers a potential avenue for cancer treatment. Among the natural products known to target the spliceosome, spliceostatins are of particular interest to us due to their potent bioactivity and intriguing biosynthesis. The other two known spliceosome inhibitor classes, pladienolides and herboxidiene, are biosynthesized by Streptomyces spp. via a modular cis-AT PKS, while spliceostatins are produced by Burkholderia spp. using the more recently described and evolutionary distinct trans-AT PKS system. Among the peculiarities of trans-AT PKSs is the large range of module varieties, including unusual domain orders, tandem domains, and atypical domain types (28). Several features of the spliceostatin PKS worth noting include (Fig. 2): (i) a TE domain in module 1 that has been recently shown to act as a dehydratase, generating a Z-configured double bond (30); (ii) two DH-like domains (termed PS) in modules 4 and 9 that are proposed to catalyze formation of the tetrahydropyran rings as demonstrated during pederin biosynthesis (29); (iii) the integrated CR domains in module 8, as observed in pederin biosynthesis, and known to catalyze dehydration and decarboxylation during β-branching; and finally (iv) the FMO domain contained in module 9 of the PKS, which has been proposed to catalyze Baeyer–Villiger oxidative release to generate hemiketal congeners (19, 20, 26).
Besides the FMO, two other oxygenase activities are encoded in the spliceostatin biosynthetic gene cluster, i.e., the cytochrome P450 fr9R and the Fe(II)/α-ketoglutarate–dependent dioxygenase fr9P as stand-alone genes. The combined evidence presented here suggests that, contrary to previous hypotheses (19, 20, 26), the FMO catalyzes epoxidation, the cytochrome P450, hydroxylation at C-4 and the dioxygenase, hydroxylation at C-1, which is followed by decarboxylation (presumably catalyzed by an as-of-yet uncharacterized decarboxylase) leading to hemiketal 4 (Fig. 3). The P450 and the FMO mutants produce hemiketal analogs, proving that they are not essential for hemiketal formation. On the other hand, the dioxygenase mutant is devoid of any hemiketal congeners and recombinant Fr9P catalyzes hydroxylation at C-1 in vitro. Because 3 is the preferred substrate of Fr9P, hydroxylation at C-4 by the P450 Fr9R is likely catalyzed before hydroxylation at C-1 by dioxygenase Fr9P. Moreover, we showed that the MSMB 43 strain, which contains a homologous spliceostatin biosynthetic gene cluster, produces hemiketal-bearing spliceostatins along with previously reported carboxylic acid congeners. Inactivation of the corresponding dioxygenase gene in MSMB 43 blocked hemiketal production in the same fashion as with FERM BP-3421, indicating that hemiketal biosynthesis follows similar logic in the two strains.
When the P450 gene was inactivated, a previously unknown 4-deoxy spliceostatin hemiketal 7 was produced along with the 4-deoxy carboxylic acid congener 2. The cytotoxicity of 7 (IC50 of 14 nM against N87, and 5 nM against BT474 cancer cell lines) is reduced ≥50-fold compared with 4-hydroxylated analog 4 (IC50 of 0.1 nM against N87 and BT474 cell lines), highlighting the significant contribution of the 4-hydroxy functional group for bioactivity. The same relationship is seen with semisynthetic analogs of the carboxylic acid pair 2/3, with the propylamide of 3 being 10-fold more active than the propylamide of 2 (22). The epoxide functional group, which is common to all three classes of splicing inhibitors (Fig. 1A), is not absolutely required for potency. However, it does contribute considerably to splicing inhibition and cytotoxicity (46, 47). For example, the epoxide-containing 2 has an IC50 range of 2–5 nM in our cell lines, whereas exocyclic methylene 1 shows an IC50 of 30 nM (22). Likewise, hemiketal 14, isolated from the FMO mutant, has an IC50 of >100 nM. It is noteworthy that both the herboxidiene and pladienolide gene clusters contain stand-alone FMO genes—herE and pldD, respectively—that have been tentatively assigned as encoding epoxidases (17, 18).
Reactions catalyzed by 2-oxoacid–dependent dioxygenases usually occur with retention of configuration (36). Accordingly, the conversion of 3→8 and 2→9 would be expected to introduce a hydroxy function with S configuration (Fig. 4). However, NMR analysis of stabilized Fr9P product analogs 12 and 13 shows the R configuration at C-1, suggesting that either the Fr9P-catalyzed hydroxylation occurred with inversion of configuration or a conversion occurred posthydroxylation. Mechanistic studies will be necessary to answer this question.
Understanding spliceostatin biosynthesis is an important step toward sustained compound production and analog generation. The analogs described here (7, 8, 9, 12, 13, and 14) helped elucidate the biosynthetic route to hemiketal formation as well as confirmed the positive contributions of the C-4 hydroxy and epoxide groups for cytotoxic potency. Because compounds 2 and 3 are desirable drug leads, the dioxygenase mutant generated in this study sets the stage for further strain development. Efforts to increase production titers of 3 will be described elsewhere.
Materials and Methods
Genome Sequencing.
Strain FERM BP-3421 was acquired from the International Patent Organism Depositary at the National Institute of Advanced Industrial Science and Technology (Tsukuba, Japan). DNA for genome sequencing was isolated using Qiagen’s DNA Maxi Kit according to the manufacturer’s protocol. Specifications of the DNA submitted for sequencing were Abs260/Abs280 = 1.90 and Abs260/Abs230 = 2.00. Sequencing was performed using 454 and Illumina technologies by 454 Life Sciences and the Beijing Genomics Institute (BGI), respectively. Assembly was performed by BGI. The 454 data were assembled with Newbler into 1,743 contigs and 1,732 scaffolds (N50, 6,416), and the Illumina data were assembled with SOAPdenovo into 1,061 contigs and 84 scaffolds (N50, 447, 469). Assembly of both datasets with SOAPdenovo resulted in 491 contigs and 84 scaffolds (N50, 447, 523). The obtained genome sequence (7.68 Mbp, 67.5% G+C) was mined for a hybrid PKS–NRPS pathway that would be in agreement with spliceostatin biosynthesis. The nucleotide sequence of the identified fr9 cluster from FERM BP-3421 has been deposited to GenBank under accession no. KJ461964.
Gene Inactivation in Burkholderia sp. FERM BP-3421.
The FMO domain was inactivated by in-frame deletion. All other mutants were generated by replacing the targeted gene with a tetracycline-resistance marker. Inactivation was performed by homologous recombination. Inactivation plasmids (see SI Materials and Methods for plasmid construction) based on pEX100T (48) or pEX18Tc (49) were transferred into FERM BP-3421 by conjugation from E. coli S17.1, i.e., FERM BP-3421 and E. coli S17-1 containing the plasmid for gene inactivation were separately cultivated in 10 mL of Luria–Bertani (LB) medium at 30 °C, 200 rpm orbital shaking, overnight (with the E. coli culture containing tetracycline, 10 µg/mL). Two milliliters of the E. coli culture was harvested by centrifugation, washed twice with 2 mL of LB (to remove antibiotics), and resuspended in 0.2 mL of fresh LB. Two milliliters of the Burkholderia culture was harvested by centrifugation and resuspended in 0.2 mL of fresh LB. The two cell suspensions were carefully mixed and spread onto LB agar without antibiotics. Two negative control plates, one containing only E. coli and the other, only FERM BP-3421, treated as above were prepared. Plates were incubated overnight at 30 °C. A “loopful” of cells from the conjugation plate was then streaked for single colonies onto selection LB plates and incubated at 30 °C for 2 d. Single colonies were streaked again on selection plates and incubated at 30 °C for 2 d. In the case of gene replacement with a tetracycline marker, selection plates contained gentamycin (10 µg/mL, for removing E. coli after conjugation), tetracycline (25 µg/mL, for selection of mutants), and sucrose [5% (wt/vol), for counterselection of the vector backbone]. Double-crossover mutants with the desired gene replacement were obtained directly. In the case of in-frame deletion (FMO domain), selection plates contained gentamycin (10 µg/mL) and tetracycline (25 µg/mL) to select for a single-crossover mutant, which was then streaked onto LB agar without supplements and incubated at 30 °C for 2 d. The single-crossover mutant was then passed three times through LB agar containing 5% (wt/vol) sucrose. Single colonies were replica plated on LB agar with or without tetracycline. Out of 38 tet-sensitive clones, only 2 were confirmed as the desired double-crossover mutants (in-frame deletion), 18 were reversion to wild type, and the others did not show any amplicon. In all cases, mutants were confirmed by colony PCR (RED Taq; Sigma).
Gene Inactivation in Burkholderia sp. MSMB 43.
Strain MSMB 43 was obtained from the Centers for Disease Control and Prevention (CDC). MSMB 43 was isolated by investigators at the Menzies School of Health Research, Charles Darwin University (Casuarina, NT, Australia) (24) and is currently being described as “Burkholderia humptydooensis.” The inactivation plasmid (SI Materials and Methods) was transferred into MSMB 43 by conjugation from E. coli S17-1 as described above for FERM BP-3421. Selection plates contained gentamicin (10 µg/mL) and tetracycline (50 µg/mL) to select for a single-crossover mutant, which was then streaked onto LB agar without supplements and incubated at 37 °C for 2 d. The single-crossover mutant was then passed three times through LB agar without supplements. A total of 219 single colonies was replica plated on LB agar with or without tetracycline (50 µg/mL). Six colonies were tetracycline sensitive; three were confirmed as the desired double-crossover mutants (in-frame deletion), and three were reversion to wild type. All mutants were confirmed by colony PCR (RED Taq; Sigma).
Cultivation Conditions and Metabolite Analysis.
Burkholderia sp. FERM BP-3421 and MSMB 43 were routinely cultured in LB broth or agar medium (Becton Dickinson) at 30 °C, unless otherwise stated. Frozen stocks were prepared with 20% (vol/vol) glycerol and kept at −80 °C. For metabolite analyses, seed medium (10 g/L polypeptone, 5 g/L yeast extract, 5 g/L sodium chloride) was inoculated with a frozen stock, and the culture was incubated at 30 °C, 220 rpm overnight. This first seed culture was used to inoculate a second stage seed at 10% (vol/vol), which was incubated as above. Production medium (10 g/L soluble starch, 10 g/L glycerol, 5 g/L glucose, 10 g/L HySoy soypeptone, 5 g/L corn steep liquor, 2 g/L ammonium sulfate, 0.06 g/L magnesium sulfate heptahydrate, 2 g/L calcium carbonate) was inoculated with seed culture at 2.5% (vol/vol) and incubated at 25 °C, 200 rpm for 3 d. When 2S4G production medium (40 g/L glycerol, 20 g/L HySoy soypeptone, 2 g/L ammonium sulfate, 0.1 g/L magnesium sulfate heptahydrate, 2 g/L calcium carbonate) was used, cultures were harvested at day 5. Seed cultures were grown either in 14-mL, polypropylene round-bottom tubes (BD) containing 5 mL of medium or in 250-mL Erlenmeyer flasks containing 50 mL of medium. Tetracycline (25–50 µg/mL) was added where appropriate. Production cultures were grown either in 250-mL Erlenmeyer flasks containing 50 mL of medium or in 2.8-L Fernbach flasks containing 0.5 L of medium. No antibiotics were added to production cultures. The fermentation was processed with 5% (wt/vol) Diaion HP-20 resin as follows. Wet, activated HP-20 was added to the culture at the end of fermentation, and the mixture was shaken for 30 min on a tilting platform or shaker. After centrifugation, the supernatant was decanted and the cell/resin pellet extracted twice with 1/2 vol ethyl acetate. After removing solvent under reduced pressure, the crude extract was dissolved in DMSO to make a 2× sample for HPLC and LC/MS analyses. Alternatively, a 0.5-mL aliquot of the production culture was centrifuged to remove cells, and the supernatant was mixed with 0.5 mL of acetonitrile. After centrifugation and filtering, the sample was analyzed by HPLC and/or LC/MS. HPLC analytic conditions were as follows: column, reversed-phase C18 YMC ODS-A, 4.6 × 150 mm, 5 µm; mobile phase A, 0.01% trifluoroacetic acid in water; mobile phase B, 0.01% trifluoroacetic acid in acetonitrile; gradient, 10–100% B over 19 min; flow rate, 1.0 mL/min; temperature, not controlled; detection, diode array detector (DAD) 230 nm; injection volume, 5–25 μL; instrument, Agilent 1100 series HPLC. LC/MS conditions were as follows: column, reversed-phase C8 Acquity UPLC BEH, 2.1 mm × 100 mm, 1.7 μm; mobile phase A, 0.1% formic acid in water; mobile phase B, 0.1% formic acid in acetonitrile; gradient, 20% B for 0.5 min, 20–80% B for 1.5 min, 80–100% B for 3 min, 100% B for 5 min, 100–20% B for 0.1 min, 20% B for 1.4 min; flow rate, 0.4 mL/min; instrument, Waters Acquity UPLC-LCT Premier TOF MS with alternating positive-ion and negative-ion full scan (100–2,000 mass units) mode.
Production of Recombinant Fr9P Enzyme in and Purification from Escherichia coli.
The codon-optimized Fr9P gene was synthesized and ligated into the NcoI–HindIII sites of pGS-21a (GenScript) to generate pAE-PF16. Recombinant, His6-GST tagged Fr9P protein was produced in and purified from E. coli BL21(DE3) after transformation with plasmid pAE-PF16. A typical purification was as follows: two 2.8-L Fernbach flasks containing 0.5 L of medium (Terrific broth with 100 mg/L ampicillin) were each inoculated with 20 mL of an overnight LB culture and incubated at 200 rpm, 25 °C. When the OD600 reached ∼0.9, cells were induced with 0.2 mM isopropyl β-d-1-thiogalactopyranoside, and incubation was resumed at 25 °C and 200 rpm. After 18–20 h, cells were harvested by centrifugation and frozen at −80 °C. The cell pellet was resuspended in ∼50 mL of ice-cold lysis buffer (10 mM phosphate buffer, pH 7.4; 500 mM NaCl; 20 mM imidazole; 10% glycerol; lysozyme, 1 mg/mL; 0.5% Tween 20; 20 mM β-mercaptoethanol) and incubated on ice for 30 min. Following sonication on ice, the cell lysate was centrifuged at 21,000 × g and 4 °C for 45 min. The supernatant was transferred to a new tube and centrifuged again at 21,000 × g and 4 °C for 30 min. Five milliliters of Ni-NTA resin slurry (Qiagen) were added to the supernatant fraction (clear lysate) contained in a small beaker on ice and gently stirred for 1 h. The suspension was transferred to a Falcon tube and centrifuged at 1,000 × g and 4 °C for 10 min. The supernatant was discarded and the resin washed three times, each with 30 mL of ice-cold wash buffer (10 mM phosphate buffer, pH 7.4; 500 mM NaCl; 40 mM imidazole; 10% glycerol; 20 mM β-ME) followed by centrifugation at 1,000 × g and 4 °C for 10 min. The resin was transferred to a disposable column and washed three more times, each with 2.5 mL of wash buffer. The enzyme was eluted with 3× 2.5-mL elution buffer (10 mM phosphate buffer, pH 7.4; 500 mM NaCl; 250 mM imidazole; 10% glycerol; 20 mM β-ME). The buffer was exchanged to 50 mM MOPS, pH 7.5, using a PD-10 column and the solution concentrated using a Vivaspin column with molecular weight cutoff of 30 kDa. Storage buffer contained 50 mM MOPS, pH 7.5, 2 mM DTT, and 10% glycerol (for storage at −80 °C) or 50% glycerol (for storage at −20 °C). The yield of purified enzyme was ∼25 mg per liter of culture.
Fr9P Enzyme Assay and Determination of Michaelis–Menten Kinetic Constants.
Fr9P assays were carried out in MOPS buffer (50 mM) at pH 7.5. Note that phosphate and Bis-Tris buffers were found to inhibit enzyme activity (Fig. S4C). For assays presented in Fig. 4, the complete reaction contained 3 (0.1 mM), α-ketoglutarate (0.2 mM), sodium ascorbate (0.02 mM), NH4Fe(II)SO4 (0.01 mM), and Fr9P (0.4–1.6 µM); reactions were incubated for 1–2 h at room temperature (21 °C). For the reaction carried out under anaerobic conditions, 0.925 mL of MOPS buffer (contained in a capped glass vial) was bubbled with nitrogen for 10 min at 37 °C. At the same time, a α-ketoglutarate stock solution (10 mM) was likewise bubbled with nitrogen for 10 min at 37 °C. After bringing the buffer to room temperature, reagents were added using a needle syringe in the following order: (i) a master mix of sodium ascorbate, Fe(II) and 3, and (ii) Fr9P. The mixture was bubbled with nitrogen for an additional 10 min at room temperature before adding α-ketoglutarate to initiate the reaction under nitrogen atmosphere (final volume, 1 mL). For determination of kinetic constants, recombinant Fr9P (0.2 µM) was incubated with spliceostatin substrate (10–500 µM), α-ketoglutarate (0.002–1 mM), sodium ascorbate (0.1 mM), and NH4Fe(II)SO4 (0.05 mM) in 50 mM MOPS buffer (pH 7.5) at 30 °C for 1 min. Reactions were then immediately transferred to ice, quenched with 1 vol of acetonitrile, filtered, and analyzed by HPLC as above (25-μL injections). Product area under the curve was used to calculate conversion (micromolar product per minute) using a 3 standard curve. Michaelis–Menten constants were calculated from Lineweaver–Burke plots.
Incubation of 8 with a Cell-Free Lysate of the NRPS− Mutant.
The NRPS− mutant of FERM BP-3421 was grown in 50 mL of 2S4G spliceostatin production medium as described above. Cells were harvested after 3 d and frozen at −80 °C. The cell pellet was defrosted and resuspended in 5 mL of MOPS buffer, pH 7.5. Cells were lyzed by sonication on ice. After centrifugation at 21,000 × g for 20 min, the supernatant was transferred to a new tube on ice. 8 was obtained using recombinant Fr9P. Twenty microliters of the cell-free lysate was added to 8 (0.1 mM) contained in 200 µL of Fr9P reaction mixture and incubated at 25 °C and 200 rpm for 2 h. Boiled cell-free lysate was used as negative control, in which case formation of 4 was not observed. After quenching with 1 vol of MeCN, centrifuging, and filtering, the reaction was analyzed by HPLC and LC/MS.
Isolation and Structure Elucidation of Hemiketal 14 from the ΔFMO Mutant.
The ∆FMO mutant of FERM BP-3421 was cultured in 1 L of 2S4G production medium as described above. After removing cells by centrifugation (20 min at 4,500 × g), wet, activated HP-20 [5% (wt/vol)] was added to the supernatant, and the mixture was mixed for 30 min on an orbital shaker. The compound-bound resin was collected by centrifugation and extracted twice with 0.5 L of ethyl acetate each. The dried, crude extract (0.5 g) was purified by preparative, reversed-phase HPLC (column: Phenomenex Luna C18, 150 × 21.2 mm, 5 μm; mobile phase A: 0.02% acetic acid in water; mobile phase B: 0.02% acetic acid in acetonitrile; gradient: 30% B for 1.5 min, 30–80% B over 8.5 min, 80–100% B over 0.5 min, 100% B for 2 min, 100–30% B over 0.5 min; flow rate: 27 mL/min). The fraction with retention time of 7.1–7.6 min was collected and freeze-dried to afford 14 (51% pure at 215 nm). This fraction was submitted to a second round of reverse-phase HPLC purification (column: Phenomenex Luna C18, 150 × 21.2 mm, 5 μm; mobile phase A: 0.1% formic acid in water; mobile phase B: 0.1% formic acid in acetonitrile; gradient: 33% B for 10 min, 33–100% B over 0.5 min, 100% B for 2 min, 100–33% B over 0.5 min; flow rate: 27 mL/min). The fraction with retention time of 5.75–6.75 min was collected and freeze-dried to afford 14 (11.8 mg, 94% pure by UV at 215 nm) as a white powder. High-resolution electrospray ionization mass spectrometry, m/z 492.2961, error −0.1 mDa for C27H42NO7 [M+H]+. For 1D and 2D NMR dataset, see Table S1.
Refer to SI Materials and Methods for further methods.
Supplementary Material
Acknowledgments
We thank Jay E. Gee (CDC) and Bart J. Currie (Menzies School of Health Research) for providing strain MSMB 43; H. P. Schweizer (Colorado State University) for vectors pEX18Tc and pEX100T; Xidong Feng and Keiko Tabei (Groton Center of Chemistry, Pfizer) for high-resolution MS and MS/MS analyses; Melissa Wagenaar (Groton Center of Chemistry, Pfizer) for purification of 7 and 14; Kathleen A. Farley, Dennis P. Anderson, and Guoyun Bai (Groton Center of Chemistry, Pfizer) for NMR support; My-Hanh Lam and Frank Loganzo (Oncology, Pfizer) for cytotoxicity data; Ellen Murphy for initial assembly of the 454 sequence; Brian Dougherty, Baohong Zhang, Paul Fracasso, and Wen He (Computational Sciences, Pfizer) for help with Illumina sequencing; Andrew Tomaras (Biotherapeutics, Pfizer) for supportive discussions on Burkholderia genetics; and Haiyin He, Li-Ping Chang, Greg Steele, Min He, Chakrapani Subramanyam, Andreas Maderna, Russell Dushin, Edmund Graziani, and many others for helpful discussions during the course of these studies.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The DNA sequence of the fr9 biosynthetic gene cluster reported in this paper has been deposited in the GenBank database (accession no. KJ461964).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1408300111/-/DCSupplemental.
References
- 1.Bonnal S, Vigevani L, Valcárcel J. The spliceosome as a target of novel antitumour drugs. Nat Rev Drug Discov. 2012;11(11):847–859. doi: 10.1038/nrd3823. [DOI] [PubMed] [Google Scholar]
- 2.Butler MS. Remediating cancer via splicing modulation. J Med Chem. 2013;56(17):6573–6575. doi: 10.1021/jm401289z. [DOI] [PubMed] [Google Scholar]
- 3.Kaida D, Schneider-Poetsch T, Yoshida M. Splicing in oncogenesis and tumor suppression. Cancer Sci. 2012;103(9):1611–1616. doi: 10.1111/j.1349-7006.2012.02356.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mizui Y, et al. Pladienolides, new substances from culture of Streptomyces platensis Mer-11107. III. In vitro and in vivo antitumor activities. J Antibiot (Tokyo) 2004;57(3):188–196. doi: 10.7164/antibiotics.57.188. [DOI] [PubMed] [Google Scholar]
- 5.Sakai T, Asai N, Okuda A, Kawamura N, Mizui Y. Pladienolides, new substances from culture of Streptomyces platensis Mer-11107. II. Physico-chemical properties and structure elucidation. J Antibiot (Tokyo) 2004;57(3):180–187. doi: 10.7164/antibiotics.57.180. [DOI] [PubMed] [Google Scholar]
- 6.Seki-Asano M, et al. Isolation and characterization of a new 12-membered macrolide FD-895. J Antibiot (Tokyo) 1994;47(12):1395–1401. doi: 10.7164/antibiotics.47.1395. [DOI] [PubMed] [Google Scholar]
- 7.Nakajima H, et al. New antitumor substances, FR901463, FR901464 and FR901465. II. Activities against experimental tumors in mice and mechanism of action. J Antibiot (Tokyo) 1996;49(12):1204–1211. doi: 10.7164/antibiotics.49.1204. [DOI] [PubMed] [Google Scholar]
- 8.Nakajima H, Takase S, Terano H, Tanaka H. New antitumor substances, FR901463, FR901464 and FR901465. III. Structures of FR901463, FR901464 and FR901465. J Antibiot (Tokyo) 1997;50(1):96–99. doi: 10.7164/antibiotics.50.96. [DOI] [PubMed] [Google Scholar]
- 9.Miller-Wideman M, et al. Herboxidiene, a new herbicidal substance from Streptomyces chromofuscus A7847. Taxonomy, fermentation, isolation, physico-chemical and biological properties. J Antibiot (Tokyo) 1992;45(6):914–921. doi: 10.7164/antibiotics.45.914. [DOI] [PubMed] [Google Scholar]
- 10.Kotake Y, et al. Splicing factor SF3b as a target of the antitumor natural product pladienolide. Nat Chem Biol. 2007;3(9):570–575. doi: 10.1038/nchembio.2007.16. [DOI] [PubMed] [Google Scholar]
- 11.Kaida D, et al. Spliceostatin A targets SF3b and inhibits both splicing and nuclear retention of pre-mRNA. Nat Chem Biol. 2007;3(9):576–583. doi: 10.1038/nchembio.2007.18. [DOI] [PubMed] [Google Scholar]
- 12.Hasegawa M, et al. Identification of SAP155 as the target of GEX1A (Herboxidiene), an antitumor natural product. ACS Chem Biol. 2011;6(3):229–233. doi: 10.1021/cb100248e. [DOI] [PubMed] [Google Scholar]
- 13.Eskens FA, et al. Phase I pharmacokinetic and pharmacodynamic study of the first-in-class spliceosome inhibitor E7107 in patients with advanced solid tumors. Clin Cancer Res. 2013;19(22):6296–6304. doi: 10.1158/1078-0432.CCR-13-0485. [DOI] [PubMed] [Google Scholar]
- 14.Ghosh AK, Chen ZH. Enantioselective syntheses of FR901464 and spliceostatin A: Potent inhibitors of spliceosome. Org Lett. 2013;15(19):5088–5091. doi: 10.1021/ol4024634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kumar VP, Chandrasekhar S. Enantioselective synthesis of pladienolide B and truncated analogues as new anticancer agents. Org Lett. 2013;15(14):3610–3613. doi: 10.1021/ol401458d. [DOI] [PubMed] [Google Scholar]
- 16.Lagisetti C, et al. Pre-mRNA splicing-modulatory pharmacophores: The total synthesis of herboxidiene, a pladienolide-herboxidiene hybrid analog and related derivatives. ACS Chem Biol. 2014;9(3):643–648. doi: 10.1021/cb400695j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Machida K, et al. Organization of the biosynthetic gene cluster for the polyketide antitumor macrolide, pladienolide, in Streptomyces platensis Mer-11107. Biosci Biotechnol Biochem. 2008;72(11):2946–2952. doi: 10.1271/bbb.80425. [DOI] [PubMed] [Google Scholar]
- 18.Shao L, Zi J, Zeng J, Zhan J. Identification of the herboxidiene biosynthetic gene cluster in Streptomyces chromofuscus ATCC 49982. Appl Environ Microbiol. 2012;78(6):2034–2038. doi: 10.1128/AEM.06904-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang F, et al. Cloning and elucidation of the FR901464 gene cluster revealing a complex acyltransferase-less polyketide synthase using glycerate as starter units. J Am Chem Soc. 2011;133(8):2452–2462. doi: 10.1021/ja105649g. [DOI] [PubMed] [Google Scholar]
- 20.Liu X, et al. Genomics-guided discovery of thailanstatins A, B, and C as pre-mRNA splicing inhibitors and antiproliferative agents from Burkholderia thailandensis MSMB43. J Nat Prod. 2013;76(4):685–693. doi: 10.1021/np300913h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakajima H, et al. New antitumor substances, FR901463, FR901464 and FR901465. I. Taxonomy, fermentation, isolation, physico-chemical properties and biological activities. J Antibiot (Tokyo) 1996;49(12):1196–1203. doi: 10.7164/antibiotics.49.1196. [DOI] [PubMed] [Google Scholar]
- 22.He H, et al. 2014. Cytotoxic spliceostatins from Burkholderia sp. and their semisynthetic analogues. J Nat Prod, in press.
- 23.Motoyoshi H, et al. Structure-activity relationship for FR901464: A versatile method for the conversion and preparation of biologically active biotinylated probes. Biosci Biotechnol Biochem. 2004;68(10):2178–2182. doi: 10.1271/bbb.68.2178. [DOI] [PubMed] [Google Scholar]
- 24.Gee JE, et al. Recovery of a Burkholderia thailandensis-like isolate from an Australian water source. BMC Microbiol. 2008;8:54–58. doi: 10.1186/1471-2180-8-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu X, Biswas S, Tang GL, Cheng YQ. Isolation and characterization of spliceostatin B, a new analogue of FR901464, from Pseudomonas sp. No. 2663. J Antibiot (Tokyo) 2013;66(9):555–558. doi: 10.1038/ja.2013.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tang MC, He HY, Zhang F, Tang GL. Baeyer-Villiger oxidation of acyl carrier protein-tethered thioester to acyl carrier protein-linked thiocarbonate catalyzed by a monooxygenase domain in FR901464 biosynthesis. ACS Catal. 2013;3(3):444–447. [Google Scholar]
- 27.Piel J. A polyketide synthase-peptide synthetase gene cluster from an uncultured bacterial symbiont of Paederus beetles. Proc Natl Acad Sci USA. 2002;99(22):14002–14007. doi: 10.1073/pnas.222481399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Piel J. Biosynthesis of polyketides by trans-AT polyketide synthases. Nat Prod Rep. 2010;27(7):996–1047. doi: 10.1039/b816430b. [DOI] [PubMed] [Google Scholar]
- 29.Pöplau P, Frank S, Morinaka BI, Piel J. An enzymatic domain for the formation of cyclic ethers in complex polyketides. Angew Chem Int Ed Engl. 2013;52(50):13215–13218. doi: 10.1002/anie.201307406. [DOI] [PubMed] [Google Scholar]
- 30.He HY, Tang MC, Zhang F, Tang GL. cis-Double bond formation by thioesterase and transfer by ketosynthase in FR901464 biosynthesis. J Am Chem Soc. 2014;136(12):4488–4491. doi: 10.1021/ja500942y. [DOI] [PubMed] [Google Scholar]
- 31.Jenner M, et al. Substrate specificity in ketosynthase domains from trans-AT polyketide synthases. Angew Chem Int Ed Engl. 2013;52(4):1143–1147. doi: 10.1002/anie.201207690. [DOI] [PubMed] [Google Scholar]
- 32.Khosla C, Kapur S, Cane DE. Revisiting the modularity of modular polyketide synthases. Curr Opin Chem Biol. 2009;13(2):135–143. doi: 10.1016/j.cbpa.2008.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meunier B, de Visser SP, Shaik S. Mechanism of oxidation reactions catalyzed by cytochrome p450 enzymes. Chem Rev. 2004;104(9):3947–3980. doi: 10.1021/cr020443g. [DOI] [PubMed] [Google Scholar]
- 34.Kudo F, Motegi A, Mizoue K, Eguchi T. Cloning and characterization of the biosynthetic gene cluster of 16-membered macrolide antibiotic FD-891: Involvement of a dual functional cytochrome P450 monooxygenase catalyzing epoxidation and hydroxylation. ChemBioChem. 2010;11(11):1574–1582. doi: 10.1002/cbic.201000214. [DOI] [PubMed] [Google Scholar]
- 35.Anzai Y, et al. Functional analysis of MycCI and MycG, cytochrome P450 enzymes involved in biosynthesis of mycinamicin macrolide antibiotics. Chem Biol. 2008;15(9):950–959. doi: 10.1016/j.chembiol.2008.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Prescott AG, Lloyd MD. The iron(II) and 2-oxoacid-dependent dioxygenases and their role in metabolism. Nat Prod Rep. 2000;17(4):367–383. doi: 10.1039/a902197c. [DOI] [PubMed] [Google Scholar]
- 37.Eichhorn E, van der Ploeg JR, Kertesz MA, Leisinger T. Characterization of α-ketoglutarate-dependent taurine dioxygenase from Escherichia coli. J Biol Chem. 1997;272(37):23031–23036. doi: 10.1074/jbc.272.37.23031. [DOI] [PubMed] [Google Scholar]
- 38.Zhang Z, et al. Structural origins of the selectivity of the trifunctional oxygenase clavaminic acid synthase. Nat Struct Biol. 2000;7(2):127–133. doi: 10.1038/72398. [DOI] [PubMed] [Google Scholar]
- 39.Walsh CT, Wencewicz TA. Flavoenzymes: Versatile catalysts in biosynthetic pathways. Nat Prod Rep. 2013;30(1):175–200. doi: 10.1039/c2np20069d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Du L, Lou L. PKS and NRPS release mechanisms. Nat Prod Rep. 2010;27(2):255–278. doi: 10.1039/b912037h. [DOI] [PubMed] [Google Scholar]
- 41.Gibson M, Nur-e-alam M, Lipata F, Oliveira MA, Rohr J. Characterization of kinetics and products of the Baeyer-Villiger oxygenase MtmOIV, the key enzyme of the biosynthetic pathway toward the natural product anticancer drug mithramycin from Streptomyces argillaceus. J Am Chem Soc. 2005;127(50):17594–17595. doi: 10.1021/ja055750t. [DOI] [PubMed] [Google Scholar]
- 42.Seo MJ, Zhu D, Endo S, Ikeda H, Cane DE. Genome mining in Streptomyces. Elucidation of the role of Baeyer-Villiger monooxygenases and non-heme iron-dependent dehydrogenase/oxygenases in the final steps of the biosynthesis of pentalenolactone and neopentalenolactone. Biochemistry. 2011;50(10):1739–1754. doi: 10.1021/bi1019786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tibrewal N, et al. Baeyer-Villiger C-C bond cleavage reaction in gilvocarcin and jadomycin biosynthesis. J Am Chem Soc. 2012;134(44):18181–18184. doi: 10.1021/ja3081154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thompson CF, Jamison TF, Jacobsen EN. FR901464: Total synthesis, proof of structure, and evaluation of synthetic analogues. J Am Chem Soc. 2001;123(41):9974–9983. doi: 10.1021/ja016615t. [DOI] [PubMed] [Google Scholar]
- 45.Albert BJ, Sivaramakrishnan A, Naka T, Czaicki NL, Koide K. Total syntheses, fragmentation studies, and antitumor/antiproliferative activities of FR901464 and its low picomolar analogue. J Am Chem Soc. 2007;129(9):2648–2659. doi: 10.1021/ja067870m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Effenberger KA, et al. Coherence between cellular responses and in vitro splicing inhibition for the anti-tumor drug pladienolide B and its analogs. J Biol Chem. 2014;289(4):1938–1947. doi: 10.1074/jbc.M113.515536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Villa R, et al. Stabilized cyclopropane analogs of the splicing inhibitor FD-895. J Med Chem. 2013;56(17):6576–6582. doi: 10.1021/jm400861t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schweizer HP, Hoang TT. An improved system for gene replacement and xylE fusion analysis in Pseudomonas aeruginosa. Gene. 1995;158(1):15–22. doi: 10.1016/0378-1119(95)00055-b. [DOI] [PubMed] [Google Scholar]
- 49.Hoang TT, Karkhoff-Schweizer RR, Kutchma AJ, Schweizer HP. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: Application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene. 1998;212(1):77–86. doi: 10.1016/s0378-1119(98)00130-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.