

Significance
Autoimmune diabetes is caused by the lymphocyte-mediated destruction of the pancreatic insulin-producing β cells. The inflammatory niche surrounding β cells prior to diabetes onset provokes death-inducing molecular changes in these cells. Unknown molecular pathways related to β-cell viability that are altered by inflammation in vivo, and therefore potential therapeutic targets, remain to be identified, because most of the previous studies took in vitro approaches. Here, we report that cyclin D3, classically related to cell proliferation, is targeted in autoimmune diabetes and exerts a protective role on β cells by promoting their survival and fitness without affecting proliferation. These findings unveil a dual, cell cycle-independent role of cyclin D3 with high potential in the areas of autoimmunity and metabolism.
Abstract
Type 1 diabetes is an autoimmune condition caused by the lymphocyte-mediated destruction of the insulin-producing β cells in pancreatic islets. We aimed to identify final molecular entities targeted by the autoimmune assault on pancreatic β cells that are causally related to β cell viability. Here, we show that cyclin D3 is targeted by the autoimmune attack on pancreatic β cells in vivo. Cyclin D3 is down-regulated in a dose-dependent manner in β cells by leukocyte infiltration into the islets of the nonobese diabetic (NOD) type 1 diabetes-prone mouse model. Furthermore, we established a direct in vivo causal link between cyclin D3 expression levels and β-cell fitness and viability in the NOD mice. We found that changes in cyclin D3 expression levels in vivo altered the β-cell apoptosis rates, β-cell area homeostasis, and β-cell sensitivity to glucose without affecting β-cell proliferation in the NOD mice. Cyclin D3-deficient NOD mice exhibited exacerbated diabetes and impaired glucose responsiveness; conversely, transgenic NOD mice overexpressing cyclin D3 in β cells exhibited mild diabetes and improved glucose responsiveness. Overexpression of cyclin D3 in β cells of cyclin D3-deficient mice rescued them from the exacerbated diabetes observed in transgene-negative littermates. Moreover, cyclin D3 overexpression protected the NOD-derived insulinoma NIT-1 cell line from cytokine-induced apoptosis. Here, for the first time to our knowledge, cyclin D3 is identified as a key molecule targeted by autoimmunity that plays a nonredundant, protective, and cell cycle-independent role in β cells against inflammation-induced apoptosis and confers metabolic fitness to these cells.
Apoptosis is the major cause of pancreatic β-cell death in autoimmune diabetes (type 1 diabetes, or T1D) (1–9). The inflammatory infiltration into pancreatic islets in T1D provokes a large number of cell death-inducing molecular changes in β cells. Proinflammatory cytokines trigger the activation and translocation of transcription factors into the nucleus, the induction of target gene transcription (10), and the posttranslational modification of proteins in β cells (3, 11).
Previously, several approaches have been taken to address differential gene expression in β cells owing to proinflammatory cytokines (10, 12, 13). Nonetheless, those studies focused on molecular targets, the expression of which was altered in islets or β-cell lines upon in vitro incubation with proinflammatory cytokines (e.g., IL-1β, IFN-γ, and TNF-α) (10, 12, 13). That type of study does not account for the in vivo microenvironment to which β cells are exposed in animals with autoimmune-prone genetic backgrounds, such as the nonobese diabetic (NOD) mouse model, which surely comprises more factors than the above-mentioned cytokines. Therefore, potential targets involved in β-cell death remain to be identified.
We found that in NOD mice, cyclin D3 (CcnD3 gene) mRNA expression was impaired in endocrine cells from heavily infiltrated pancreatic islets (Table S1). The cyclin D3 promoter contains binding sequences for the NF-κB transcription factor (14). IL-1β and TNF-α are key cytokines causing β-cell damage in T1D, and NF-κB is linked to the action of both cytokines (3). Therefore, cyclin D3 could be down-regulated in β cells by the action of NF-κB.
Cell-cycle entry in mammal eukaryotic cells is coordinated by D-type cyclins D1, D2, and D3 (15, 16). Cyclins D1 and D3 are involved in cell-cycle progression in a cyclin-dependent kinase (CDK)-dependent manner, in CDK-independent activation of transcription, and in metabolic control (17–21). Therefore, cyclin D3 might play unknown roles in pancreatic β cells, which could explain why cyclin D3 down-regulation is associated with T1D.
Cyclin D3-deficient mice with a non-autoimmune-prone genetic background do not exhibit pancreatic β-cell hypoplasia, which implies that cyclin D3 is not required for β-cell mass generation in adult individuals (22, 23). Moreover, two previous reports using other non-autoimmune-prone mouse strains did not detect significant cyclin D3 expression in pancreatic islets (23, 24). To address whether a causal link exists between cyclin D3 down-regulation in autoimmune diabetes in NOD mice upon insulitic attack and autoimmune diabetes, we first assessed cyclin D3 expression levels in pancreatic β cells from the autoimmune-prone NOD and NOD/SCID genetic backgrounds. Next, we developed NOD mice ubiquitously deficient in cyclin D3, NOD mice that overexpressed cyclin D3 locally in β cells, and a combination of both (i.e., cyclin D3-deficient NOD mice overexpressing cyclin D3 solely in β cells). We characterized these mice with regard to autoimmune diabetes and β-cell function.
Results
Cyclin D3 Is Down-Regulated in β Cells by Insulitic Attack Without Impairing β-Cell Proliferative Activity.
Previous results from microarray studies from our laboratory (Table S1) found that cyclin D3 mRNA expression levels were almost threefold reduced in pancreatic endocrine cells from prediabetic (11-wk-old) NOD mice compared with those in the lymphocyte-deficient (25) NOD/SCID genetic background. Prediabetic NOD mice exhibit heavily infiltrated pancreatic islets, whereas islets from age-matched NOD/SCID mice are infiltration-free. We confirmed these results by quantitative real-time PCR (qRT-PCR) (Fig. 1A) and found that pancreatic endocrine islet cells from predibetic NOD exhibited a dramatic reduction in cyclin D3 mRNA expression levels compared with those from age-matched NOD/SCID (1.00 ± 0.95 vs. 27.98 ± 18.30, respectively). To address whether cyclin D3 expression was also down-regulated at the protein level we isolated islets from 6-wk-old NOD (mild infiltration), 11-wk-old NOD (heavy infiltration), 6-wk-old and 11-wk-old NOD/SCID (no infiltration) mice, and 11-wk-old NOD/SCID T mice (nondiabetic NOD/SCID mice that had been adoptively transferred 10 million NOD total spleen cells from prediabetic NOD donors 2 wk earlier to promote islet infiltration in these otherwise infiltration-free recipients) and assessed cyclin D3 protein expression by flow cytometry (Fig. 1B). We used CD45 as a hematopoietic marker to exclude the immune cells infiltrating islets and the Glut-2 glucose transporter as a β-cell marker.
Fig. 1.

In NOD mice, cyclin D3 mRNA and protein expression are down-regulated by islet infiltration in a dose-dependent manner, without affecting the proliferative activity of β cells (A) Pancreatic islet cells from either 11-wk-old NOD mice (N; n = 4) and 11-wk-old NOD/SCID mice (NS; n = 4) were extracted, and the CD45− cell subset (endocrine cells) was selected by magnetic sorting to perform qRT-PCR analysis to assess cyclin D3 expression. (B) Pancreatic islet cells were extracted from 6- (N-6w; n = 33) and 11-wk-old (N-11w, n = 9) NOD mice, 6-wk-old (NS-6w, n = 13) NOD/SCID mice, 11-wk-old (NS-11w, n = 4) NOD/SCID mice, and 11-wk-old NOD/SCID mice that had received 10 million adoptively transferred NOD total splenocytes at 9 wk of age (NST-11w; n = 11). The intensity of median fluorescence (IMF) of cyclin D3 staining on β (CD45−Glut-2+) cells was measured by flow cytometry. All comparisons were performed relative to the N-6w values; otherwise comparisons are plotted as horizontal lines above the bars being statistically different. (C) Six-week-old NOD/SCID mice were adoptively transferred i.v. with 107 NOD total spleen cells (T, n = 6) or injected with saline solution (S.S., n = 7), and the endocrine cells were sorted (CD45− fraction) from the total pool of islet cells by magnetic separation. Cyclin D3 (CcnD3) mRNA expression levels were addressed by qRT-PCR analysis. (D) Assessment of cyclin D3 expression levels in β cells from NOD/SCID mice that were adoptively transferred with different amounts (×106) of NOD total splenocytes (TSC; numbers of recipient mice per condition were as follows: 0, n = 6; 1, n = 6; 3, n = 7; 5, n = 8, and 10, n = 11) 2 wk before the islet extraction at 11 wk of age. The IMF of cyclin D3 staining on β cells was measured by flow cytometry. All comparisons were related to TSC (0). Linear regression was performed according to the equation y = −101.69 + 4.64x, P = 0.0004. (E) Proliferation assessment in β cells from 11-wk-old NOD/SCID mice that had received adoptive transfers of different amounts of total splenocytes 2 wk before the islet extraction, measured as percentages of CD45−Glut-2+ Ki67+ cells. The numbers of mice used per condition were as follows: 0, n = 7; 1, n = 7; 3, n = 6; 5, n = 4, and 10, n = 9. Data are represented as the mean ± SEM in A, B, C, and E; in D, the data are plotted as the individual obtained values. In A the value of the N group was used as reference and set at 1; in C the value of the S.S. group was used as reference and set at 1. *P ≤ 0.05, **P ≤ 0.01.
The cyclin D3 expression levels in β (CD45−Glut-2+) cells from 11-wk-old NOD mice were 1.3 times lower than those in 6-wk-old NOD mice (77.18 ± 9.24 vs. 103.3 ± 4.89, respectively) and 2.3 times lower than those observed in age-matched NOD/SCID mice (77.18 ± 9.24 vs. 177.6 ± 38.00, respectively). No significant differences were detected between β cells from either 6- or 11-wk-old NOD/SCID mice (Fig. 1B).
To confirm the causal relationship between islet infiltration and cyclin D3 down-regulation we performed adoptive transfer experiments in which 6-wk-old NOD/SCID mice were adoptively transferred i.v. with either 107 NOD total spleen cells or injected with saline solution and 2 wk later cyclin D3 RNA expression levels in islet endocrine cells (CD45− fraction) were assessed by qRT-PCR (Fig. 1C). An approximate sixfold reduction in the CcnD3 mRNA expression levels was observed upon adoptive transfer of splenocytes into NOD/SCID recipients compared with the saline control group (1.00 ± 0.608 vs. 0.165 ± 0.038, respectively). Furthermore, we addressed whether the cyclin D3 protein levels in pancreatic β cells were also affected by leukocyte infiltration in a dose-dependent manner. We found that there was a significant inverse correlation between the cyclin D3 protein expression levels and the dose of NOD splenocytes that were adoptively transferred into the NOD/SCID recipients (Fig. 1D).
Regarding the other D-type cyclins, D1 and D2, we found that cyclin D1 mRNA was equally expressed in pancreatic endocrine cells from both 11-wk-old NOD and NOD/SCID islets, whereas cyclin D2 expression in NOD/SCID endocrine cells was significantly higher (around 14 times higher) than in those from age-matched NOD mice (1.01 ± 0.17 in NOD vs. 13.78 ± 5.56 in NOD/SCID) (Fig. S1 A–C). However, contrary to what occurs with cyclin D3, neither cyclin D1 nor D2 mRNA expression levels were altered in islet endocrine cells from NOD/SCID mice upon adoptive transfer of NOD splenocytes (Fig. S1 D–F). This result evidences the uniqueness of cyclin D3 among D-type cyclins in being the only one down-regulated by inflammation.
To determine whether cyclin D3 down-regulation hampered the NOD β-cell replicative activity, we evaluated the proliferation rates of β cells (CD45−Glut-2+Ki67+) from 11-wk-old NOD/SCID mice that received different doses of adoptively transferred splenocytes from NOD donors at 9 wk of age (Fig. 1E). The apparent differences in β-cell proliferation rates among the experimental groups were not statistically significant.
Cyclin D3 Overexpression Protects NIT-1 β Cells from Spontaneous and Cytokine-Induced Apoptosis.
To address whether cyclin D3 overexpression could protect the NIT-1 NOD insulinoma cell line from apoptosis upon exposure to proinflammatory cytokines we stably transfected NIT-1 cells with a plasmid (pBSKNeoRIP-mCcnD3-Eα) that contained the mouse cyclin D3 cDNA under the control of the rat insulin promoter (RIP) (26) and a neomycin selection marker. As a mock control, we used NIT-1 cells transfected with the empty pBSKNeo vector. Cyclin D3 expression was assessed by flow cytometry in stably transfected NIT-1 cells (Fig. S2A), and the cells were assayed for early apoptosis (Fig. S2B). Interestingly, NIT-1 cells that overexpressed cyclin D3 were more resistant to both basal and IL-1β–induced apoptosis compared with the mock pBSKNeo transfectants (Fig. S2B). In contrast, neither IFN-γ nor IFN-γ + IL-1β–induced NIT-1 cell apoptosis were affected by cyclin D3 overexpression. Cyclin D3 overexpression only increased NIT-1 cell proliferation in the presence of IFN-γ; however, at basal condition or in the presence of IL-1β (Fig. S2C) there was no effect.
Cyclin D3 Deficiency Enhances β-Cell Apoptosis and Diabetes in Vivo, Whereas Cyclin D3 Overexpression in β Cells Protects Against Both of Them.
We further investigated the role of cyclin D3 in NOD β cells in vivo to ascertain whether cyclin D3 deficiency would enhance β-cell apoptosis, hence exacerbating diabetes, and conversely whether cyclin D3 overexpression in β cells would protect from diabetes.
First, we obtained cyclin D3-deficient NOD mice (N-KO) (Fig. 2A and Fig. S3A; see Fig. S3B for the assessment of the anti-mouse cyclin D3 antibody binding specificity). The lack of cyclin D3 was associated with a smaller β-cell area per islet that was nearly half the size of that in WT (N-WT) littermate mice (6,533 ± 1,286 μm2 per islet in N-KO vs. 13,797 ± 1,437 μm2 per islet in N-WT; Fig. 2B). We also introduced the CcnD3 null mutation into the NOD/SCID strain (NS-KO).The deficiency in cyclin D3 in the NOD/SCID background (NS-KO) also was associated to a smaller β-cell area compared with that exhibited by NS-WT islets (3,937 ± 478.3 in the NS-KO compared with 6,100 ± 647.5 in the NS-WT littermates) (Fig. 2B). Interestingly, the β-cell area of NS-WT islets was also significantly smaller than that observed in N-WT (6,100 ± 647.5 in NS-WT vs. 13,793 ± 1,437 in N-WT) (Fig. 2B).
Fig. 2.

Generation of the N-KO mouse model. (A) Pancreatic islet cells were isolated from 6-wk-old N-WT (n = 24) and N-KO (n = 12) mice. The IMF of cyclin D3 staining on β cells was measured by flow cytometry and was expressed as the percent relative to the IMF detected in β cells from N-KO islets, which was set as 1%. (B) β-cell area per islet in 6-wk-old mice (the number of islets analyzed was N-WT, n = 104; N-HTZ, n = 54; N-KO, n = 58; NS-WT, n = 101; and NS-KO, n = 101). (C) Body weight of 12-wk-old mice (N-WT, n = 23; N-HTZ, n = 12; and N-KO, n = 9) is represented. (D) Representative picture showing respective body sizes from either 6-wk-old N-KO (Left) or N-WT (Right) mice individuals. (E) Pancreas weight and (F) pancreas-to-body weight ratio of 10- to 12-wk-old NS-WT, n = 4; NS-HTZ, n = 5; and, NS-KO, n = 4. Data are represented as mean ± SEM in A, C, E, and F. Statistical comparisons are referred to the N-WT values in A–C and NS-WT in E and F; otherwise, comparisons are plotted as horizontal lines above the bars being statistically different. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
Body weight was also affected by cyclin D3 expression levels (Fig. 2 C and D). However, pancreas weight was not diminished in the NS-KO mice compared with NS-WT littermates (Fig. 2E). The pancreas-to-body weight ratio was significantly augmented in both NS-HTZ (NOD/SCID mice heterozygous for the deficiency in cyclin D3) and NS-KO, compared with NS-WT littermates, owing to the smaller body size exhibited by both genotypes (Fig. 2F).
We assessed pancreatic lipase (PL) mRNA expression levels in the exocrine pancreatic tissue from 8- to 12-wk-old NS-WT and NS-HTZ mice and we observed that PL expression was up-regulated in exocrine pancreas from NS-HTZ mice (Fig. S4).
We studied the spontaneous diabetes incidence in N-KO mice (Fig. 3A) and found that N-KO mice developed exacerbated diabetes, characterized by an earlier diabetes onset and higher cumulative disease incidence (89.3%) compared with N-WT littermates (64.5%), whereas N-HTZ (NOD mice heterozygous for the deficiency in cyclin D3) presented an intermediate phenotype, characterized by an increased cumulative disease incidence (87.5%) without accelerated disease onset (Fig. 3A). Furthermore, to assess both the autoimmune cause of the diabetic phenotype and the severity of the insulitic infiltrate, we scored the infiltration degree associated with either the N-WT or the N-KO genotype in 6-wk-old mice (Fig. 3B). We observed that the N-KO genotype, along with exhibiting the worst diabetes course (Fig. 3A), also exhibited a more severe insulitic assault (Fig. 3B). No differences in β-cell proliferation were associated with a lack of cyclin D3 expression in the β cells (Fig. 3C).
Fig. 3.
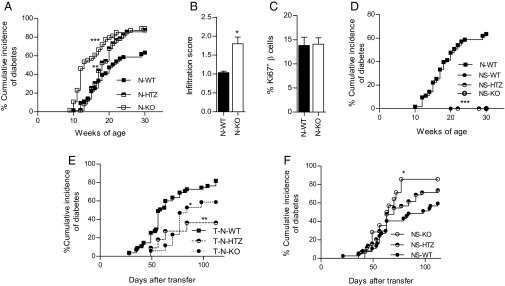
Lack of cyclin D3 expression exacerbates T1D onset and insulitis by rendering β cells more susceptible to autoimmune attack, without altering the β-cell proliferation rates. (A) The cumulative diabetes incidence in N-KO (n = 56), N-HTZ (n = 62), and N-WT (n = 63) mice was assessed. (B) Pancreata from 6-wk-old mice (n = 4 mice per experimental group) were extracted and levels of infiltration scored. (C) Proliferative activity was assessed in pancreatic β cells. The percentages of proliferating CD45−Glut2+Ki67+ β cells from 6-wk-old N-WT (n = 24) and N-KO (n = 10) mice were plotted. (D) The cumulative incidence of spontaneous diabetes in N-WT (n = 63), NS-WT (n = 9), NS-HTZ (n = 11), and NS-KO (n = 3) mice was monitored. (E) NOD/SCID recipient mice (3 to 5 wk old) were adoptively transferred with 10 million total splenocytes from N-KO (T-N-KO, n = 17), N-HTZ (T-N-HTZ, n = 11), or N-WT (T-N-WT, n = 26) donors. (F) NS-KO (n = 15), NS-HTZ (n = 44), and NS-WT (n = 37) recipient mice (3 to 5 wk old) were adoptively transferred with 10 million total splenocytes from NOD donors. The cumulative incidence of adoptively transferred diabetes is plotted. Data are shown as the mean ± SEM in B and C and as the percentages of cumulative diabetic mice at a particular time point in A, D, E, and F. In A and D the comparisons are referred to the N-WT group, in E to the T-N-WT group, and in F to the NS-WT group. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
In the absence of a NOD lymphocytic repertoire, the deficiency in cyclin D3 expression did not induce spontaneous diabetes in NOD/SCID mice (Fig. 3D), thus implying that lymphocytic infiltration triggers a mechanism(s) in addition to cyclin D3 down-regulation that is also required for diabetes onset.
To exclude the possibility that cyclin D3 deficiency in the immune compartment was responsible for the exacerbated diabetes observed in N-KO mice, we performed adoptive transfer experiments with 10 million total splenocytes from N-KO, N-HTZ, and N-WT donor mice into NOD/SCID recipient mice (Fig. 3E). The diabetes incidence was monitored for 16 wk after transfer, and we observed that splenocytes from N-KO donors were less diabetogenic than those from N-WT donors (58.8% vs. 81.6%; Fig. 3E). Therefore, cyclin D3 deficiency in the immune system did not account for the exacerbated diabetes observed in the N-KO mice.
To further restrict the role of cyclin D3 deficiency to β cells, we performed reverse adoptive transfers wherein total splenocytes from NOD donors were injected into NS-KO, NS-HTZ, or NS-WT mice recipients. The diabetes incidence was monitored for 16 wk after transfer (Fig. 3F), and in accordance to our previous results the NS-KO mice were more susceptible to diabetes onset than their NS-WT littermates (80% vs. 59.5%, respectively), confirming that cyclin D3 expression in β cells is relevant to their viability upon exposure to the autoimmune insult.
Because N-HTZ and N-KO exhibited very similar phenotypes, we assessed whether cyclin D3 hemideficiency in the NS-HTZ β cells had any effect on the expression of the other two D-type cyclins in islet cells by qRT-PCR (Fig. S5). We chose the NOD/SCID background to avoid the insulitis occurring in the NOD background that was likely to alter the expression results obtained. The hemideficiency of cyclin D3 caused a sixfold reduction in cyclin D1 mRNA expression levels in islet cells (1.00 ± 0.43 in NS-WT vs. 0.1575 ± 0.028 in NS-HTZ), whereas it had no effect on cyclin D2 expression.
Five transgenic NOD founder lines were produced by directly microinjecting the RIPCcnD3Eα transgene into fertilized NOD oocytes with the goal of overexpressing cyclin D3 in the β cells alone. Among these five transgenic lines (6876, 6877, 6880, 6889, and 6896), only founder line 6896 exhibited significant cyclin D3 protein overexpression in the β cells compared with transgene negative littermates (Fig. 4A). Hence, only transgenic line 6896 was used for further experiments (N-TG, transgene positive individuals; N-WT, transgene-negative littermates).
Fig. 4.

Generation of NOD mice that overexpress cyclin D3 in β cells (NODRIPCcnD3). (A) Pancreatic islets cells from 6-wk-old CcnD3 transgenic mice were stained to assess cyclin D3 expression levels in β cells by flow cytometry. The IMF of cyclin D3 staining was determined (number of different mice used per line: 6876-WT, n = 11; 6876-TG, n = 16; 6877-WT, n = 16; 6877-TG, n = 5; 6880-WT, n = 28; 6880-TG, n = 21; 6889-WT, n = 10; 6889-TG, n = 5; 6896-WT, n = 22; and 6896-TG, n = 13). (B) β-cell area per islet (the number of islets analyzed was N-TG, n = 94; N-WT, n = 104; N-HTZ, n = 54; N-KO, n = 58; NS-WT, n = 101; and NS-KO, n = 101). (C) Representative immunofluorescence images of pancreatic sections from 6-wk-old 6896 founder line N-TG or N-WT mice, nuclei (blue), cyclin D3 (red), and insulin (green). (Scale bar, 50 μm.) Data are represented as the mean ± SEM in A and B. The comparisons took as reference the values obtained in the transgene negative group for each founder line (A) or the N-WT group (B); otherwise, comparisons are plotted as horizontal lines above the bars being statistically different. *P ≤ 0.05, ***P ≤ 0.001.
No differences were observed regarding the β-cell area per islet (Fig. 4B) between the N-TG and N-WT mice. In Fig. 4B the β-cell areas of all of the genotypes studied is represented. Cyclin D3 and insulin expression were examined by immunofluorescence staining and confocal microscopy (Fig. 4C). In N-TG mice cyclin D3 was highly abundant in the cytoplasm.
The kinetics of disease onset was delayed in N-TG individuals, implying a protective role for cyclin D3 against spontaneous diabetes (Fig. 5A). There was no significant difference either in the insulitis scores (Fig. 5B) or in β-cell proliferation associated with cyclin D3 overexpression in the β cells (Fig. 5C). The restoration of cyclin D3 expression solely in β cells from N-KO mice had no effect on the body weight (Fig. 5D), but it significantly diminished the basal glycemia values (Fig. 5E).
Fig. 5.

Cyclin D3 overexpression only in pancreatic β cells protects NOD mice from T1D and confers protection against apoptosis and T1D to β cells from N-KO mice. (A) The cumulative diabetes incidence was monitored in N-TG (n = 29) and N-WT (n = 30) mice. (B) Pancreata from 6-wk-old mice (n = 4 for each, N-TG and N-WT) were used to assess the level of infiltration. (C) Islet cells from 6-wk-old N-WT (n = 12) and N-TG (n = 5) mice were stained to assess β-cell proliferative activity; the percentages of CD45−Glut2+Ki67+ proliferating cells are plotted. (D) Body weight of 12-wk-old mice (N-TG, n = 13; N-KO-TG, n = 12; and N-KO-WT, n = 9) is represented. (E) Nondiabetic, 11-wk-old mice (N-WT, n = 21; N-TG, n = 13; N-KO, n = 11; and N-KO-TG, n = 3) were fasted overnight, administered anesthesia, and glycemia was measured. (F) Either 3- to 5-wk-old NS-WT (n = 37), NS-KO-TG (n = 11), or NS-KO-WT (n = 15) mice were adoptively transferred with 10 million total splenocytes from NOD donors and diabetes onset was monitored. (G) Comparative graph summarizing the different cumulative diabetes incidences relative to cyclin D3 expression levels in β cells (N-WT, n = 63; N-HTZ, n = 62; N-KO, n = 56; N-TG, n = 29; NS-WT, n = 9; NS-HTZ, n = 11; and NS-KO, n = 3). (H) TUNEL assessment of apoptotic β cells was performed on pancreatic sections from 6-wk-old N-KO (n = 4), N-TG (n = 4), and N-WT (n = 4) mice and quantified using confocal microscopy. The numbers of TUNEL-positive nuclei per insulin-positive area were measured. Data are represented as mean ± SEM in B–E and H and as percentages of diabetic mice at a particular time point (cumulative diabetes incidence) in A, F, and G. In A–C, E, G, and H the comparisons were performed taking the N-WT values as reference; in D the N-TG values and in F the NS-KO values were taken as reference, respectively, to perform statistical comparisons (otherwise comparisons are plotted as horizontal lines above the bars being statistically different). *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
To fully confirm the causal relationship between cyclin D3 deficiency and β-cell death in T1D, we introduced the 6896 transgene into the NS-KO mice (NS-KO-TG). This experimental approach allowed us to examine whether the restoration of cyclin D3 expression limited to β cells from NOD/SCID mice fully deficient in cyclin D3 protected them from adoptively transferred diabetes. We transferred 10 million splenocytes from NOD mice into individuals with the NS-KO-WT and NS-KO-TG genotypes and monitored the diabetes incidence (Fig. 5F). Diabetes incidence exhibited by NS-KO-TG mice (45.5%) was approximately half of that shown by NS-KO-WT littermates (80%) (Fig. 5F).
A summary of the cumulative incidence of spontaneous diabetes onset in the different genotypes studied is shown in Fig. 5G. Those NOD genotypes with the lowest cyclin D3 expression levels in β cells (N-KO and N-HTZ) exhibited the most severe diabetic phenotype, whereas the genotype with the highest cyclin D3 expression levels (N-TG) was associated to the mildest diabetic phenotype. Based on our results, we concluded that cyclin D3 plays a relevant role in the promotion of β-cell survival that does not depend on changes in β-cell proliferation.
We further explored whether cyclin D3 acted to protect β cells from apoptosis. We performed a TdT UTP nick end labeling (TUNEL) staining assay to detect late apoptotic events in pancreatic sections from 6-wk-old N-WT, N-KO, and N-TG mice (Fig. 5H). We observed the highest apoptotic levels in N-KO β cells (1.6-fold increase compared with N-WT β cells) and the lowest apoptotic rate in N-TG β cells (1.9-fold decrease compared with N-WT β cells). This finding correlates with the spontaneous diabetes incidence exhibited by these three genotypes.
Furthermore, because apoptotic cells are naturally removed by infiltrating phagocytes, N-KO islets should bear the highest ratio per square millimeter of phagocytes compared with the other genotypes studied. To assess the level of phagocyte infiltration in N-KO islets, we chose as phagocyte markers Mac-3 (expressed in macrophages) and CD11b (expressed in both types of phagocytes: macrophages and activated neutrophils and also in other immune cell subsets such as natural killer cells and dendritic cells). We examined the number of CD11b+ and Mac-3+ cells per square millimeter of β-cell area in 6-wk-old mice with the N-WT, N-KO, and N-TG genotypes (Fig. S6). We found that, as expected, N-KO islets exhibited the largest Mac-3+ load, followed by N-TG islets, whereas N-WT had the smallest load. The reported wide heterogeneity in cell types expressing CD11b that also comprehend nonphagocytic cell subsets could explain the lack of differences found for CD11b staining in the N-KO islets compared with N-WT islets.
This finding confirmed a dose-dependent role of cyclin D3 in the prevention of β-cell apoptosis.
Cyclin D3 Causes a Significant Improvement in Glucose Responsiveness in Vivo.
We examined the effects of cyclin D3 perturbation on glucose tolerance tests in vivo. To assess the in vivo β-cell fitness in N-KO vs. N-WT mice, we performed i.p. glucose tolerance tests (IPGTTs) on nondiabetic 11-wk-old mice.
N-KO mice showed deficient responses to glucose at 15 and 30 min poststimulus compared with N-WT littermates (Fig. 6A). This result very probably reflects the more advanced state of infiltration of the N-KO islets compared with those from N-WT littermates. The basal blood glucose levels in 11-wk-old N-KO mice were significantly lower than those observed in the N-WT littermates (55.91 ± 5.8 in the N-KO vs. 81.2 ± 3.77 in the N-WT) (Fig. 6B, Upper), despite exhibiting the same basal insulinemia values (Fig. 6B, Lower).
Fig. 6.

The loss of cyclin D3 leads to impaired β-cell responses to blood glucose in NOD mice, whereas cyclin D3 overexpression in β cells improves glucose responsiveness in vivo. Nondiabetic 11-wk-old N-KO (n = 11), N-WT (n = 23), N-TG (n = 13), and N-WT (n = 20) mice were intraperitoneally administered glucose and glycemia (A and C) and insulinemia (E and F) were measured at different time points. As a negative control, a group of mice was submitted only to the anesthesia (N-WT-P) without any glucose stimulation (n = 3). In A, B, and E, results are shown for the N-KO and N-WT mice. In C, D, and F, results are shown for the N-TG and N-WT mice. (B) Nondiabetic 11-wk-old NOD-KO and N-WT mice were fasted overnight and administered anesthesia and glycemia (Upper: N-KO, n = 11 and N-WT, n = 21) and insulinemia (Lower: N-KO, n = 6 and N-WT, n = 9) were measured. (D) Nondiabetic 11-wk-old N-TG and N-WT mice were fasted overnight and administered anesthesia and glycemia (Upper: N-WT, n = 20 and N-TG, n = 13) and insulinemia (Lower: N-WT, n = 6 and N-TG, n = 7) were measured. (G) Islet insulin content was measured by ELISA and expressed as insulin content from a pool of 20 islets per milliliter. Data are represented as the mean ± SEM in A–G. All comparisons took the N-WT values as reference. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
We also tested whether cyclin D3 overexpression would confer an improved sensitivity to glucose challenges by submitting 11-wk-old N-TG and N-WT littermates to an i.p. glucose challenge (Fig. 6C). Notably, N-TG mice exhibited improved glucose responses compared with N-WT littermates at 15, 30, and 60 min poststimulus, thus confirming that cyclin D3 expression leads to improved β-cell fitness. Both genotypes showed no differences in basal glycemia values (Fig. 6D, Upper); however, N-TG mice exhibited significantly lower basal insulinemia values compared with transgene negative littermates (0.762 ± 0.115 in N-WT vs. 0.529 ± 0.022 in N-TG) (Fig. 6D, Lower).
These phenotypical differences with regard to in vivo glucose clearance observed among the five genotypes were not attributable to differences in insulinemia values at the glycemia measurement time points (Fig. 6 E and F) or to differences in islet insulin content (Fig. 6G). We further pursued an in vitro analysis of the islet physiology to discriminate between pancreatic islet fitness and insulin sensitivity in peripheral tissues.
Cyclin D3 Is Required for Proper Intracellular Calcium [Ca2+]i Fluxes in β Cells in Response to Glucose Challenges.
We further assessed β-cell fitness ex vivo by examining changes in the intracellular calcium concentrations upon islet exposure to increasing extracellular glucose concentrations. We chose islets from young mice (6 wk old) to avoid damaged islets owing to the heavy infiltration observed at older ages.
Pancreatic islets from 6-wk-old N-TG, N-WT, N-HTZ, and N-KO mice were isolated and loaded with 5 μM Fura-2:00 AM to detect increases in intracellular [Ca2+]i concentrations (Fig. 7). Similar calcium analysis results were obtained for N-TG and N-WT islets, which were different from those obtained for N-HTZ and N-KO islets (Fig. 7).
Fig. 7.
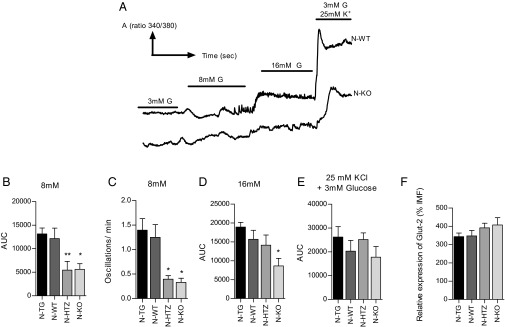
Cyclin D3 is required for glucose-stimulated intracellular calcium [Ca2+]i fluxes in β cells. Freshly isolated pancreatic islets from 6-wk-old N-WT (n = 20), N-TG (n = 18), N-HTZ (n = 30), and N-KO (n = 18) mice were loaded with 5 mM Fura-2:00 AM and perfused with different glucose concentrations (3 , 8, and 16 mM glucose and 25 mM KCl in 3 mM glucose) in modified Ringer’s solution. (A) A representative plot that compares the behaviors of the N-WT and N-KO islets with regard to Ca2+ influx is shown. (B) A graph plotting the AUC for a 5-min period (AUC/5) in response to 8 mM glucose (C) Frequency of [Ca2+]i oscillations in pancreatic islets from each genotype in response to 8 mM glucose. (D) Graph plotting the AUC/5 min in response to 16 mM glucose. (E) Graph plotting the AUC/5 min in response to 3 mM glucose and 25 mM K+. (F) Assessment of Glut-2 expression levels in pancreatic β cells by MFI. Data are represented as the mean ± SEM in B–F. All comparisons performed used the N-WT values as reference. *P ≤ 0.05, **P ≤ 0.01.
At 8 mM glucose, N-WT islets exhibited the expected calcium transient influx followed by rapid oscillations. These oscillations were almost absent in islets from N-KO and N-HTZ mice (Fig. 7 A and C). In addition, N-KO and N-HTZ islets were much less responsive to 8 mM glucose than N-WT and N-TG islets, as can be observed by comparing the area under the curve (AUC) plot that represents the quantification of the total calcium entry into the islet (Fig. 7B). No differences were observed between AUC plots from N-WT and N-TG islets. At 16 mM glucose only the N-KO islets exhibited abnormal changes in [Ca2+]i (Fig. 7D).
The altered responses to glucose exhibited by N-KO and N-HTZ islets at 8 mM glucose and by N-KO at 16 mM glucose, measured as changes in [Ca2+]i, were not attributable to an impaired islet sensitivity to high K+ concentrations (Fig. 7E) or to differences in the expression levels of the β cell-specific glucose transporter Glut-2 (Fig. 7F).
In conclusion, we have found that cyclin D3 is targeted by the autoimmune attack against β cells in T1D and that cyclin D3 plays a novel dual role that is independent of its classical role in initiating cell proliferation. On one hand, cyclin D3 promotes β-cell survival by protecting β cells from apoptosis, and on the other it guarantees β-cell metabolic fitness by regulating changes in [Ca2+]i in response to glucose stimulation.
Discussion
Cyclin D3 Down-Regulation Due to Inflammation.
Our results demonstrate that cyclin D3 has a nonredundant and cell cycle-independent role in the protection of β cells against apoptosis and maintenance of fitness that cannot be fulfilled by the other D-type cyclins expressed in these cells in the absence of cyclin D3.
Islet inflammation leads to cyclin D3 down-regulation in pancreatic β cells from NOD mice, causing enhanced β-cell apoptosis (4). Although there is an inverse correlation between the severity of insulitic attack and cyclin D3 expression, there were no differences in the β-cell proliferative activity related to cyclin D3 down-regulation, presumably because cyclin D2 might compensate for cyclin D3 down-regulation in cell cycle progression. In this sense, although the expression of cyclin D1 mRNA is impaired in cyclin D3-hemideficient endocrine cells, cyclin D2 mRNA is not altered, which suggests that cyclin D2 could be the D-type cyclin responsible for keeping the β-cell cycle intact in the absence of cyclin D3. Thus, the three D-type cyclins seem to follow different regulation patterns in the NOD endocrine cells, cyclin D3 being the only D-type cyclin negatively regulated by inflammation.
The results obtained in the NIT-1 insulinoma model may reflect the protective mechanism exerted by cyclin D3 in β cells in vivo, by rendering them more resistant to inflammation-induced apoptosis. The mechanism accounting for cyclin D3 protection against IL-1β–induced apoptosis in NIT-1 cells very probably obeys the negative interference between cyclin D3 and either NF-κB, the AP1-transcription factor, or the MAPK signaling (27, 28). In this case, cyclin D3 and NF-κB would negatively feed back each other, because the cyclin D3 promoter has binding sites for the NF-κB transcription factor (14) and cyclin D3 would hinder the NF-κB–mediated IL-1β signaling. This would not be surprising, because cyclin D3 has also been reported to bind certain transcription factors such as PPAR-γ, ATF-5 factors (18, 19, 29), or nuclear receptors (30).
It is noteworthy that cyclin D3 overexpression does not protect NIT-1 cells from IFN-γ−induced apoptosis. At any rate, the extent to which IFN-γ is able to induce apoptosis in NIT-1 cells is very limited in comparison with that exhibited by IL-1β. These observations, in agreement with what has already been described, evidence that both cytokines signal by two different pathways to promote apoptosis, one of which, the one triggered by IL-1β, is severely inhibited by cyclin D3, whereas the one related to IFN-γ is not affected by changes in cyclin D3 expression levels. Although IFN-γ signals through the JAK2/STAT1 pathway and IL-1β through the NF-κB pathway, both pathways can cross-talk through the MAPK kinase cascade (27, 28). Moreover, both TNF-α and IFN-γ have been reported to potentiate IL-1β effects on β cells (28). Signaling through IL-1β receptor causes induction of Fas, the extrinsic proapoptotic pathway, and iNOS, involved in β-cell necrosis, among other genes in mouse β cells (2, 31). In addition, IL-1β has been reported to cause β-cell dysfunction (32).
We also find that cyclin D3 causes IFN-γ to enhance NIT-1 cell proliferation. These divergent actions of cyclin D3 on IL-1β (protecting from IL-1β–induced apoptosis) and IFN-γ (promoting proliferation) signaling pathways, respectively, imply a complex biochemical network in which cyclin D3 exerts a dominant role over the otherwise deleterious effects of these cytokines on β cells.
Cyclin D3 Role in β-Cell Viability, Proliferation, and Immunogenicity.
Here, we report that the apoptosis rates and the diabetes incidences for the different genotypes studied regarding cyclin D3 expression show parallel behaviors, evidencing a direct relationship between cyclin D3 expression in the β cells and protection against diabetes. At the same time, no changes in β-cell proliferation are observed. It has been reported that the adenoviral transfer of cyclin D3 into human pancreatic islets is followed by the translocation of this protein to the cell nucleus, and subsequently β-cell replication is promoted (33). We have not observed this outcome in β cells from transgenic NOD mice overexpressing cyclin D3. A possible explanation for these two apparently contradictory outcomes could be that most of the overexpressed cyclin D3 in transgenic mice remains in the cytoplasm of β cells. Hence, the sole overexpression of cyclin D3 in β cells is not sufficient to lead to enhanced β-cell replication, but the translocation to the nucleus is required. Moreover, an increase in the expression of other cell cycle rate-limiting factors distinct from cyclin D3 (e.g., Cdk4 in mice) may be required together with cyclin D3 overexpression to promote β-cell proliferation.
According to our findings, cyclin D3 is not relevant for β-cell replication in adult NOD mice. However, the smaller islet size in both NOD and NOD/SCID mice deficient in cyclin D3 suggests that cyclin D3 would be involved in regulating β-cell mass generation at earlier phases of life. In both cases, cyclin D3 deficiency renders β cells more susceptible to either spontaneous (NOD background) or transferred (NOD/SCID background) autoimmune attack. In this sense, the reduction in size of the target organ may facilitate the onset of diabetes in both cases. However, the sole reduction in β-cell area does not account for diabetes, because NS-KO mice do not develop diabetes spontaneously.
The loss of cyclin D3 enhances β-cell apoptosis, leading to an increased need of autoantigen engulfment by macrophages to avoid inflammation. Furthermore, the reported inefficient macrophage scavenger activity in the NOD strain (34, 35) may lead to an enhanced proinflammatory environment, promoting the recruitment of a larger number of effector immune cells to the apoptotic foci in the islets. This would explain the higher infiltration score observed in pancreatic islets from cyclin D3-deficient NOD mice, despite exhibiting a less diabetogenic immune repertoire. In this sense, by promoting a more proinflammatory environment cyclin D3 deficiency would yield β cells that are more immunogenic.
Although cyclin D3 interaction with caspase-2 has been reported to enhance apoptosis in some cell lines in a cell cycle-related manner (36) we have observed that cyclin D3 prevents apoptosis in a cell-cycle independent fashion, thus suggesting the existence of other cyclin D3 partners distinct from caspase-2 with anti-apoptotic action.
Therefore, the higher apoptotic rates, enhanced proinflammatory environment, and smaller size of the target tissue would result in the exacerbation of the disease observed in NOD mice deficient in cyclin D3.
Cyclin D3 in β-Cell Metabolic Fitness.
In addition to higher apoptotic susceptibility, cyclin D3-deficient β cells exhibit a reduced responsiveness to glucose stimulus, which is translated into delayed glucose clearance in N-KO mice. No abnormalities were observed in either insulinemia or islet insulin content in the absence of cyclin D3, implying that other adaptive peripheral mechanisms along with those provided by cyclin D3 are present. The apparent contradiction between the lower basal glycemia levels and impaired glucose clearance upon glucose challenge in the N-KO mice reflects a dichotomy in the action of cyclin D3. On one side, cyclin D3 negatively modulates the sensitivity of peripheral tissues to insulin (18), and on the other cyclin D3 exerts a positive effect on glucose sensing by pancreatic β cells. The final balance between both driving forces, sensitivity to insulin and insulin secretion, will determine blood glucose levels. The lower insulinemia values in mice overexpressing cyclin D3 in their islets, despite exhibiting normal glycemia compared with WT littermates, are an intriguing outcome, because a priori the insulin sensitivity in peripheral tissues is expected to be the same in both experimental groups. In addition, glucose sensitivity of β cells regarding calcium fluxes are not improved when cyclin D3 is overexpressed in these cells. These observations suggest that cyclin D3, by an unknown mechanism, may exert a distal influence on peripheral tissues when overexpressed in β cells, modulating glucose clearance. In agreement with this supposition, NOD mice deficient in cyclin D3 that overexpress cyclin D3 limited to β cells exhibit lower blood glucose levels (improved glucose clearance) than NOD mice ubiquotously deficient in cyclin D3.
In addition, cyclin D3 action extends beyond the endocrine pancreas, because its partial deficiency in the exocrine pancreas is associated to a significant up-regulation of the pancreatic lipase mRNA, very probably obeying a compensatory mechanism to avoid the accumulation of triglycerides.
Concluding Remarks
Cyclin D3, although traditionally classified as a protein involved in the cell cycle, interacts physically with proteins that have functions and structures distinct from CDKs. All of the evidence suggests a scenario in which the function of cyclin D3 in β cells would encompass roles that coordinate glucose metabolism, survival, and cell replication in a complex manner. Further work needs to be conducted to unveil potential checkpoints at which cyclin D3 intervenes to protect β cells from apoptosis and regulates their physiology.
In conclusion, cyclin D3 down-regulation in pancreatic β cells before diabetes onset in NOD mice leads to β-cell dysfunction and death in a cell cycle-independent manner. Here, we identify cyclin D3 as a key molecule involved in preserving β-cell mass in optimal physiological conditions, highlighting the potential implications for future therapeutic interventions in T1D.
Materials and Methods
Mice.
Mice were housed in specific pathogen-free conditions. All animal experimentation procedures performed in this study were overseen and approved by the Institutional Ethical Committee for Animal Experimentation of the University of Lleida (CEEA), in accordance with the European and US regulations on animal experimentation. All results involving animal experimentation were obtained with female mice, because in the NOD strain males exhibit lower diabetes incidence and delayed kinetics of the disease in comparison with their female littermates (37). Mice homozygous for cyclin D3 deficiency in the mixed 129SvxC57BL/6 genetic background (22) were generously provided by P. Sicinski, Harvard University, Cambridge, MA. We back-crossed these mice onto the NOD background for 12 generations, checking for Idd susceptibility loci (38). Cyclin D3 null mutation was detected by PCR as described (22). We generated transgenic NOD mice by microinjecting the RIP-CcnD3Eα transgene (4.2-kb NotI fragment) into fertilized NOD oocytes (Xenogen). The RIP CcnD3Eα construct comprised the rat insulin promoter (RIP) (26) and the murine cyclin D3 cDNA fragment, a generous gift of M. Roussel, St. Jude Children's Research Hospital, Memphis, TN (39). The progenies were analyzed for insertion of the transgene by PCR with the forward-RIP2, CAAGACTCCAGGGATTTGAGGGA and reverse-D3R1, GACGCAGGACAGGTAGCGATCCAG primers. The PCR product size was 460 bp.
Diabetes assessment, adoptive transfer experiments, and pancreatic islet isolation were performed as previously described (38). For adoptive transfer experiments all of the spleen donors used were older than 8 wk of age.
Immunohistochemical analysis of pancreatic infiltration was performed as previously reported (38). Briefly, paraffin sections were counterstained with hematoxylin/eosin (Sigma-Aldrich) for infiltration studies (Leica). To score islet infiltration the assigned values were as follows: 0 for noninfiltrated islets, 1 for peri-insulitic lesions, 2 for intrainsular insulitis with less than 50% of islet area infiltrated, and 3 for insulitis with more than 50% islet area infiltrated. A minimum of eight sections per mouse at four different levels (>150 islets per genotype) were examined. For each individual mouse the formula below was applied to calculate the infiltration score, and then for each genotype the mean ± SEM was plotted:
qRT-PCR.
Total RNA was extracted from a fraction of CD45− cells from either endocrine (islets) or exocrine pancreas with TRIzol reagent (Invitrogen) and 500 ng of the total RNA in 20 μL was reverse-transcribed into cDNA using the iScript cDNA Synthesis Kit (Bio-Rad). The cDNAs were used as templates for qRT-PCR. The qRT-PCR mixture contained 5 μL of iTaq Universal Sybr Green Supermix (Bio-Rad), 4 μL of cDNA (diluted 1:5), and 0.5 μL each of the forward and reverse primers in a final volume of 10 μL. Primers used for detection of mouse cyclin D1 and D2 were those described previously in ref. 23. For cyclin D3, the primers used were mCcnD3Fw-CAGTGACCGGCAGGCTTTG and mCcnD3Rev-CACTTCAGTGCCTGTGATCC; for mouse pancreatic lipase the primers used were mPLFw-CTACCAGCTAATTACTTCAGATGC and mPLRev- CTGTGGCCAATCAGGTGGACATTG; and for β-actin, β-actinFw-TGGAATCCTGTGGCATCCATGAAA and β-actinRev-TAAAACGCAGCTCAGTAAGAGTCC. The qRT-PCR procedure was as follows: 95 °C for 3 min, 95 °C for 10 s, 63 °C for for 40 cycles, and a melting curve from 65 to 95 °C in a CFX 96 real-time system (Bio-Rad). Data were analyzed using CFX Manager Software version 3.1 (Bio-Rad). β-actin was used as a control for normalizing gene expression. The data obtained were calculated by the 2−ΔΔCt method.
Magnetic Cell Separation.
Once islets were isolated and disaggregated by trypsin digestion, the resulting cell suspension was incubated with a purified anti-mouse CD45 antibody (Becton Dickinson) at 4 °C for 30 min, subsequently washed, and incubated with an anti-rat secondary antibody coupled to magnetic beads (BioMags; Qiagen Iberia) for 20 min at 4 °C. The magnetic separation was carried out using a magnet following the BioMags manufacturer’s instructions (Qiagen).
IPGTTs.
After fasting for 16 h, mice were anesthetized with sodium pentobarbital (60 mg/kg). The mice received an i.p. injection of 2 g of glucose/kg body weight. Glycemia and insulinemia were measured at different time points after the injection. Insulinemia was determined by ELISA (Mercodia).
Morphometric Studies.
Pancreatic paraffin sections were stained for insulin (A0564; Dako). The β-cell area was quantified in a blinded fashion with optic microscopy (Leica) and ImageJ software (National Institutes of Health).
Western Blot Analysis.
Tissue samples were homogenized in RIPA buffer containing protease inhibitors (Sigma-Aldrich). SDS/PAGE was performed under reducing conditions, and cyclin D3 was detected with anti-mouse cyclin D3 (610280; Becton Dickinson) and anti–β-actin (A5316; Sigma-Aldrich).
Immunofluorescence Analysis.
Paraffin sections were stained with anti-mouse cyclin D3, CD11b, and Mac-3 antibodies (610280, 01712D, and 553323, respectively; Becton Dickinson), anti-insulin (A0564; Dako), and Hoechst 333421 for nuclear staining (Sigma-Aldrich).
Islet Insulin Content.
Islets were immersed in an acid–alcohol solution, sonicated, and stored overnight at 4 °C. After centrifugation and protein purification from the supernatant the insulin content was determined by ELISA (Mercordia).
Islet Cell Staining.
Isolated islets were trypsinized and were fixed and permeabilized before cellular staining with anti-mouse Glut-2 (β-cell marker, BAM1440; R&D), anti-mouse Ki67 (proliferation marker, M7248; Dako), anti-mouse cyclin D3, and phycoerythrin-conjugated CD45 antibodies (610280 and 553081, respectively; Becton Dickinson). Sample data were acquired on a FACSCanto II flow cytometer (Becton Dickinson).
Determination of Early (Annexin V) or Late (TUNEL) Apoptotic Events.
Apoptosis assays were performed using the respective apoptosis detection kits (Becton Dickinson). For apoptosis analysis in NIT-1 cells by annexin V staining, the 7AAD reagent was used as a cell death marker (Becton Dickinson).
NIT-1 Cell Transfection.
The NIT-1 stable transfectants were selected with 0.4 mg/mL of G418. For cytokine-induced cell apoptosis, 105 cells per well were seeded into 12-well plates on day 0; on day 1, cytokines were added at final concentrations of 50 U/mL for IL-1β and 200 U/mL for IFN-γ (Prospec); on day 2, the cells were harvested and assayed for either annexin V staining or Ki67 staining.
Recording [Ca2+]i.
Freshly isolated islets loaded with 5 μM Fura-2:00 AM (Invitrogen) were sequentially perfused with 3, 8, and 16 mM glucose and at the end with 25 mM KCl in 3 mM glucose and individual measurements for each islet were taken as reported before (40). The time interval for each glucose concentration condition was 10 min.
Statistical Analysis.
A Kruskal–Wallis test was used to assess overall statistical differences when more than two experimental groups were compared and subsequent Mann–Whitney tests were applied to evaluate differences between pairs of groups. A linear regression model was performed to fit cyclin D3 expression levels depending on the dose of injected NOD splenocytes. For assessing the difference between diabetes cumulative incidence curves the log-rank test was applied. A two-way ANOVA test was used to analyze the differences in glycemia and insulinemia levels in glucose tolerance tests. All analyses were obtained with the SPSS statistical package. The threshold for statistical significance was set at 0.05.
Supplementary Material
Acknowledgments
We thank P. Sicinski for generously providing us with cyclin D3-deficient mice on the Sv-129/C57BL/6 background; M. Roussel for giving us the mouse cyclin D3 cDNA; Anaïs Panosa, Ainhoa García, Jordi Altirriba, and Montse Ortega for their technical assistance; Alana Azebedo for experimental assistance with the TdT UTP nick end labeling studies; X. Gasull and G. Cabeza for assessments during the intracellular calcium flux experiments; the Animal Facility personnel at the University of Lleida for remarkable NOD mice husbandry; and Joan Valls for his guidance regarding the statistical analysis. This work was supported by Spanish Ministerio de Sanidad y Consumo ISCIII Grants PI041310 (to C.M.) and FIS 06/1104 (to D.M.); Spanish Ministerio de Ciencia y Tecnología Grants SAF2006-07757, SAF2007-31050-E, and SAF2008-02536 (to C.M.) and SAF2011-29319 (to T.S.); the 2009 SGR-DGR grant from the Generalitat de Catalunya (to J.V., C.M., and D.M.) and TR265 Grant A001E-12132/2009 from the University of Lleida (to J.V., C.M., and D.M.); the 2012 Basic Research grant from the Academia de Ciencias Médicas y de la Salud de Cataluña y Baleares Foundation (to C.M.); and Generalitat Valenciana Grant PROMETEO/2011/080 and Ministerio de Economía y Competitividad Grant BFU2011-28358 (to Á.N.). N.A.S.-Á. was the recipient of both University of Lleida and Instituto de Ciencia y Tecnología del Distrito Federal predoctoral fellowships, respectively, and U.S. and E.S. are recipients of L’Agència de Gestió d’Ajuts Universitaris i de Recerca predoctoral fellowships from the Generalitat de Catalunya. C.M. and J.V. are assistant professors of immunology in the Serra Hunter Program (University of Lleida) and investigators at the same institution and the Institut de Recerca Biomèdica of Lleida.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1323236111/-/DCSupplemental.
References
- 1.Itoh N, et al. Requirement of Fas for the development of autoimmune diabetes in nonobese diabetic mice. J Exp Med. 1997;186(4):613–618. doi: 10.1084/jem.186.4.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eizirik DL, Mandrup-Poulsen T. A choice of death—the signal-transduction of immune-mediated beta-cell apoptosis. Diabetologia. 2001;44(12):2115–2133. doi: 10.1007/s001250100021. [DOI] [PubMed] [Google Scholar]
- 3.Mauricio D, Mandrup-Poulsen T. Apoptosis and the pathogenesis of IDDM: A question of life and death. Diabetes. 1998;47(10):1537–1543. doi: 10.2337/diabetes.47.10.1537. [DOI] [PubMed] [Google Scholar]
- 4.Watanabe A, et al. Quantitative determination of apoptosis of pancreatic β-cells in a murine model of type 1 diabetes mellitus. J Nucl Med. 2012;53(10):1585–1591. doi: 10.2967/jnumed.111.102459. [DOI] [PubMed] [Google Scholar]
- 5.Moriwaki M, et al. Fas and Fas ligand expression in inflamed islets in pancreas sections of patients with recent-onset Type I diabetes mellitus. Diabetologia. 1999;42(11):1332–1340. doi: 10.1007/s001250051446. [DOI] [PubMed] [Google Scholar]
- 6.Mandrup-Poulsen T. Beta cell death and protection. Ann N Y Acad Sci. 2003;1005:32–42. doi: 10.1196/annals.1288.005. [DOI] [PubMed] [Google Scholar]
- 7.Pakala SV, Kurrer MO, Katz JD. T helper 2 (Th2) T cells induce acute pancreatitis and diabetes in immune-compromised nonobese diabetic (NOD) mice. J Exp Med. 1997;186(2):299–306. doi: 10.1084/jem.186.2.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O’Brien BA, Harmon BV, Cameron DP, Allan DJ. Apoptosis is the mode of beta-cell death responsible for the development of IDDM in the nonobese diabetic (NOD) mouse. Diabetes. 1997;46(5):750–757. doi: 10.2337/diab.46.5.750. [DOI] [PubMed] [Google Scholar]
- 9.Stassi G, et al. Nitric oxide primes pancreatic beta cells for Fas-mediated destruction in insulin-dependent diabetes mellitus. J Exp Med. 1997;186(8):1193–1200. doi: 10.1084/jem.186.8.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cardozo AK, Kruhøffer M, Leeman R, Orntoft T, Eizirik DL. Identification of novel cytokine-induced genes in pancreatic beta-cells by high-density oligonucleotide arrays. Diabetes. 2001;50(5):909–920. doi: 10.2337/diabetes.50.5.909. [DOI] [PubMed] [Google Scholar]
- 11.Wägner AM, et al. Post-translational protein modifications in type 1 diabetes: a role for the repair enzyme protein-L-isoaspartate (D-aspartate) O-methyltransferase? Diabetologia. 2007;50(3):676–681. doi: 10.1007/s00125-006-0556-1. [DOI] [PubMed] [Google Scholar]
- 12.Kacheva S, Lenzen S, Gurgul-Convey E. Differential effects of proinflammatory cytokines on cell death and ER stress in insulin-secreting INS1E cells and the involvement of nitric oxide. Cytokine. 2011;55(2):195–201. doi: 10.1016/j.cyto.2011.04.002. [DOI] [PubMed] [Google Scholar]
- 13.Chen MC, Schuit F, Eizirik DL. Identification of IL-1beta-induced messenger RNAs in rat pancreatic beta cells by differential display of messenger RNA. Diabetologia. 1999;42(10):1199–1203. doi: 10.1007/s001250051292. [DOI] [PubMed] [Google Scholar]
- 14.Wang Z, Sicinski P, Weinberg RA, Zhang Y, Ravid K. Characterization of the mouse cyclin D3 gene: Exon/intron organization and promoter activity. Genomics. 1996;35(1):156–163. doi: 10.1006/geno.1996.0334. [DOI] [PubMed] [Google Scholar]
- 15.Sherr CJ. Mammalian G1 cyclins. Cell. 1993;73(6):1059–1065. doi: 10.1016/0092-8674(93)90636-5. [DOI] [PubMed] [Google Scholar]
- 16.Ma Y, Yuan J, Huang M, Jove R, Cress WD. Regulation of the cyclin D3 promoter by E2F1. J Biol Chem. 2003;278(19):16770–16776. doi: 10.1074/jbc.M212702200. [DOI] [PubMed] [Google Scholar]
- 17.Stamatakos M, et al. Cell cyclins: Triggering elements of cancer or not? World J Surg Oncol. 2010;8:111. doi: 10.1186/1477-7819-8-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sarruf DA, et al. Cyclin D3 promotes adipogenesis through activation of peroxisome proliferator-activated receptor gamma. Mol Cell Biol. 2005;25(22):9985–9995. doi: 10.1128/MCB.25.22.9985-9995.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wei Y, et al. ATF5 increases cisplatin-induced apoptosis through up-regulation of cyclin D3 transcription in HeLa cells. Biochem Biophys Res Commun. 2006;339(2):591–596. doi: 10.1016/j.bbrc.2005.11.054. [DOI] [PubMed] [Google Scholar]
- 20.Aguilar V, Fajas L. Cycling through metabolism. EMBO Mol Med. 2010;2(9):338–348. doi: 10.1002/emmm.201000089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coqueret O. Linking cyclins to transcriptional control. Gene. 2002;299(1-2):35–55. doi: 10.1016/s0378-1119(02)01055-7. [DOI] [PubMed] [Google Scholar]
- 22.Sicinska E, et al. Requirement for cyclin D3 in lymphocyte development and T cell leukemias. Cancer Cell. 2003;4(6):451–461. doi: 10.1016/s1535-6108(03)00301-5. [DOI] [PubMed] [Google Scholar]
- 23.Kushner JA, et al. Cyclins D2 and D1 are essential for postnatal pancreatic beta-cell growth. Mol Cell Biol. 2005;25(9):3752–3762. doi: 10.1128/MCB.25.9.3752-3762.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Georgia S, et al. Cyclin D2 is essential for the compensatory β-cell hyperplastic response to insulin resistance in rodents. Diabetes. 2010;59(4):987–996. doi: 10.2337/db09-0838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blunt T, et al. Identification of a nonsense mutation in the carboxyl-terminal region of DNA-dependent protein kinase catalytic subunit in the scid mouse. Proc Natl Acad Sci USA. 1996;93(19):10285–10290. doi: 10.1073/pnas.93.19.10285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guerder S, Picarella DE, Linsley PS, Flavell RA. Costimulator B7-1 confers antigen-presenting-cell function to parenchymal tissue and in conjunction with tumor necrosis factor alpha leads to autoimmunity in transgenic mice. Proc Natl Acad Sci USA. 1994;91(11):5138–5142. doi: 10.1073/pnas.91.11.5138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103(2):239–252. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 28.Andersen NA, Larsen CM, Mandrup-Poulsen T. TNFalpha and IFNgamma potentiate IL-1beta induced mitogen activated protein kinase activity in rat pancreatic islets of Langerhans. Diabetologia. 2000;43(11):1389–1396. doi: 10.1007/s001250051544. [DOI] [PubMed] [Google Scholar]
- 29.Liu W, et al. Cyclin D3 interacts with human activating transcription factor 5 and potentiates its transcription activity. Biochem Biophys Res Commun. 2004;321(4):954–960. doi: 10.1016/j.bbrc.2004.07.053. [DOI] [PubMed] [Google Scholar]
- 30.Knudsen KE, Cavenee WK, Arden KC. D-type cyclins complex with the androgen receptor and inhibit its transcriptional transactivation ability. Cancer Res. 1999;59(10):2297–2301. [PubMed] [Google Scholar]
- 31.Yamada K, et al. Mouse islet cell lysis mediated by interleukin-1-induced Fas. Diabetologia. 1996;39(11):1306–1312. doi: 10.1007/s001250050574. [DOI] [PubMed] [Google Scholar]
- 32.Mandrup-Poulsen T. The role of interleukin-1 in the pathogenesis of IDDM. Diabetologia. 1996;39(9):1005–1029. doi: 10.1007/BF00400649. [DOI] [PubMed] [Google Scholar]
- 33.Fiaschi-Taesch NM, et al. Cytoplasmic-nuclear trafficking of G1/S cell cycle molecules and adult human β-cell replication: A revised model of human β-cell G1/S control. Diabetes. 2013;62(7):2460–2470. doi: 10.2337/db12-0778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.O’Brien BA, Huang Y, Geng X, Dutz JP, Finegood DT. Phagocytosis of apoptotic cells by macrophages from NOD mice is reduced. Diabetes. 2002;51(8):2481–2488. doi: 10.2337/diabetes.51.8.2481. [DOI] [PubMed] [Google Scholar]
- 35.Marée AF, et al. Quantifying macrophage defects in type 1 diabetes. J Theor Biol. 2005;233(4):533–551. doi: 10.1016/j.jtbi.2004.10.030. [DOI] [PubMed] [Google Scholar]
- 36.Mendelsohn AR, Hamer JD, Wang ZB, Brent R. Cyclin D3 activates Caspase 2, connecting cell proliferation with cell death. Proc Natl Acad Sci USA. 2002;99(10):6871–6876. doi: 10.1073/pnas.072290599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kikutani H, Makino S. The murine autoimmune diabetes model: NOD and related strains. Adv Immunol. 1992;51:285–322. doi: 10.1016/s0065-2776(08)60490-3. [DOI] [PubMed] [Google Scholar]
- 38.Marzo N, et al. Cyclin-dependent kinase 4 hyperactivity promotes autoreactivity in the immune system but protects pancreatic cell mass from autoimmune destruction in the nonobese diabetic mouse model. J Immunol. 2008;180(2):1189–1198. doi: 10.4049/jimmunol.180.2.1189. [DOI] [PubMed] [Google Scholar]
- 39.Inaba T, et al. Genomic organization, chromosomal localization, and independent expression of human cyclin D genes. Genomics. 1992;13(3):565–574. doi: 10.1016/0888-7543(92)90126-d. [DOI] [PubMed] [Google Scholar]
- 40.Soriano S, et al. Rapid regulation of K(ATP) channel activity by 17beta-estradiol in pancreatic beta-cells involves the estrogen receptor beta and the atrial natriuretic peptide receptor. Mol Endocrinol. 2009;23(12):1973–1982. doi: 10.1210/me.2009-0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.