

Significance
Inflammation often complicates diseases associated with oxidative stress. This study shows that inflammatory macrophages release proteins with specific forms of cysteine oxidation to disulfides, particularly glutathionylation. Redox proteomics identified peroxiredoxin 2 (PRDX2) as a protein released in glutathionylated form by inflammation both in vivo and in vitro. Extracellular PRDX2 then triggers the production of TNF-α. These data indicate that redox-dependent mechanisms, in an oxidative cascade, can induce inflammation.
Keywords: cysteine oxidation, thiol oxidation, redox proteomics
Abstract
The mechanism by which oxidative stress induces inflammation and vice versa is unclear but is of great importance, being apparently linked to many chronic inflammatory diseases. We show here that inflammatory stimuli induce release of oxidized peroxiredoxin-2 (PRDX2), a ubiquitous redox-active intracellular enzyme. Once released, the extracellular PRDX2 acts as a redox-dependent inflammatory mediator, triggering macrophages to produce and release TNF-α. The oxidative coupling of glutathione (GSH) to PRDX2 cysteine residues (i.e., protein glutathionylation) occurs before or during PRDX2 release, a process central to the regulation of immunity. We identified PRDX2 among the glutathionylated proteins released in vitro by LPS-stimulated macrophages using mass spectrometry proteomic methods. Consistent with being part of an inflammatory cascade, we find that PRDX2 then induces TNF-α release. Unlike classical inflammatory cytokines, PRDX2 release does not reflect LPS-mediated induction of mRNA or protein synthesis; instead, PRDX2 is constitutively present in macrophages, mainly in the reduced form, and is released in the oxidized form on LPS stimulation. Release of PRDX2 is also observed in human embryonic kidney cells treated with TNF-α. Importantly, the PRDX2 substrate thioredoxin (TRX) is also released along with PRDX2, enabling an oxidative cascade that can alter the –SH status of surface proteins and thereby facilitate activation via cytokine and Toll-like receptors. Thus, our findings suggest a model in which the release of PRDX2 and TRX from macrophages can modify the redox status of cell surface receptors and enable induction of inflammatory responses. This pathway warrants further exploration as a potential novel therapeutic target for chronic inflammatory diseases.
Inflammation, an essential component of the innate immune response to pathogens, is also clearly implicated in the pathogenesis of many diseases, including rheumatoid arthritis, multiple sclerosis, ischemia-reperfusion injury, and complications of sepsis. Most of these diseases are also associated with increased production of reactive oxygen species (ROS), which results in oxidative stress (1, 2) and causes various forms of protein oxidation (3). Here we present evidence indicating that protein oxidation results in the release of inflammatory signals and identify peroxiredoxin-2 (PRDX2) as such a signal.
The tripeptide glutathione (GSH) is the main intracellular thiol antioxidant. Decreased GSH levels result in increased ROS, impaired immune responses, inflammation, and increased susceptibility to infection (4). Recent studies point to a role for GSH, as well as its oxidized form (GSSG), in the regulation of cellular function and gene expression, activities that go well beyond the free radical scavenger action commonly associated with GSH (5–7). Importantly, GSH participates in the redox regulation of immunity (8, 9) via the formation of mixed disulfides between protein cysteines and glutathione. This process, commonly referred to as glutathionylation, is known to regulate signaling proteins and transcription factors. Thus, in earlier studies in which we introduced the term redox proteomics for methods similar to those used here, we identified several glutathionylated proteins in human mitogen-activated T lymphocytes (10, 11).
In this study, we used redox proteomics to demonstrate that immune activation induces macrophages to release glutathionylated proteins, most likely via a nonclassical pathway. Thus, we show that the glutathionylated (oxidized) form of the intracellular redox enzyme PRDX2 is among the proteins released in response to inflammatory stimuli.
We also show that recombinant PRDX2 induces macrophages to produce TNF-α in the absence of any other stimuli. Thus, we demonstrate that the thioredoxin peroxidase PRDX2 is appropriately classified as a danger signal, meaning a molecule that is released by cells undergoing stress and acts as an immune mediator (12).
Finally, we show that PRDX2 is released together with its substrate, thioredoxin 1 (TRX1). TRX1 in turn can alter the redox state of several proteins, including membrane proteins. Thus, we propose a model in which inflammatory signals trigger an oxido-reduction cascade of secreted redox enzymes that act in concert as soluble immune regulators in inflammatory responses.
Results
Glutathionylated Proteins Released by LPS-Stimulated Macrophages: Identification of PRDX2 Release.
When cells are preloaded with biotinylated glutathione (BioGEE), the glutathionylated proteins become labeled with biotin and can be affinity-purified on streptavidin and detected by nonreducing Western blot analysis using streptavidin-peroxidase. As shown in Fig. 1, LPS-stimulated RAW264 macrophages release several glutathionylated proteins into the culture supernatant. The fact that reducing mixed disulfides with DTT results in loss of the majority of the bands confirms that the labeling is specifically attributed to glutathionylation.
Fig. 1.
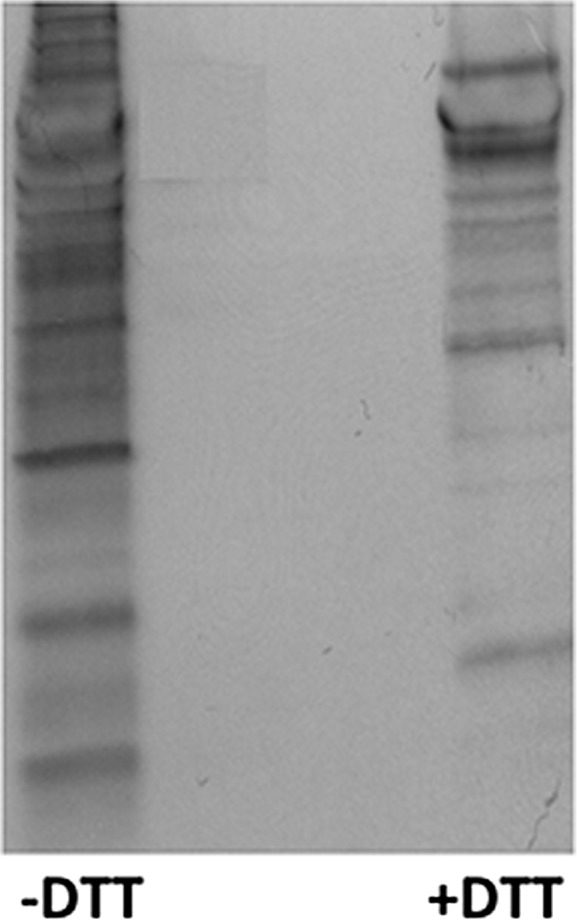
Glutathionylated proteins are released by LPS-stimulated RAW264 cells. Supernatants from BioGEE-preloaded cells exposed to 100 ng/mL LPS for 24 h were analyzed by Western blot analysis with streptavidin peroxidase. Left lane, nonreduced sample; right lane, sample reduced with 10 mM DTT before loading on the gel.
Having established that LPS-treated cells release glutathionylated proteins, we then identified those proteins by redox proteomics. Proteins from supernatants of BioGEE-preloaded, LPS-stimulated RAW264 cells were affinity-purified on streptavidin beads. Shotgun proteomics identified several proteins, including PRDX2. We decided to focus on PRDX2 despite the low peptide coverage with which it was identified (13.6% in a first experiment and 30% in a second experiment; Tables S1 and S2), mainly because we previously demonstrated a cytokine-like function for TRX, another glutathionylated, releasable thiol-disulfide oxidoreductase (13). The proteomics results were validated by Western blot analysis or ELISA, to confirm LPS-induced PRDX2 secretion. We also immunoprecipitated PRDX2, followed by Western blot analysis with an anti-GSH antibody to confirm its glutathionylation.
Western blot analysis of the supernatants from RAW264 cells showed that LPS induces release of PRDX2 (Fig. 2A). Analysis of the corresponding cellular lysates showed that LPS does not increase the intracellular PRDX2 levels (Fig. 2B). In fact, there was a small decrease in intracellular levels of PRDX2, consistent with the hypothesis that LPS induces the release of preexisting PRDX2 and that intracellular PRDX2 levels are not rapidly replenished. The induction of PRDX2 release by LPS was dose-dependent and detectable at an LPS concentration as low as 0.01 ng/mL (Fig. 2C). Again, this was not attributed to induction of total intracellular PRDX2 (Fig. 2D). Time-course experiments showed that PRDX2 is released relatively late (24 h) after LPS stimulation (Fig. 2E).
Fig. 2.

LPS induces PRDX2 release. (A) Western blot of supernatants from RAW264.7 cells treated for 24 h with and without 100 ng/mL LPS. PRDX2 was measured in triplicate samples each derived from an independent well. (Upper) Western blot with anti-PRDX2. (Lower) Ponceau red staining. (B) Intracellular PRDX2 in the cell lysates of the experiment in A. (Upper) Western blot with anti-PRDX2. (Lower) Western blot with anti-GAPDH. (C) PRDX2 in supernatant from RAW264 cells treated for 24 h with different concentrations of LPS. (D) PRDX2 in the corresponding lysates. Percentages of viable cells, as mean ± SD of quadruplicate samples, were 98 ± 2% with 0.01 ng/mL LPS, 95 ± 7% with 0.1 ng/mL LPS, 92 ± 4% with 1 ng/mL LPS, 91 ± 3% with 10 ng/mL LPS, and 90 ± 2% with 100 ng/mL LPS. (E) Time course of PRDX2 release in supernatants from cells treated with 100 ng/mL LPS for 8, 16, or 24 h.
All of the electrophoreses in the foregoing experiments were performed under nonreducing conditions. It can be seen that the PRDX2 released was almost exclusively in the form of a dimer (∼40 kD), whereas intracellular PRDX2 was present both as a monomer and as a dimer. These results are consistent with the knowledge that PRDX2 can form disulfide-linked dimers that represent the catalytically active form of the protein (14).
To gain better insight into the release of PRDX2 from inflammatory cells, we next investigated whether its release is associated with induction of its transcription, and also whether PRDX2 is released by other cells, including primary cells. LPS-stimulated release of PRDX2 was not accompanied by induction of PRDX2 mRNA at either 4 h or 24 h after stimulation (Fig. 3A). In contrast, in the same 4-h or 24-h samples, mRNA encoding TNF-α, a classical inflammatory cytokine, increased dramatically (ratio to control, 20 ± 1.1 at 4 h and 29 ± 3.7 at 24 h; n = 3). We also investigated the release of PRDX2 by LPS-stimulated primary human macrophages and peripheral blood mononuclear cells (PBMCs) from different donors. These experiments, in which human PRDX2 was measured in the 24-h supernatant by ELISA, confirmed the induction of its release by LPS (Fig. 3B).
Fig. 3.

PRDX2 release in different experimental conditions. (A) PRDX2 mRNA in RAW264 cells at 4 h or 24 h after stimulation with 100 ng/mL LPS. PRDX2 mRNA expressed in arbitrary units (normalized to GAPDH as described in Materials and Methods), representing the fold change relative to one of the control samples at 4 h. Results are the mean ± SD of triplicate samples analyzed in duplicate. (B) Release of PRDX2 by human macrophages (Left) or PBMCs stimulated with 100 ng/mL LPS for 24 h. PRDX2 was measured by ELISA in triplicate samples of macrophages from four donors or PBMCs from five donors. Mean ± SD values for macrophages were control, 3.22 ± 1.92 ng/mL and LPS, 4.97 ± 2.69 ng/mL. Mean ± SD values for PBMCs were control, 2.90 ± 2.54 ng/mL and LPS, 8.10 ± 6.31 ng/mL. Increases were statistically significant for both macrophages and PBMCs (P < 0.05, t test for paired data). (C) Release of PRDX2 by PBMCs at physiological oxygen concentration. PBMCs were stimulated with 100 ng/mL LPS for 24 h under physiological (5%; Left) or atmospheric (20%; Right) oxygen levels. PRDX2 was measured by ELISA in triplicate PBMC samples from three donors. *P < 0.05; **P < 0.01; ***P < 0.001 vs. respective control (without LPS), Student t test. (D) PRDX2 released by HEK 293T cells stimulated with 50 ng/mL TNF-α for 24 h. Each lane represents an independent biological sample.
All of these experiments were performed in standard cell culture incubators kept at atmospheric oxygen concentration (20%). However, cells are normally exposed to physiological oxygen levels that are twofold to fourfold lower, and several immune responses are dependent on oxygen concentration (15). We have investigated whether atmospheric oxygen might have artifactually increased PRDX2 release because, for instance, oxidative stress might alter the redox state of PRDX2. We have compared the release of PRDX2 by PBMCs cultured under atmospheric (20%) or physiological (5%) oxygen concentration. As shown in Fig. 3C, oxygen concentration did not affect either basal PRDX2 release or the release in response to LPS. Finally, the release of PRDX2 was not restricted to LPS-stimulated monocytes/macrophages, but was observed in HEK 293 cells treated with TNF-α as well (Fig. 3D).
To prove that released PRDX2 is glutathionylated, we immunoprecipitated PRDX2 from the supernatant of LPS-stimulated RAW264 cells, then performed Western blot analysis with an anti-GSH antibody. Fig. 4 shows a glutathionylated band compatible with the PRDX2 dimer (indicated by an arrow), but nothing corresponding to the PRDX2 monomer. The same band was also visible on staining with anti-PRDX2 antibodies, thus confirming its identity as PRDX2. Of note, in the immunoprecipitated sample (lane 1), two bands were stained similarly by anti-PRDX2 antibodies, but the upper band was more intensely stained by anti-GSH antibodies, suggesting that the doublet may represent microheterogeneity owing to glutathionylation.
Fig. 4.

PRDX2 in supernatants from LPS-treated cells is glutathionylated. The sample was immunoprecipitated with anti-PRDX2. Lane 1, immunoprecipitate; lane 2, total supernatants before immunoprecipitation. (Left) Western blot with anti-GSH. (Right) Western blot with anti-PRDX2.
Proinflammatory Activity of Human Recombinant (hr)PRDX2.
We then investigated whether hrPRDX2 induces TNF-α production. As shown in Fig. 5A, PRDX2 at 10 µg/mL stimulated TNF-α release by RAW264 cells to a comparable extent as the danger signal HMGB1 at 1 µg/mL or LPS at 50 pg/mL. The hrPRDX2 preparation used was cleaned of potential LPS contamination using Detoxi-gel columns as described in Materials and Methods, and was found to be free of LPS contamination by the limulus amoebocyte lysate assay (limit of sensitivity, 0.06 EU endotoxin/mL). However, to completely rule out the possibility that residual LPS might account for the effect of hrPRDX2, we also tested the protein after preincubation with the LPS inhibitor polymixin B at 10 µg/mL. Polymixin B (Fig. 5A, closed bars), while inhibiting TNF-α release induced by LPS, did not affect that induced by hrPRDX2. The effect of hrPRDX2, like that of LPS, was augmented by preincubation of the cells for 18 h with 100 U/mL IFN-γ (Fig. 5B). PRDX2 also induced TNF-α production by human primary macrophages from four donors (Fig. 5C).
Fig. 5.

hPRDX2 induces TNF-α production. (A) hrPRDX2 was added at 10 µg/mL to RAW264 cells, and TNF-α production was measured 24 h later by ELISA in triplicate samples. Solid bars represent PRDX2 preincubated with polymixin B, as described in the text; open bars represent PRDX2 preincubated without polymyxin B. (B) TNF-α induction by hrPRDX2 in RAW cells pretreated with 100 U/mL IFN-γ or not pretreated. *P < 0.01 vs. control (A) or vs. PRDX2 alone (B). (C) hrPRDX2 was added to human primary macrophage cultures from four donors, and TNF-α production was measured by ELISA 24 h later in triplicate.
Thioredoxin-1 Is Secreted Along with PRDX2.
Because PRDX2 is a TRX peroxidase, we deemed it important to evaluate whether its substrate is released under the same experimental conditions. As shown in Fig. 6, TRX1 was also released by PBMCs from all donors tested, and LPS significantly augmented its release. As with PRDX2, oxygen concentration had no significant effect on TRX1 secretion.
Fig. 6.

LPS induces the release of TRX1 by PBMCs. Cells were stimulated with 100 ng/mL LPS for 24 h under physiological (5%; Left) or atmospheric (20%; Right) oxygen levels. TRX1 was measured by ELISA in triplicate PBMC samples from three donors. *P < 0.05; **P < 0.01; ***P < 0.001 vs. respective control (without LPS), Student t test.
Discussion
We document here that glutathionylated proteins, previously known to be mainly cytoplasmic (10, 11), are also released under inflammatory conditions. In particular, we found that LPS stimulates the release of PRDX2 in both human and mouse macrophages and in human PBMCs. PRDX2 is also released by HEK 293T cells treated with TNF-α, indicating that its secretion is also induced by a well-known inflammatory cytokine, not just in response to LPS.
The characteristics of PRDX2 production are completely different from those of classical proinflammatory cytokines. Unlike TNF-α, PRDX2 is constitutively expressed in the cells and is released on stimulation with LPS. Also in contrast to TNF-α, PRDX2 mRNA is not up-regulated by LPS. These features, and the kinetics of PRDX2 release shown in Fig. 2E, are similar to those of HMGB1, which is considered a late mediator of inflammation (16).
Moreover, like HMGB1, PRDX2 does not have a signal peptide, suggesting that its release does not occur via the classical secretory pathway. Because cysteine oxidation has been shown to be involved in the nucleocytoplasmic transport of HMGB1 (17), our findings suggest that the cysteine oxidation we have demonstrated enables the release of PRDX2. In fact, nonclassical release pathways are not uncommon among inflammatory mediators; for example, IL-1α, IL-1β, and TRX are known to be released through nonclassical pathways (18, 19).
Interestingly, whereas intracellular PRDX2 is both monomeric and dimeric, PRDX2 is released only as a dimer. Like other PRDXs, PRDX2 is known to form disulfide-linked homodimers during its catalytic cycle (14). Interestingly, glutathionylation seems to target a cysteine not involved in dimer formation. In fact, Western blot analysis with anti-GSH antibodies after immunoprecipitation for PRDX2 revealed glutathionylation of the dimer.
Like HMGB1, PRDX2 induces production of TNF-α (16); thus, both proteins may function to amplify signals of the TNF-α–induced inflammatory response. This is germane to a report indicating that PRDX1 has proinflammatory activity in vitro (20). Although the present study focuses on its release by macrophages stimulated with a classical inflammatory agent, LPS, which acts through TLR4, any necrotic cell will release PRDX2. In agreement with this, a recent paper reported that various PRDXs are released and mediate inflammation in mouse brain following ischemia induced by middle cerebral artery occlusion (21). All of these data suggest that PRDX2 may act as a danger signal, a term that identifies inflammatory molecules released by stressed or inflammatory cells (12, 22). Various studies have reported the presence of PRDXs in body fluids including plasma and bronchoalveolar lavage fluids, their secretion by cancer cells, and detection in sera from cancer patients, suggesting that PRDXs may be a link between inflammation and cancer (23).
PRDX2 is not the only oxidoreductase to be released and act as a short-range stimulus, i.e., as a cytokine. TRX is also released by monocytes (24), and we previously reported that extracellular TRX is a chemoattractant for inflammatory cells (13). A truncated form of TRX is also released and has immunomodulating activities (25).
The observation that TRX1 is released along with PRDX2 following LPS stimulation raises an interesting issue in terms of the mechanism of cytokine and chemokine-like actions of extracellular PRDX2 and TRX1. In fact, PRDX2 is a TRX peroxidase, using reduced TRX as the electron donor in the reduction of H2O2. Our findings show that both the enzyme (PRDX2) and the substrate (TRX1) are released by activated macrophages, also known to release H2O2 (26). Given that TRX is also an oxidoreductase, it can oxidoreduce surface thiols, thereby inducing dimerization or deoligomerization of receptors, possibly changing their structure and thus causing activation or inhibition in a cascade fashion, as outlined in Fig. 7. In fact, although TRX is often considered only a reducing enzyme, several studies have shown that it can oxidize protein thiols in an oxidizing environment and in the absence of TRX reductase. This has been demonstrated in yeast where TRX reductase was knocked out (27) and in bacteria where TRX was expressed in the periplasm (28). The presence of PRDX in the secretome, along with the absence of NADPH and the presence of H2O2, might favor the action of TRX as an oxidant of membrane proteins in target cells. Thus, depending on the presence and concentrations of thiol antioxidants and H2O2 in the secretome, TRX could act as an oxidizing or reducing agent.
Fig. 7.

Metabolic pathways of redox signaling by released PRDX2 and TRX1.
A number of cytokine receptors can be activated by disulfide-mediated dimerization. For instance, homodimerization of the erythropoietin receptor through disulfide linkage causes its activation (29), reduction of a labile disulfide bond of CD132 inhibits IL-2 signaling (30), and, finally, disulfide-linked receptor heterodimerization is essential for the signaling of IL-3, IL-5, and GM-CSF (31). We thus propose a mechanism by which TRX and PRDX2 could regulate immunity through oxidoreduction of membrane thiols/disulfides.
Other oxidoreductases are released during infection or inflammation and participate in a more complex metabolic pathway. We previously reported that xanthine oxidase, a superoxide-generating enzyme, is released into the circulation on injection of mice with LPS or IFN inducers, such as double-stranded RNA (32). One of the first cytokines identified, migration-inhibitory factor, has a TRX redox active domain (33) and is released as a cysteinylated protein (34). Protein disulfide isomerase, complexed with TRX, is present on the cell surface and associates with TNF receptors (35).
These enzymes that have cytokine-like activities after release can be termed “redoxkines” and may be important mediators of sterile inflammation in diseases associated with redox imbalance. Their redox state and oxidative posttranslational modifications, including glutathionylation, likely play roles in regulating their activity, stability, or release, thereby providing a mechanism for redox regulation of immunity. Many chronic inflammatory diseases (e.g., rheumatoid arthritis, Crohn’s disease, multiple sclerosis) critically involve cytokine activation and oxidative stress, and the pathways described here might be of importance and worth exploring for possibly defining new therapeutic approaches.
Materials and Methods
Reagents.
LPS, Escherichia coli 055:B5, was obtained from Sigma-Aldrich. HMGB-1 was a gift from K.J. Tracey (North Shore University Hospital, Manhasset, NY). Rat recombinant IFN-γ and human recombinant TNF-α were obtained from R&D Systems. Human recombinant PRDX2 was expressed in E. coli as described elsewhere (36) and cleared from LPS contamination using Detoxi-Gel columns (Pierce). Purified PRDX2 was then tested for LPS contamination using the limulus amoebocyte lysate assay (Pyrogent Plus; Lonza).
Cell Culture.
RAW264.7 cells were cultured in RPMI medium 1640 (PAA Laboratories) with 10% FBS (Life Technologies). GSH labeling and LPS stimulation were performed in serum-free Opti-MEM I medium (Life Technologies) (16). Cell viability was measured in 96-well microtiter plates using CellTiterBlue (Promega). HEK 293T cells were cultured in DMEM (PAA Laboratories) with 10% FBS (Life Technologies). Stimulation for 24 h with 50 ng/mL TNF-α was performed in serum-free DMEM and supernatants used for Western blot analysis.
Human peripheral blood was purchased from the National Health Service Blood and Transplant. All donors provided written informed consent. The study was approved by the local Research Governance and Ethics Committee. PBMC- and M-CSF–derived macrophages were prepared as described previously (37). Selected experiments were performed with PBMCs cultured at atmospheric (20%) or physiological (5%) oxygen concentration, as described elsewhere (15).
Identification of Glutathionylated Proteins and Biochemical Determination.
RAW264.7 cells (1.4 × 106 in 3 mL) were plated in six-well plates. One day later, the medium was changed to 1 mL of Opti-MEM, and BioGEE (Life Technologies) was added to a final concentration of 200 µM. One hour later, LPS was added at a concentration of 100 ng/mL unless specified otherwise. After 24 h, supernatant was collected, N-ethylmaleimide (NEM) was added to a final concentration of 40 mM to prevent thiol-disulfide exchange, and samples were clarified by centrifugation. Supernatants were then desalted and concentrated using 5-kDa Vivaspin columns (GE Healthcare). In some experiments, supernatant was analyzed by SDS/PAGE (12% acrylamide) under nonreducing conditions, followed by Western blot analysis. Biotinylated proteins were visualized using streptavidin peroxidase (Roche) at 1:25,000 dilution. For staining with anti-GSH, a monoclonal anti-GSH primary antibody (Virogen) was used at 1:1,000 dilution, followed by an anti-mouse secondary antibody (Stressgen) at 1:5,000 dilution.
Western blot analysis for PRDX2 was performed using a polyclonal anti-PRDX2 (Sigma-Aldrich) at 1:1,000 dilution, followed by an anti-rabbit secondary antibody (Sigma-Aldrich) at 1:100,000 dilution. When intracellular proteins were analyzed for PRDX2 by Western blot analysis, membranes were then stripped and reprobed with rabbit monoclonal anti-GAPDH antibodies (1:1,000 dilution; Cell Signaling Technology) as a loading control. A similar control was not possible for cell culture supernatants, being in serum-free medium; in that case, membranes were stained with Ponceau red before Western blot analyses. The only visible band is shown in the figures as a loading control. Blots were developed using the ECL WB Analysis System (GE Healthcare).
For the identification of glutathionylated proteins, desalted and concentrated supernatants were incubated with 70 µL of streptavidin beads (streptavidin immobilized on agarose CL-4B; Sigma-Aldrich), after which the beads were washed first with PBS and then with NH4CO3. The beads were then subjected to tryptic digestion, performed with 5 µL of 25 ng/mL trypsin and 45 µL of 25 mM NH4HCO3 on ice for 45 min, incubated overnight at 37 °C. The resulting tryptic peptides were extracted by 5% formic acid /50% acetonitrile and concentrated using a SpeedVac (Thermo Scientific). The tryptic peptide extract was analyzed by liquid chromatography-mass spectrometry using a reverse-phase chromatograph coupled to a tandem mass spectrometer. Peptide and proteins identifications were obtained using the database search engine SEQUEST (BioWorks version 3.3; Thermo Scientific) against an IPI mouse database (version 3.72).
Immunoprecipitation.
Supernatant from LPS-treated RAW264 cells, treated with NEM and concentrated as described above, was incubated with anti-PRDX2 antibody (Sigma-Aldrich) at 1:60 dilution at 4 °C for 4 h, followed by precipitation with 40 µL of protein G agarose (Sigma-Aldrich) for 18 h at 4 °C. Agarose beads were washed three times with PBS and then eluted by boiling in sample buffer (without reductants) for 4 min. Both the supernatant and the eluate were analyzed by SDS/PAGE (12% acrylamide) under nonreducing conditions followed by Western blot analysis with anti-GSH as described above. The membrane was then stripped and probed with anti-PRDX2.
Immunoassays for PRDX2, TRX1, and TNF-α.
When indicated, human PRDX2 was measured using an ELISA kit for human PRDX2 from Antibodies Online. Human TNF-α was assayed by ELISA, using a DuoSet Kit (R&D Systems). Human TRX1 was measured by ELISA as described elsewhere (24).
qRT-PCR.
Total RNA was extracted with TRIzol (Life Technologies), and RNA quality and concentration were measured with a NanoDrop ND-1000 spectrophotometer. Reverse-transcription and quantitative PCR (qPCR) were performed as reported previously (38) using Taqman gene expression assays (Applied Biosystems) for mouse PRDX2, TNF-α, and GAPDH (housekeeping). Relative gene expression was quantified using the comparative threshold cycle method (∆∆Ct), according to Applied Biosystems guidelines. Results were normalized to GAPDH expression and expressed as arbitrary units, representing gene expression versus one of the control samples at 4 h.
Supplementary Material
Acknowledgments
This work is dedicated to the memory of Leonard A. Herzenberg, 1931–2013. This work was supported by the Brighton and Sussex Medical School, the R.M. Phillips Charitable Trust, European Development Fund, Project TC2N, the Deutsche Forschungsgemeinschaft [DFG SFB593-N01 (to C.H.L.)], and the von Behring–Röntgen Foundation (to C.H.L.). P.C. was supported by a fellowship from the Institute Pasteur/Fondazione Cenci-Bolognetti, Rome. A preliminary account of this work was presented at the annual meeting of the Federation of American Societies for Experimental Biology, San Diego, CA, April 25, 2012. This work is part of a thesis submitted by S. Salzano in partial fulfillment of the requirements of the University of Brighton and the University of Sussex for the degree of Doctor of Philosophy by the Brighton and Sussex Medical School.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1401712111/-/DCSupplemental.
References
- 1.Freeman BA, Crapo JD. Biology of disease: Free radicals and tissue injury. Lab Invest. 1982;47(5):412–426. [PubMed] [Google Scholar]
- 2.McCord JM. Oxygen-derived free radicals in postischemic tissue injury. N Engl J Med. 1985;312(3):159–163. doi: 10.1056/NEJM198501173120305. [DOI] [PubMed] [Google Scholar]
- 3.Berlett BS, Stadtman ER. Protein oxidation in aging, disease, and oxidative stress. J Biol Chem. 1997;272(33):20313–20316. doi: 10.1074/jbc.272.33.20313. [DOI] [PubMed] [Google Scholar]
- 4.Ghezzi P. Role of glutathione in immunity and inflammation in the lung. Int J Gen Med. 2011;4:105–113. doi: 10.2147/IJGM.S15618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schafer FQ, Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med. 2001;30(11):1191–1212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- 6.Sen CK, Packer L. Antioxidant and redox regulation of gene transcription. FASEB J. 1996;10(7):709–720. doi: 10.1096/fasebj.10.7.8635688. [DOI] [PubMed] [Google Scholar]
- 7.Fratelli M, et al. Gene expression profiling reveals a signaling role of glutathione in redox regulation. Proc Natl Acad Sci USA. 2005;102(39):13998–14003. doi: 10.1073/pnas.0504398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ghezzi P. Regulation of protein function by glutathionylation. Free Radic Res. 2005;39(6):573–580. doi: 10.1080/10715760500072172. [DOI] [PubMed] [Google Scholar]
- 9.Ghezzi P. Protein glutathionylation in health and disease. Biochim Biophys Acta. 2013;1830(5):3165–3172. doi: 10.1016/j.bbagen.2013.02.009. [DOI] [PubMed] [Google Scholar]
- 10.Ghezzi P, et al. Protein glutathionylation: Coupling and uncoupling of glutathione to protein thiol groups in lymphocytes under oxidative stress and HIV infection. Mol Immunol. 2002;38(10):773–780. doi: 10.1016/s0161-5890(01)00114-6. [DOI] [PubMed] [Google Scholar]
- 11.Fratelli M, et al. Identification by redox proteomics of glutathionylated proteins in oxidatively stressed human T lymphocytes. Proc Natl Acad Sci USA. 2002;99(6):3505–3510. doi: 10.1073/pnas.052592699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gallucci S, Matzinger P. Danger signals: SOS to the immune system. Curr Opin Immunol. 2001;13(1):114–119. doi: 10.1016/s0952-7915(00)00191-6. [DOI] [PubMed] [Google Scholar]
- 13.Bertini R, et al. Thioredoxin, a redox enzyme released in infection and inflammation, is a unique chemoattractant for neutrophils, monocytes, and T cells. J Exp Med. 1999;189(11):1783–1789. doi: 10.1084/jem.189.11.1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schröder E, et al. Crystal structure of decameric 2-Cys peroxiredoxin from human erythrocytes at 1.7 Å resolution. Structure. 2000;8(6):605–615. doi: 10.1016/s0969-2126(00)00147-7. [DOI] [PubMed] [Google Scholar]
- 15.Atkuri KR, Herzenberg LA, Niemi AK, Cowan T, Herzenberg LA. Importance of culturing primary lymphocytes at physiological oxygen levels. Proc Natl Acad Sci USA. 2007;104(11):4547–4552. doi: 10.1073/pnas.0611732104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang H, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285(5425):248–251. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- 17.Hoppe G, Talcott KE, Bhattacharya SK, Crabb JW, Sears JE. Molecular basis for the redox control of nuclear transport of the structural chromatin protein Hmgb1. Exp Cell Res. 2006;312(18):3526–3538. doi: 10.1016/j.yexcr.2006.07.020. [DOI] [PubMed] [Google Scholar]
- 18.Rubartelli A, Bajetto A, Allavena G, Wollman E, Sitia R. Secretion of thioredoxin by normal and neoplastic cells through a leaderless secretory pathway. J Biol Chem. 1992;267(34):24161–24164. [PubMed] [Google Scholar]
- 19.Tarantini F, et al. The precursor but not the mature form of IL1alpha blocks the release of FGF1 in response to heat shock. J Biol Chem. 2001;276(7):5147–5151. doi: 10.1074/jbc.C000714200. [DOI] [PubMed] [Google Scholar]
- 20.Riddell JR, Wang XY, Minderman H, Gollnick SO. Peroxiredoxin 1 stimulates secretion of proinflammatory cytokines by binding to TLR4. J Immunol. 2010;184(2):1022–1030. doi: 10.4049/jimmunol.0901945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shichita T, et al. Peroxiredoxin family proteins are key initiators of post-ischemic inflammation in the brain. Nat Med. 2012;18(6):911–917. doi: 10.1038/nm.2749. [DOI] [PubMed] [Google Scholar]
- 22.Foell D, Wittkowski H, Roth J. Mechanisms of disease: A “DAMP” view of inflammatory arthritis. Nat Clin Pract Rheumatol. 2007;3(7):382–390. doi: 10.1038/ncprheum0531. [DOI] [PubMed] [Google Scholar]
- 23.Ishii T, Warabi E, Yanagawa T. Novel roles of peroxiredoxins in inflammation, cancer and innate immunity. J Clin Biochem Nutr. 2012;50(2):91–105. doi: 10.3164/jcbn.11-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sahaf B, Rosén A. Secretion of 10-kDa and 12-kDa thioredoxin species from blood monocytes and transformed leukocytes. Antioxid Redox Signal. 2000;2(4):717–726. doi: 10.1089/ars.2000.2.4-717. [DOI] [PubMed] [Google Scholar]
- 25.Pekkari K, Holmgren A. Truncated thioredoxin: Physiological functions and mechanism. Antioxid Redox Signal. 2004;6(1):53–61. doi: 10.1089/152308604771978345. [DOI] [PubMed] [Google Scholar]
- 26.Nathan CF, Root RK. Hydrogen peroxide release from mouse peritoneal macrophages: Dependence on sequential activation and triggering. J Exp Med. 1977;146(6):1648–1662. doi: 10.1084/jem.146.6.1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.García-Santamarina S, et al. Is oxidized thioredoxin a major trigger for cysteine oxidation? Clues from a redox proteomics approach. Antioxid Redox Signal. 2013;18(13):1549–1556. doi: 10.1089/ars.2012.5037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Debarbieux L, Beckwith J. The reductive enzyme thioredoxin 1 acts as an oxidant when it is exported to the Escherichia coli periplasm. Proc Natl Acad Sci USA. 1998;95(18):10751–10756. doi: 10.1073/pnas.95.18.10751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Watowich SS, et al. Homodimerization and constitutive activation of the erythropoietin receptor. Proc Natl Acad Sci USA. 1992;89(6):2140–2144. doi: 10.1073/pnas.89.6.2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Metcalfe C, Cresswell P, Barclay AN. Interleukin-2 signalling is modulated by a labile disulfide bond in the CD132 chain of its receptor. Open Biol. 2012;2(1):110036. doi: 10.1098/rsob.110036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stomski FC, et al. Identification of a Cys motif in the common beta chain of the interleukin 3, granulocyte-macrophage colony-stimulating factor, and interleukin 5 receptors essential for disulfide-linked receptor heterodimerization and activation of all three receptors. J Biol Chem. 1998;273(2):1192–1199. doi: 10.1074/jbc.273.2.1192. [DOI] [PubMed] [Google Scholar]
- 32.Ghezzi P, Bianchi M, Mantovani A, Spreafico F, Salmona M. Enhanced xanthine oxidase activity in mice treated with interferon and interferon inducers. Biochem Biophys Res Commun. 1984;119(1):144–149. doi: 10.1016/0006-291x(84)91630-9. [DOI] [PubMed] [Google Scholar]
- 33.Kleemann R, et al. Disulfide analysis reveals a role for macrophage migration inhibitory factor (MIF) as thiol-protein oxidoreductase. J Mol Biol. 1998;280(1):85–102. doi: 10.1006/jmbi.1998.1864. [DOI] [PubMed] [Google Scholar]
- 34.Watarai H, et al. Posttranslational modification of the glycosylation inhibiting factor (GIF) gene product generates bioactive GIF. Proc Natl Acad Sci USA. 2000;97(24):13251–13256. doi: 10.1073/pnas.230445397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Söderberg A, Hossain A, Rosén A. A protein disulfide isomerase/thioredoxin-1 complex is physically attached to exofacial membrane tumor necrosis factor receptors: Overexpression in chronic lymphocytic leukemia cells. Antioxid Redox Signal. 2013;18(4):363–375. doi: 10.1089/ars.2012.4789. [DOI] [PubMed] [Google Scholar]
- 36.Hanschmann EM, et al. Both thioredoxin 2 and glutaredoxin 2 contribute to the reduction of the mitochondrial 2-Cys peroxiredoxin Prx3. J Biol Chem. 2010;285(52):40699–40705. doi: 10.1074/jbc.M110.185827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sacre S, Medghalchi M, Gregory B, Brennan F, Williams R. Fluoxetine and citalopram exhibit potent antiinflammatory activity in human and murine models of rheumatoid arthritis and inhibit Toll-like receptors. Arthritis Rheum. 2010;62(3):683–693. doi: 10.1002/art.27304. [DOI] [PubMed] [Google Scholar]
- 38.Mengozzi M, et al. Erythropoietin-induced changes in brain gene expression reveal induction of synaptic plasticity genes in experimental stroke. Proc Natl Acad Sci USA. 2012;109(24):9617–9622. doi: 10.1073/pnas.1200554109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.