

Abstract
Mutations in the gene for rhodopsin, RHO, cause autosomal dominant retinitis pigmentosa, a disease characterized by death of rod photoreceptor cells. At the end stage, when most rods are gone, cones die too, taking central vision with them. One goal of gene therapy, therefore, is to preserve central vision by promoting rod survival in the vicinity of the macula. Dominance in RHO mutations is associated with two phenomena: interference with the function of normal rhodopsin and intrinsic toxicity of the mutant protein. In the case of interference, increased production of the wild-type protein may be therapeutic, but in the case of toxicity, suppression of the mutant protein may also be needed. RHO augmentation has made use of advances in gene delivery to the retina using adeno-associated virus (AAV). Several strategies have been developed for suppression of rhodopsin expression, but because of the heterogeneity of RHO mutations they are not specific for the mutant allele: They suppress both mutant and wild-type RHO. Experiments in autosomal dominant retinitis pigmentosa (adRP) mouse models suggest that both RHO augmentation and supplementation plus suppression preserve the survival of rod cells.
Mutations in the rhodopsin (RHO) gene cause rod photoreceptor cells to die. Increased production of the wild-type protein plus suppression of the mutant protein may preserve rod cell survival.
As many as 40% of retinitis pigmentosa cases display autosomal dominant inheritance, and approximately one quarter of these are attributable to mutations in RHO, the gene for rod cell opsin (Sullivan et al. 2006). A complicating feature of RHO-based autosomal dominant retinitis pigmentosa (adRP) is its alleleic diversity: More than 100 disease-causing mutations have been mapped to RHO, and the vast majority are dominant (see https://sph.uth.edu/retnet/home.htm). These mutations can be classified both by their clinical presentation and by their impact on the cellular distribution of rhodopsin. Such distinctions are critical because they determine how, or even whether, a particular patient should be considered for gene therapy.
Based on clinical observations, Cideciyan and coworkers (1998) described two classes of adRP. Class A patients lost retinal function over the entire retina and reported the onset of night blindness early in life. In contrast, class B patients exhibited a milder phenotype, normal rod activation kinetics, and preserved rod outer segment length, with anomalies in the rod visual cycle that were mutation specific. Photoreceptor degeneration in subclass B1 was not homogenous, and some patients showed an inferior to superior progression of disease. These authors conclude that the “catastrophic loss of rod function” in class A patients may not be correctable, so that therapy should be focused on preserving cones; whereas, in class B patients, rods have the potential to be rescued, and preserving rods is the best hope for protecting cones. The question of why cones die in retinitis pigmentosa is also crucial, because the answer will determine how many rods must be rescued to preserve central vision.
Two classes of opsin mutations were also described as a result of studies in tissue culture cells (Sung et al. 1991; Kaushal and Khorana 1994). Class I opsin mutants were expressed at high levels in cells, reconstituted rhodopsin by binding 11-cis-retinal and partitioned to the plasma membrane. Class II mutants were expressed at low levels, did not bind 11-cis-retinal and remained in the endoplasmic reticulum. The relationship between the cellular behavior of mutant opsin proteins and the clinical impact of opsin mutations is not always clear, but a few generalizations can be made. Most of the mutations that lead to opsin misfolding and instability (i.e., class II) lead to a milder disease phenotype (class B). In contrast, mutations affecting the carboxyl-terminus of opsin, such as V345L and P347S, are expressed at high levels. These mutated proteins form rhodopsin, but lead to early onset, severe retinal degeneration (class A).
Transgenic animal models of adRP have provided some clarification and some confusion. For example, rhodopsin with carboxyl-terminal chain terminating mutations (Q344X and S334X) is mislocalized to the plasma membrane of the inner segment (Sung et al. 1994; Li et al. 1996; Green et al. 2000), and this improper membrane insertion is toxic to rod photoreceptors. In contrast, class II mutations (P23H and T17M), which cause opsin to be retained in the endoplasmic reticulum, may cause cell death by continued activation of the unfolded protein response (Lin et al. 2007; Gorbatyuk et al. 2010; Kunte et al. 2012). Mendes and colleagues (2005) have refined the classification of RHO mutants, synthesizing results obtained in cell and transgenic models of adRP. One risk of transgenic models is related to the potential for overexpression of rhodopsin, which in itself may be toxic (Olsson et al. 1992a; Tan et al. 2001). Sakami and coworkers described a knock in mouse model of P23H Rho (the mouse version of RHO). Unlike transgenic P23H models, this line displayed the slow retinal degeneration characteristic of type B patients, and opsin did not accumulate in the endoplasmic reticulum but led to the disruption of disc membranes in rod outer segments (Sakami et al. 2011).
Two critical questions must be answered when considering rod-directed gene therapy: (1) Is the degeneration progressing too quickly for rescue by gene delivery to rod photoreceptors? If rod cells have begun their cell death program when the diagnosis is made, then therapy must be directed at macular cones (Yang et al. 2009; Punzo et al. 2009; Usui et al. 2009; Murakami et al. 2012), or alternative strategies such as stem cell therapy, retinal prosthesis or gene delivery of light sensitive channels to cells of the inner retina may be indicated. (2) How does mutant opsin kill the cell? RHO mutations may be dominant for either of two reasons (Wilson and Wensel 2003; Mendes et al. 2005). Rhodopsin forms dimeric complexes in the disc membrane (Fotiadis et al. 2003), and mutant proteins might interfere with the function of normal rhodopsin or its assembly in the membrane, thereby exerting dominant negative effects. Alternatively, gain-of-function mutations could cause rhodopsin to be intrinsically damaging to the rod cell. It may be possible to treat dominant negative mutations by increasing the level of the normal protein (supplementation). For mutations that cause rhodopsin to be injurious, however, suppressing the expression of the mutant proteins may also be required.
RHODOPSIN SUPPLEMENTATION
Whether or not the production of mutated opsin should be blocked, the conditions for delivery of a normal human opsin gene must be established for gene supplementation. Delivery of a wild-type copy of the defective gene is the most direct approach for gene therapy and is being tested both animal models and human clinical trials of recessive retinal degenerations, as described in many of the other reviews in this collection. Key issues that must be addressed are delivery to photoreceptors, specificity and the appropriate level of expression.
DELIVERY
For gene delivery to photoreceptors, adeno-associated virus (AAV) is currently the best-developed vehicle. Recombinant AAV leads to long-term (probably lifelong) expression of the delivered gene in nondividing cells, and because the delivered DNA is maintained as an episome (Song et al. 2004), there is little danger of insertional mutagenesis (Kaeppel et al. 2013). AAV does not cause disease and does not provoke an inflammatory response following subretinal injection, although preexisting antibodies to the virus can limit its usefulness for some applications (Mingozzi and High 2013). In three trials of gene therapy for Leber congenital amaurosis type II, AAV serotype 2 has been shown to be safe for human use (Bainbridge et al. 2008; Maguire et al. 2008; Cideciyan et al. 2009). In the eye, AAV efficiently infects both photoreceptors and the retinal pigment epithelium following subretinal injections. In rodents, a single subretinal injection of 1 or 2 µL can detach the entire retina and infect 80% of photoreceptors before the neural retina reattaches to the retinal pigment epithelium. In larger animals (such as dogs) and in humans, larger volumes on the order of 100–300 µL are delivered, and viral infection is restricted to the area of detachment. A limitation of AAV is its limited carrying capacity: 4.7 kb inserts in single stranded AAV and half of that in self-complementary AAV. Recently, genetic modifications to the viral capsid proteins have permitted transduction of photoreceptor cells from the vitreous chamber, but efficiency is currently too low, certainly in primates, to use this approach for effective replacement of rhodopsin in rods (Dalkara et al. 2013; Kay et al. 2013).
Other approaches to gene delivery to photoreceptors include the use of nanoparticles and lentivirus-based vectors (Han et al. 2012; Binley et al. 2013). Each has shown some promise, and vectors based on equine infectious anemia virus are in clinical trials for treatment of retinal disease (see http://clinicaltrials.gov, NCT01367444; NCT01301443; NCT01736592). The user communities for these systems are small, however, and for the purpose of delivering short cDNAs such as that of rhodopsin, the limited carrying capacity of AAV is not an impediment.
SPECIFICITY
AAV vectors with multiple tyrosine-to-phenylalanine modifications infect many cell types following intravitreal or subretinal injection (Petrs-Silva et al. 2011). Photoreceptor specific expression of transgenes can be achieved using several different promoters, including that of rhodopsin kinase and interphotoreceptor retinoid binding protein. The proximal rod opsin promoter of 485 bp leads to efficient transgene expression in rod photoreceptors, but delivered genes are also expressed in cone photoreceptors (Glushakova et al. 2006; Mussolino et al. 2011a). Whether production of rhodopsin in cone photoreceptors will affect cone function and vision is not known. Palfi and colleagues (2010) described a longer rod opsin promoter construct containing a 1.7 kb segment of the mouse rhodopsin promoter and two additional conserved elements from the rhodopsin promoter. In an AAV vector, this promoter led to 50% greater expression of human RHO mRNA than a virus using the 500-bp proximal promoter following subretinal injection in mice. Injection of this vector just after birth led to partial protection of rod cell structure and function for up to 12 wk in Rho knockout mice.
LEVEL OF EXPRESSION
Expression of wild-type human rhodopsin at high levels in transgenic mice leads to progressive death of photoreceptor cells (Olsson et al. 1992b; Sung et al. 1994). This phenomenon has been examined in some detail by Price et al. (2012), who expressed one or two copies of a normal human RHO transgene in mice with one or two copies of wild-type mouse Rho. Two copies of the human RHO gene on a Rho−/− background produced approximately the same level of rhodopsin protein as the two endogenous copies of mouse Rho in wild-type mice. Two copies of mouse Rho plus one copy of human RHO led to a 30% decrease in the thickness of the outer nuclear layer (ONL) by 6 mo of age, with most of the decline occurring in the first month of life, i.e., during photoreceptor development. A double dose of opsin genes (two Rho plus two RHO) led to a 60% decrease in ONL thickness, with a sharp decline in the first month but continued thinning over the next 5 mo. Increased expression of rhodopsin led to an increase in the volume of rod outer segments, and the authors concluded that loss of photoreceptor cell bodies (thinning of the ONL) with three copies of the gene may be caused by cellular crowding. Increased diameter of rod outer segments was also reported by Wen et al. (2009), who used transgenic mice expressing a chimeric bovine-murine opsin gene. These authors suggest that without an increase in peripherin/rds (retinal degeneration slow) that disc membranes become unstable in larger outer segments.
Because transgenic overexpression of rhodopsin leads to death of photoreceptors, it is logical that viral delivery of RHO cDNA could be harmful if too much rhodopsin is produced. Indeed, that is what we found using AAV to deliver a cDNA encoding mouse rhodopsin under the control of its own promoter (Mao et al. 2011). Subretinal delivery of this vector in wild-type mice led to a 45% increase in the level of rhodopsin monomer (similar to the presence of a single copy of a RHO transgene), and this level of expression caused a 70% reduction in electroretinogram (ERG) a-wave and b-wave amplitudes over the 6 mo after injection (Fig. 1). This finding suggests that rhodopsin expression from viral vectors should be carefully titrated.
Figure 1.

Virally mediated overexpression of rhodopsin leads to retinal degeneration in C57Bl/6J mice. Reduction in ERG a-wave and b-wave amplitudes in eyes injected with AAV-Rho (white bars) compared with the uninjected contralateral eyes (black bars) over a time course of 6 mo after injection of virus (n = 7). For the a-wave amplitudes, ** p < 0.005 at 2, 3, and 6 mo postinjection. For b-wave amplitudes, * p < 0.05 at 1 mo and ** p < 0.005 at 2, 3, and 6 mo postinjection. (From Mao et al. 2011; reprinted, with permission, from Mary Ann Liebert, Inc. © 2011.)
Nevertheless, excess rhodopsin may not be a risk in gene therapy for adRP, because RHO genes will not be delivered to people with two normal copies of RHO. Patients harboring the P23H mutation in RHO have less than the normal amount of rhodopsin (Kemp et al. 1992), and this reduction is probably characteristic of many class B patients (Kemp et al. 1988). Likewise, the best-characterized transgenic mouse line expressing P23H Rho exhibits about one half of the normal level of rhodopsin, based on spectroscopic measurements on 1-mo-old mice (Wu et al. 1998), and the P23H RHO line that we developed exhibit a significant reduction in rhodopsin levels at the same age (Noorwez et al. 2009). Diminished levels of rhodopsin can be attributed to retention of the mutant protein in the endoplasmic reticulum and enhanced degradation of the wild-type protein in the presence of mutant opsin (Frederick et al. 2001; Rajan and Kopito 2005). In both people and mice, reduction in rhodopsin levels is revealed by a delayed recovery from photobleaching (Goto et al. 1995; Kemp et al. 1992). Consequently, individuals who stand to benefit from gene augmentation with rhodopsin can be identified, and those at risk of rhodopsin excess can be excluded from treatment.
PHOTORECEPTOR RESCUE BY RHODOPSIN SUPPLEMENTATION
In mice bearing a P23H Rho transgene, increased expression of normal rhodopsin reduces the rate of retinal degeneration. Frederick and colleagues (Frederick et al. 2001) found that in transgenic mice with no wild-type Rho, 80% of the photoreceptor cell bodies (ONL) were gone by postnatal day 30. In mice with one copy of the endogenous mouse gene, there was only a 50% reduction at the same time point, and in mice with a normal component of Rho, there was no retinal degeneration at day 30, although by postnatal day 90 photoreceptor loss was complete on all genetic backgrounds. Price and coworkers (2012) took this analysis one step further: In mice with the same P23H transgene, three wild-type rhodopsin genes (two Rho plus one RHO) retained ∼70% of the ONL at 1 mo, and nearly half of the ONL at 6 mo. These results support the hypothesis of Wilson and Wensel (2003) that class II mutations such as P23H may result in a dominant negative form of opsin that is not inherently injurious to photoreceptors.
AAV-mediated delivery of a cDNA encoding wild-type rhodopsin also reduced retinal degeneration in P23H transgenic mice (Mao et al. 2011). We used a mouse line with one copy of a human P23H RHO gene on a wild-type background (Rho+/+). Such mice lose 60% of the ONL and 70% of their scotopic a-wave response by 6 mo of age, but eyes injected with AAV-Rho retained 80% of the ONL in the superior retina and 90% in the inferior hemisphere (Fig. 2A). ERG a-wave and b-wave amplitudes also remained at ∼80% of those of wild-type C57Bl/6 mice, and the decrease in ERG response seen in untreated eyes was arrested by 2 mo of age in injected eyes (Fig. 2B). These results suggest that rhodopsin supplementation by viral delivery of a wild-type RHO cDNA may be a viable therapeutic approach in cases of adRP associated with class II mutations in RHO.
Figure 2.

AAV-Rho protects the retinas of P23H transgenic mice for up to 6 mo. A. ONL from AAV-Rho injected eyes (black bars) were 80%–85% thicker than those of the untreated group (white bars) (* p < 0.05) and were slightly thinner but not significantly different from the wild-type mice at 6 mo of age (gray bars). B. The ERG a-wave and b-wave amplitudes of AAV-Rho injected eyes were elevated (white bars) in P23H transgenic mice at 1, 2, 3, and 6 mo following injection compared with uninjected eyes (black bars) (* p < 0.05 at 1 and 2 mo postinjection; * p < 0.005 at 3 and 6 mo; n = 9). (From Mao et al. 2011; reprinted, with permission, from Mary Ann Liebert, Inc. © 2011.)
That being said, models of adRP containing carboxyl-terminal mutations in rhodopsin transgenes often exhibit rapid degeneration of the outer retina even when two copies of the wild-type mouse Rho are present at the endogenous locus, depending on the level of expression of the transgene (Li et al. 1995, 1996; Petters et al. 1997; Pennesi et al. 2008). Millington-Ward and colleagues (2011) reported that AAV delivery of a wild-type human RHO cDNA driven by a 1.7 kb opsin promoter did not protect photoreceptor function (ERG b-wave) in P347S RHO transgenic mice, supporting the view that gene supplementation by itself may not be sufficient for treatment of toxic gain-of-function mutations.
PHOTORECEPTOR RESCUE BY RNA REPLACEMENT
To overcome the damage caused by mutated rhodopsin in adRP, rhodopsin gene delivery can be accompanied by inhibition of synthesis of the mutant protein. Such suppression can be achieved by blockade of transcription, using zinc-finger transcriptional repressors, or can occur at the RNA level through the use of antisense techniques including antisense oligodeoxynucleotides, RNA enzymes (ribozymes) or RNA interference using short interfering RNA (siRNA). Although allele specific knockdown of P23H mRNA reduced the rate of retinal degeneration in P23H Rho transgenic rats over a prolonged period (LaVail et al. 2000), the large number of different adRP mutations in RHO make this approach challenging, because a different inhibitor would be needed for each mutation. An alternative approach is to block the expression of all endogenous rhodopsin, mutant and wild-type, and to deliver a replacement gene to support the structure and function of photoreceptors (Ader et al. 2005).
ZINC-FINGER REPRESSORS
Zinc-finger domains are features of many DNA binding proteins including transcriptional activators and repressors. The manner in which these domains interact with DNA has been studied in detail, and the rules have been established for modifying the amino acid sequence to bind a specific DNA sequence (Liu et al. 2002; Segal et al. 1999). These zinc finger domains can then be associated with transcriptional activator domains, transcriptional repressor domains or with site-specific nucleases. Mussolino and colleagues (2011b) produced a series of transcriptional repressors that silence RHO by combining six zinc-finger domains with the Krüppel-associated box repressor domain, screening them first in tissue culture and then in mice. Their most active repressor was specific for the human RHO promoter and, when delivered by subretinal injection of AAV to wild-type mice, there was no impact on ERG amplitudes or on the structure of the retinas. When injected into mice bearing one copy of the P347S RHO transgene and two copies of endogenous Rho, expression of the human transgene was reduced by 26% and ERG b-wave amplitudes were increased by nearly 30% in treated eyes 2 mo after gene transfer. In this model, normal rhodopsin was provided by the mouse gene, so that no additional supplementation was necessary.
RNA REPLACEMENT THERAPY
Combining genes for RNA inhibitors such as ribozymes or siRNAs in the same viral vector as the rhodopsin cDNA is straightforward because both genes are small (1046 nt for RHO cDNA, 19 bp for siRNA, 35 nt for a hammerhead ribozyme) and can be driven from short promoter sequences. For this system to operate, the RHO cDNA must be made resistant to the siRNA (or ribozyme) by the introduction of “silent” mutations. Although single mismatches within 4 nt of the cleavage site can inhibit cutting by hammerhead ribozymes (Fedor and Uhlenbeck 1990), three or more changes may be required to prevent the siRNA from directing digestion by the RNA-induced silencing complex, and both the position and the nature of these changes is important to prevent cleavage (Du et al. 2005). Potential problems may arise from mutations that change a commonly used codon to a less commonly used one or mutations that affect RNA secondary structure. Both phenomena can impact the rate of translation and therefore the folding of opsin (Duan et al. 2003; Cannarozzi et al. 2010).
Short interfering RNA can be produced in two ways: as short hairpin RNAs under the control of an RNA polymerase III (pol III) promoter (Paddison et al. 2002) or as artificial microRNAs (miRNA) under the control of pol II promoters (Zeng et al. 2005). Short hairpin RNA (shRNA) is processed to siRNA by the endonuclease Dicer in the cytoplasm, and has the advantage of high expression level and therefore more rhodopsin repression. However, pol III promoters are promiscuous, so that any AAV infected cell will produce the shRNA. Because of the well-known off-target effects of siRNAs, it is probably wise to restrict expression to the target cells, in this case photoreceptors. An artificial miRNA uses the processing sequences of a bona fide miRNA but replaces the sequence of the mature miRNA with that of an siRNA, in this case targeting the coding portion of RHO. Its synthesis can be directed by a photoreceptor-specific promoter. In fact, the gene for the artificial miRNA can be in the same transcript as the rhodopsin cDNA, either upstream in an intron sequence (Fig. 3A) or downstream in the 3′ untranslated region (Fig. 3B). Such an organization will ensure delivery of both components to the same cells, but processing of the miRNA from the RHO transcript is likely to reduce the level of mature mRNA either by interfering with splicing or polyadenylation. Nevertheless, delivery of artificial miRNAs with a resistant replacement gene has been successfully employed in a mouse model of liver failure associated with the PiZ allele of α-1 antitrypsin (Mueller et al. 2012).
Figure 3.
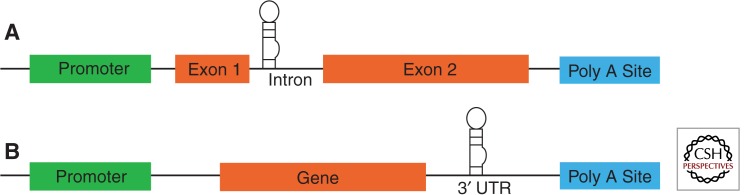
Constructs for mRNA suppression and replacement. Possible locations for miRNA insertion include (A) the intron region or (B) the 3′ UTR. In either case, the same promoter would be used to direct the synthesis of the artificial miRNA and the replacement gene. In the case of RHO adRP mutations the miRNA would be engineered to express an siRNA targeting the coding region of RHO, and the replacement RHO gene would contain synonymous changes in the third positions of codons to remove the siRNA binding site.
O’Reilly and colleagues attempted the RNA replacement approach in transgenic mice bearing a P23H RHO gene with limited success (O’Reilly et al. 2007). Their AAV serotype 5 vector contained a pol III promoter driving the expression of an shRNA that they showed to be effective in suppressing human rhodopsin mRNA in transgenic mice. The shRNA gene was combined in the same vector with a RHO cDNA containing seven silent mutations to block binding of the siRNA driven by a 1.7 kb mouse Rho promoter. In P23H RHO mice injected as newborns and measured at postnatal day 10 they observed partial protection of ONL thickness (33%) compared to eyes injected with a virus expressing GFP, but only by separating their highest values from their lowest values. The transgenic mice used in these experiments exhibited rapid retinal degeneration: In untreated eyes, all photoreceptors were lost by 2 wk, so this was an extremely stringent test of therapy.
In contrast, we tested RNA replacement in a slow, progressive model of adRP based on the presence of a single human P23H transgene on the background of heterozygous disruption of the mouse Rho gene (Rho+/−) (Mao et al. 2012). In these mice, maximum ERG amplitudes were decreased by 50% at 1 mo of age, and ONL thickness was half that of nontransgenic littermates by 2 mo of age. We employed an AAV5 virus expressing both an shRNA that targeted mouse Rho and human RHO and an siRNA-resistant Rho cDNA containing three silent mutations. Injection of this virus at postnatal day 15 led to 80% improvement in ONL thickness at 3 mo postinjection, and this difference persisted for up to 9 mo postinjection. Similarly, ERG a-wave and b-wave amplitudes remained more or less stable over the 9 mo of the experiment in injected eyes but decreased by 85% in untreated eyes (Fig. 4). Retinal degeneration was faster in this model than in the P23H RHO, mouse Rho+/+ mice used for supplementation without suppression (Mao et al. 2011), and the knockdown of endogenous rhodopsin mRNA was necessary to achieve this level of ONL preservation.
Figure 4.

A single AAV RNA replacement vector preserves retinal function for 9 mo in P23H RHO transgenic mice. The combination vector was designated RS301. Left eyes of both strains were untreated and right eyes were injected with AAV-RS301. ERG amplitudes of the untreated, nontransgenic eyes were set at 100%. (At 9 mo, the average a-wave amplitude of these untreated eyes was 180 µV and the average b-wave amplitude was 565 µV.) By 9 mo the untreated P23H transgenic eyes showed a significantly diminished response in both a- and b-wave amplitudes, <20% of that of the untreated, nontransgenic eyes (n = 7; ** p < 0.005). P23H eyes injected with AAV-RS301 maintained 80% of the a-wave amplitude of untreated, nontransgenic eyes and 95% of the b-wave amplitude. (From Mao et al. 2012; reprinted, with permission, from Mary Ann Liebert, Inc., © 2012.)
Results of Millington-Ward and colleagues (2011) suggest that RNA replacement may also work for class I mutations in rhodopsin. After using marker genes (EGFP and dsRed) to verify that photoreceptors could be infected with two AAV vectors following subretinal injection, they coinjected RHO P347S, Rho+/− mice at postnatal day 5 with AAV serotype 5 expressing an shRNA from the H1 promoter and a separate virus expressing a replacement RHO gene. The authors had earlier shown that delivery of the shRNA vector alone efficiently suppressed human RHO mRNA levels and reduced the rate of thinning of the ONL in a mouse line that carried the human P347S RHO gene on a mouse Rho+/+ background (Chadderton et al. 2009). With the combined injection, they observed a 35% increase in ONL thickness compared to control injected eyes at 6 wk of age. Measurement of the ERG b-wave amplitudes suggested that the protective effect lasted up to 20 wk postinjection (Fig. 5). Although b-wave amplitudes were diminished threefold compared to the 6 wk time point, they were three times greater in the RNA-replacement eyes than in control-treated eyes.
Figure 5.

ERG response following suppression and replacement of P347S RHO. At postnatal day 5, mice were injected in one eye with a 1:30 mixture of AAV expressing the shRNA (S) and AAV expressing the siRNA-resistant replacement gene (R). Contralateral eyes were injected with a control virus expressing EGFP. Six weeks or 20 wk later, mice were dark-adapted overnight and ERG responses recorded from both eyes. (A,B) Overlays of the ERG recordings; gray and black lines represent recordings from combined suppression and replacement therapy (S+R) and control (Ctr)-injected eyes, respectively. RE, right eyes; LE, left eyes. (C) Mean b-wave amplitudes. Gray and black columns represent values corresponding to S+R and control-injected eyes, respectively. Error bars represent standard deviation [*** p < 0.001]. (From Millington-Ward et al. 2011; reprinted, with permission, from Nature Publishing Group © 2011.)
The choice of two viruses, one for mRNA suppression and the other for mRNA replacement, is appealing in that the ratio of the two viruses can be varied depending on the level of expression of the mutant RHO gene in the animal model or the patient. Nevertheless, there are technical and regulatory barriers to this approach. Despite fluorescence micrographs indicating that photoreceptors can be coinfected with two viruses, it is likely that many photoreceptors will be infected with one or the other, leading to rhodopsin deficiency or excess in those cells. It is probable that a mixture of different recombinant viruses could be tested for safety and granted regulatory approval, but each variation in the ratio of viruses would be a different test compound and might require its own safety studies and regulatory review. Given the fact that a single virus expressing the shRNA and the replacement gene was effective in promoting long-term survival of photoreceptors in P23H RHO transgenic mice, using one vector for RNA replacement seems advisable.
CONCLUDING REMARKS: MOVING RHODOPSIN AUGMENTATION TO THE CLINIC
Several questions must be answered before rhodopsin supplementation can be developed for clinical application. These include: How much rhodopsin is too much in a primate? Which RHO mutations can be treated with supplementation alone and which require supplementation plus suppression? Does this approach work in a large animal model of adRP? Where in the retina should the vector be delivered?
Although overexpression of wild-type rhodopsin, either by transgenesis or viral gene-delivery, has been demonstrated to cause photoreceptor degeneration in mice, this phenomenon has not been reported in larger animals. If the damage to photoreceptors is cell-autonomous, then there is reason to suspect that excess rhodopsin would also be harmful in people, whereas too little expression would be without benefit. Although novel capsid variants may permit infection of photoreceptor cells following intravitreal injection (Dalkara et al. 2013; Kay et al. 2013), the inner limiting membrane would prevent transduction of rod cells in the peripheral retina. Therefore, subretinal injections would be required. Because single injections of AAV will transduce photoreceptors only in the region of retinal detachment, harm or benefit should be confined to that area. To determine a therapeutic window for opsin expression, a dose escalation study should be performed in nonhuman primates using subretinal injections with AAV serotype 5 or serotype 8 expressing human RHO cDNA (Vandenberghe et al. 2011; Boye et al. 2012). Monitoring of retinal dimensions with spectral domain optical coherence tomography and retinal function with focal ERG should then be continued for a year or more. Some animals would have to be sacrificed after a shorter interval to assess expression levels of rhodopsin and other marker proteins.
In rodents, it is important to test AAV augmentation in as many valid models of adRP based on rhodopsin mutations as are currently available. These would include transgenic mice and rats as well as mice carrying mutations in the mouse Rho gene (Budzynski et al. 2010; Sakami et al. 2011). To recapitulate adRP, the disease phenotype should be slowly progressive, so that therapy can be initiated in mature animals and after the onset of retinal degeneration. In mice and rats, it may be of value to employ self-complementary AAV for the delivery of RHO, because the onset of rhodopsin expression will be more rapid compared to that from single stranded AAV (McCarty et al. 2003). The goal would be to determine which, if any, of these models should be successfully treated with RHO augmentation and which may require supplementation plus suppression of the endogenous genes. Outcome measurements would include spectral domain optimal coherence tomography, ERG and behavioral assays such as the optokinetic response measured longitudinally in the same animals (Prusky et al. 2004).
Before moving to clinical trials, it is essential to test gene therapy by rhodopsin augmentation or replacement in large animal models of adRP. Several porcine models have been characterized, including transgenic pigs with both class I and class II mutations (Petters et al. 1997; Tso et al. 1997; Li et al. 1998; Huang et al. 2000; Ross et al. 2012). Unfortunately, all of the models, including P23H minipigs, exhibit relatively rapid retinal degeneration, so that viral delivery of RHO would have to be attempted soon after birth for any hope of efficacy. There is one naturally occurring canine model of RHO adRP that has been characterized in some detail (Kijas et al. 2002; Cideciyan et al. 2005; Gu et al. 2007). At 3 mo of age, dogs heterozygous for the T4R mutation have nearly normal ERG amplitudes, but by 13 mo, both rod and cone responses are attenuated. T4R RHO dogs do not recover normally from photobleaching, and are particular sensitive to light damage from clinical levels of light exposure, such as fundus imaging. The mechanism of this light damage is not known, but the mechanism appears to be different from intense light damage in albino mice, because inhibiting AP-1 activation does not block light injury (Gu et al. 2009). The extreme sensitivity to light damage is probably caused by a toxic gain-of-function by T4R rhodopsin, so that it is unlikely that RHO supplementation will suffice for therapy—suppression of endogenous rhodopsin may also be required. The advantage of the light damage phenotype is that success of RNA replacement can be tested simply by light exposure a few weeks after gene transfer; treated eyes should be less sensitive to intense light. A disadvantage is that retinal injections must be performed under dim red light (Komaromy et al. 2008). In contrast, it is possible that supplementation alone would prevent the slow, underlying retinal degeneration of dogs exposed only to ambient light, assuming that the mechanism for the slow retinal degeneration is distinct from the light injury response.
The large animal models of adRP will be of great use in determining the dose and distribution of gene therapy vectors. The retina of both species is cone-rich relative to rodents, particularly in the area centralis of the dog (Mowat et al. 2008) and the retinal streak of the pig (Hendrickson and Hicks 2002). Although the immediate objective of rhodopsin augmentation is the rescue of rod photoreceptors, the ultimate goal is preventing the loss of cones, and thus of central vision. Presumably, subretinal injections in humans would be placed in the perifoveal region to sustain rods in the vicinity of the fovea and thus promote the survival of foveal cones. Treatment near the central retina in canine and porcine models of adRP might predict whether rod-based therapies can protect central vision in this way.
ACKNOWLEDGMENTS
The authors acknowledge support from the Foundation Fighting Blindness, and from the National Eye Institute (grant 1R24EY022012, training grant T32 EY007132, vision core grant P30EY021721).
Footnotes
Editors: Eric A. Pierce, Richard H. Masland, and Joan W. Miller
Additional Perspectives on Retinal Disorders: Genetic Approaches to Diagnosis and Treatment available at www.perspectivesinmedicine.org
REFERENCES
- Ader M, Kenna PF, Millington-Ward S, Clark G, Kennan A, O’Reilly M, Tam LC, Aherne A, McNally N, Humphries P, et al. 2005. Toward a gene therapy for dominant disease: Validation of an RNA interference-based mutation-independent approach. Mol Ther 12: 555–561 [DOI] [PubMed] [Google Scholar]
- Bainbridge JW, Smith AJ, Barker SS, Robbie S, Henderson R, Balaggan K, Viswanathan A, Holder GE, Stockman A, Tyler N, et al. 2008. Effect of gene therapy on visual function in Leber’s congenital amaurosis. N Engl J Med 358: 2231–2239 [DOI] [PubMed] [Google Scholar]
- Binley K, Widdowson P, Loader J, Kelleher M, Iqball S, Ferrige G, de Belin J, Carlucci M, Angell-Manning D, Hurst F, et al. 2013. Transduction of photoreceptors with equire infectious anemia virus Lentiviral Vectors: Safety and biodistribution of StarGen for Stargardt disease. Invest Ophthal Vis Sci 54: 4061–4071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boye SE, Alexander JJ, Boye SL, Witherspoon CD, Sandefer KJ, Conlon TJ, Erger K, Sun J, Ryals R, Chiodo VA, et al. 2012. The human rhodopsin kinase promoter in an AAV5 vector confers rod- and cone-specific expression in the primate retina. Hum Gene Ther 23: 1101–1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budzynski E, Gross AK, McAlear SD, Peachey NS, Shukla M, He F, Edwards M, Won J, Hicks WL, Wensel TG, et al. 2010. Mutations of the Opsin gene (Y102H and I307N) lead to light-induced degeneration of photoreceptors and constitutive activation of phototransduction in mice. J Biol Chem 285: 14521–14533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannarozzi G, Schraudolph NN, Faty M, von RP, Friberg MT, Roth AC, Gonnet P, Gonnet G, Barral Y 2010. A role for codon order in translation dynamics. Cell 141: 355–367 [DOI] [PubMed] [Google Scholar]
- Chadderton N, Millington-Ward S, Palfi A, O’Reilly M, Tuohy G, Humphries MM, Li T, Humphries P, Kenna PF, Farrar GJ 2009. Improved retinal function in a mouse model of dominant retinitis pigmentosa following AAV-delivered gene therapy. Mol Ther 17: 593–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cideciyan AV, Hood DC, Huang Y, Banin E, Li ZY, Stone EM, Milam AH, Jacobson SG 1998. Disease sequence from mutant rhodopsin allele to rod and cone photoreceptor degeneration in man. Proc Natl Acad Sci 95: 7103–7108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cideciyan AV, Jacobson SG, Aleman TS, Gu D, Pearce-Kelling SE, Sumaroka A, Acland GM, Aguirre GD 2005. In vivo dynamics of retinal injury and repair in the rhodopsin mutant dog model of human retinitis pigmentosa. Proc Natl Acad Sci 102: 5233–5238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cideciyan AV, Hauswirth WW, Aleman TS, Kaushal S, Schwartz SB, Boye SL, Windsor EA, Conlon TJ, Sumaroka A, Roman AJ, et al. 2009. Vision 1 year after gene therapy for Leber’s congenital amaurosis. N Engl J Med 361: 725–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalkara D, Byrne LC, Klimczak RR, Visel M, Yin L, Merigan WH, Flannery JG, Schaffer DV 2013. In vivo-directed evolution of a new adeno-associated virus for therapeutic outer retinal gene delivery from the vitreous. Sci Transl Med 5: 189ra76. [DOI] [PubMed] [Google Scholar]
- Du Q, Thonberg H, Wang J, Wahlestedt C, Liang Z 2005. A systematic analysis of the silencing effects of an active siRNA at all single-nucleotide mismatched target sites. Nucleic Acids Res 33: 1671–1677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan J, Wainwright MS, Comeron JM, Saitou N, Sanders AR, Gelernter J, Gejman PV 2003. Synonymous mutations in the human dopamine receptor D2 (DRD2) affect mRNA stability and synthesis of the receptor. Hum Mol Genet 12: 205–216 [DOI] [PubMed] [Google Scholar]
- Fedor MJ, Uhlenbeck OC 1990. Substrate sequence effects on “hammerhead” RNA catalytic efficiency. Proc Natl Acad Sci 87: 1668–1672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fotiadis D, Liang Y, Filipek S, Saperstein DA, Engel A, Palczewski K 2003. Atomic-force microscopy: Rhodopsin dimers in native disc membranes. Nature 421: 127–128 [DOI] [PubMed] [Google Scholar]
- Frederick JM, Krasnoperova NK, Hoffmann K, Church-Kopish J, Ruther K, Howes K, Lem J, Baehr W 2001. Mutant rhodopsin transgene expression on a null background. Invest Ophthalmol Vis Sci 42: 826–833 [PubMed] [Google Scholar]
- Glushakova LG, Timmers AM, Issa TM, Cortez NG, Pang J, Teusner JT, Hauswirth WW 2006. Does recombinant adeno-associated virus-vectored proximal region of mouse rhodopsin promoter support only rod-type specific expression in vivo? Mol Vis 12: 298–309 [PubMed] [Google Scholar]
- Gorbatyuk MS, Knox T, LaVail MM, Gorbatyuk OS, Noorwez SM, Hauswirth WW, Lin JH, Muzyczka N, Lewin AS 2010. Restoration of visual function in P23H rhodopsin transgenic rats by gene delivery of BiP/Grp78. Proc Natl Acad Sci 107: 5961–5966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto Y, Peachey NS, Ripps H, Naash MI 1995. Functional abnormalities in transgenic mice expressing a mutant rhodopsin gene. Invest Ophthalmol Vis Sci 36: 62–71 [PubMed] [Google Scholar]
- Green ES, Menz MD, LaVail MM, Flannery JG 2000. Characterization of rhodopsin mis-sorting and constitutive activation in a transgenic rat model of retinitis pigmentosa. Invest Ophthalmol Vis Sci 41: 1546–1553 [PubMed] [Google Scholar]
- Gu D, Beltran WA, Li Z, Acland GM, Aguirre GD 2007. Clinical light exposure, photoreceptor degeneration, and AP-1 activation: A cell death or cell survival signal in the rhodopsin mutant retina? Invest Ophthal Vis Sci 48: 4907–4918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu D, Beltran WA, Pearce-Kelling S, Li Z, Acland GM, Aguirre GD 2009. Steroids do not prevent photoreceptor degeneration in the light-exposed T4R rhodopsin mutant dog retina irrespective of AP-1 inhibition. Invest Ophthalmol Vis Sci 50: 3482–3494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Z, Conley SM, Makkia R, Guo J, Cooper MJ, Naash MI 2012. Comparative analysis of DNA nanoparticles and AAVs for ocular gene delivery. PLoS ONE 7: e52189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrickson A, Hicks D 2002. Distribution and density of medium- and short-wavelength selective cones in the domestic pig retina. Exp Eye Res 74: 435–444 [DOI] [PubMed] [Google Scholar]
- Huang Y, Cideciyan AV, Aleman TS, Banin E, Huang J, Syed NA, Petters RM, Wong F, Milam AH, Jacobson SG 2000. Optical coherence tomography (OCT) abnormalities in rhodopsin mutant transgenic swine with retinal degeneration [letter]. Exp Eye Res 70: 247–251 [DOI] [PubMed] [Google Scholar]
- Kaeppel C, Beattie SG, Fronza R, van LR, Salmon F, Schmidt S, Wolf S, Nowrouzi A, Glimm H, von KC, et al. 2013. A largely random AAV integration profile after LPLD gene therapy. Nat Med 19: 889–891 [DOI] [PubMed] [Google Scholar]
- Kaushal S, Khorana HG 1994. Structure and function in rhodopsin. 7. Point mutations associated with autosomal dominant retinitis pigmentosa. Biochem 33: 6121–6128 [DOI] [PubMed] [Google Scholar]
- Kay CN, Ryals RC, Aslanidi GV, Min SH, Ruan Q, Sun J, Dyka FM, Kasuga D, Ayala AE, Van VK, et al. 2013. Targeting photoreceptors via intravitreal delivery using novel, capsid-mutated AAV vectors. PLoS ONE 8: e62097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp CM, Jacobson SG, Faulkner DJ 1988. Two types of visual dysfunction in autosomal dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci 29: 1235–1241 [PubMed] [Google Scholar]
- Kemp CM, Jacobson SG, Roman AJ, Sung CH, Nathans J 1992. Abnormal rod dark adaptation in autosomal dominant retinitis pigmentosa with proline-23-histidine rhodopsin mutation. Am J Ophthalmol 113: 165–174 [DOI] [PubMed] [Google Scholar]
- Kijas JW, Cideciyan AV, Aleman TS, Pianta MJ, Pearce-Kelling SE, Miller BJ, Jacobson SG, Aguirre GD, Acland GM 2002. Naturally occurring rhodopsin mutation in the dog causes retinal dysfunction and degeneration mimicking human dominant retinitis pigmentosa. Proc Natl Acad Sci 99: 6328–6333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komaromy AM, Acland GM, Aguirre GD 2008. Operating in the dark: A night-vision system for surgery in retinas susceptible to light damage. Arch Ophthalmol 126: 714–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunte MM, Choudhury S, Manheim JF, Shinde VM, Miura M, Chiodo VA, Hauswirth WW, Gorbatyuk OS, Gorbatyuk MS 2012. ER stress is involved in T17M rhodopsin-induced retinal degeneration. Invest Ophthalmol Vis Sci 53: 3792–3800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaVail MM, Yasumura D, Matthes MT, Drenser KA, Flannery JG, Lewin AS, Hauswirth WW 2000. Ribozyme rescue of photoreceptor cells in P23H transgenic rats: Long-term survival and late-stage therapy. Proc Natl Acad Sci 97: 11488–11493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Franson WK, Gordon JW, Berson EL, Dryja TP 1995. Constitutive activation of phototransduction by K296E opsin is not a cause of photoreceptor degeneration. Proc Natl Acad Sci 92: 3551–3555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Snyder WK, J Olsson JE, Dryja TP 1996. Transgenic mice carrying the dominant rhodopsin mutation P347S: Evidence for defective vectorial transport of rhodopsin to the outer segments. Proc Natl Acad Sci 93: 14176–14181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li ZY, Wong F, Chang JH, Possin DE, Hao Y, Petters RM, Milam AH 1998. Rhodopsin transgenic pigs as a model for human retinitis pigmentosa. Invest Ophthalmol Vis Sci 39: 808–819 [PubMed] [Google Scholar]
- Lin JH, Li H, Yasumura D, Cohen HR, Zhang C, Panning B, Shokat KM, LaVail MM, Walter P 2007. IRE1 signaling affects cell fate during the unfolded protein response. Science 318: 944–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Xia Z, Zhong X, Case CC 2002. Validated zinc finger protein designs for all 16 GNN DNA triplet targets. J Biol Chem 277: 3850–3856 [DOI] [PubMed] [Google Scholar]
- Maguire AM, Simonelli F, Pierce EA, Pugh EN Jr, Mingozzi F, Bennicelli J, Banfi S, Marshall KA, Testa F, Surace EM, et al. 2008. Safety and efficacy of gene transfer for Leber’s congenital amaurosis. N Engl J Med 358: 2240–2248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao H, James T, Schwein A, Shabashvili AE, Hauswirth WW, Gorbatyuk MS, Lewin AS 2011. AAV delivery of wild-type rhodopsin preserves retinal function in a mouse model of autosomal dominant retinitis pigmentosa. Hum Gene Ther 22: 567–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao H, Gorbatyuk MS, Rossmiller B, Hauswirth WW, Lewin AS 2012. Long-term rescue of retinal structure and function by rhodopsin RNA replacement with a single adeno-associated viral vector in P23H RHO transgenic mice. Hum Gene Ther 23: 256–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarty DM, Fu H, Monahan PE, Toulson CE, Naik P, Samulski RJ 2003. Adeno-associated virus terminal repeat (TR) mutant generates self-complementary vectors to overcome the rate-limiting step to transduction in vivo. Gene Ther 10: 2112–2118 [DOI] [PubMed] [Google Scholar]
- Mendes HF, van der SJ, Chapple JP, Cheetham ME 2005. Mechanisms of cell death in rhodopsin retinitis pigmentosa: Implications for therapy. Trends Mol Med 11: 177–185 [DOI] [PubMed] [Google Scholar]
- Millington-Ward S, Chadderton N, O’Reilly M, Palfi A, Goldmann T, Kilty C, Humphries M, Wolfrum U, Bennett J, Humphries P, et al. 2011. Suppression and replacement gene therapy for autosomal dominant disease in a murine model of dominant retinitis pigmentosa. Mol Ther 19: 642–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mingozzi F, High KA 2013. Immune responses to AAV vectors: Overcoming barriers to successful gene therapy. Blood 122: 23–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mowat FM, Petersen-Jones SM, Williamson H, Williams DL, Luthert PJ, Ali RR, Bainbridge JW 2008. Topographical characterization of cone photoreceptors and the area centralis of the canine retina. Mol Vis 14: 2518–2527 [PMC free article] [PubMed] [Google Scholar]
- Mueller C, Tang Q, Gruntman A, Blomenkamp K, Teckman J, Song L, Zamore PD, Flotte TR 2012. Sustained miRNA-mediated knockdown of mutant AAT with simultaneous augmentation of wild-type AAT has minimal effect on global liver miRNA profiles. Mol Ther 20: 590–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami Y, Matsumoto H, Roh M, Suzuki J, Hisatomi T, Ikeda Y, Miller JW, Vavvas DG 2012. Receptor interacting protein kinase mediates necrotic cone but not rod cell death in a mouse model of inherited degeneration. Proc Natl Acad Sci 109: 14598–14603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mussolino C, Della CM, Rossi S, Viola F, Di VU, Marrocco E, Neglia S, Doria M, Testa R, Giovannoni R, et al. 2011a. AAV-mediated photoreceptor transduction of the pig cone-enriched retina. Gene Ther 18: 637–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mussolino C, Sanges D, Marrocco E, Bonetti C, Di VU, Marigo V, Auricchio A, Meroni G, Surace EM 2011b. Zinc-finger-based transcriptional repression of rhodopsin in a model of dominant retinitis pigmentosa. EMBO Mol Med 3: 118–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noorwez SM, Sama RR, Kaushal S 2009. Calnexin improves the folding efficiency of mutant rhodopsin in the presence of pharmacological chaperone 11-cis-retinal. J Biol Chem 284: 33333–33342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Reilly M, Palfi A, Chadderton N, Millington-Ward S, Ader M, Cronin T, Tuohy T, Auricchio A, Hildinger M, Tivnan A, et al. 2007. RNA interference-mediated suppression and replacement of human rhodopsin in vivo. Am J Hum Genet 81: 127–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson JE, Gordon JW, Pawlyk BS, Roof D, Hayes A, Molday RS, Mukai S, Cowley GS, Berson EL, Dryja TP 1992a. Transgenic mice with a rhodopsin mutation (Pro23His): A mouse model of autosomal dominant retinitis pigmentosa. Neuron 9: 815–830 [DOI] [PubMed] [Google Scholar]
- Olsson JE, Gordon JW, Pawlyk BS, Roof D, Hayes A, Molday RS, Mukai S, Cowley GS, Berson EL, Dryja TP 1992b. Transgenic mice with a rhodopsin mutation (Pro23His): A mouse model of autosomal dominant retinitis pigmentosa. Neuron 9: 815–830 [DOI] [PubMed] [Google Scholar]
- Paddison PJ, Caudy AA, Bernstein E, Hannon GJ, Conklin DS 2002. Short hairpin RNAs (shRNAs) induce sequence-specific silencing in mammalian cells. Genes Dev 16: 948–958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palfi A, Millington-Ward S, Chadderton N, O’Reilly M, Goldmann T, Humphries MM, Li T, Wolfrum U, Humphries P, Kenna PF, et al. 2010. Adeno-associated virus-mediated rhodopsin replacement provides therapeutic benefit in mice with a targeted disruption of the rhodopsin gene. Hum Gene Ther 21: 311–323 [DOI] [PubMed] [Google Scholar]
- Pennesi ME, Nishikawa S, Matthes MT, Yasumura D, LaVail MM 2008. The relationship of photoreceptor degeneration to retinal vascular development and loss in mutant rhodopsin transgenic and RCS rats. Exp Eye Res 87: 561–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrs-Silva H, Dinculescu A, Li Q, Deng WT, Pang JJ, Min SH, Chiodo V, Neeley AW, Govindasamy L, Bennett A, et al. 2011. Novel properties of tyrosine-mutant AAV2 vectors in the mouse retina. Mol Ther 19: 293–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petters RM, Alexander CA, Wells KD, Collins EB, Sommer JR, Blanton MR, Rojas G, Hao Y, Flowers WL, Banin E, et al. 1997. Genetically engineered large animal model for studying cone photoreceptor survival and degeneration in retinitis pigmentosa [see comments]. Nat Biotechnol 15: 965–970 [DOI] [PubMed] [Google Scholar]
- Price BA, Sandoval IM, Chan F, Nichols R, Roman-Sanchez R, Wensel TG, Wilson JH 2012. Rhodopsin gene expression determines rod outer segment size and rod cell resistance to a dominant-negative neurodegeneration mutant. PLoS ONE 7: e49889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusky GT, Alam NM, Beekman S, Douglas RM 2004. Rapid quantification of adult and developing mouse spatial vision using a virtual optomotor system. Invest Ophthalmol Vis Sci 45: 4611–4616 [DOI] [PubMed] [Google Scholar]
- Punzo C, Kornacker K, Cepko CL 2009. Stimulation of the insulin/mTOR pathway delays cone death in a mouse model of retinitis pigmentosa. Nat Neurosci 12: 44–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajan RS, Kopito RR 2005. Suppression of wild-type rhodopsin maturation by mutants linked to autosomal dominant retinitis pigmentosa. J Biol Chem 280: 1284–1291 [DOI] [PubMed] [Google Scholar]
- Ross JW, Fernandez de Castro JP, Zhao J, Samuel M, Walters E, Rios C, Bray-Ward P, Jones BW, Marc RE, Wang W, et al. 2012. Generation of an inbred miniature pig model of retinitis pigmentosa. Invest Ophthalmol Vis Sci 53: 501–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakami S, Maeda T, Bereta G, Okano K, Golczak M, Sumaroka A, Roman AJ, Cideciyan AV, Jacobson SG, Palczewski K 2011. Probing mechanisms of photoreceptor degeneration in a new mouse model of the common form of autosomal dominant retinitis pigmentosa due to P23H opsin mutations. J Biol Chem 286: 10551–10567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal DJ, Dreier B, Beerli RR, Barbas CF III 1999. Toward controlling gene expression at will: Selection and design of zinc finger domains recognizing each of the 5′-GNN-3′ DNA target sequences. Proc Natl Acad Sci 96: 2758–2763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song S, Lu Y, Choi YK, Han Y, Tang Q, Zhao G, Berns KI, Flotte TR 2004. DNA-dependent PK inhibits adeno-associated virus DNA integration. Proc Natl Acad Sci 101: 2112–2116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan LS, Bowne SJ, Birch DG, Hughbanks-Wheaton D, Heckenlively JR, Lewis RA, Garcia CA, Ruiz RS, Blanton SH, Northrup H, et al. 2006. Prevalence of disease-causing mutations in families with autosomal dominant retinitis pigmentosa: A screen of known genes in 200 families. Invest Ophthalmol Vis Sci 47: 3052–3064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung CH, Schneider B, Agarwal N, Papermaster DS, Nathans J 1991. Functional heterogeneity of mutant rhodopsins responsible for autosomal dominant retinitis pigmentosa. Proc Natl Acad Sci 88: 8840–8844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung CH, Makino C, Baylor D, Nathans J 1994. A rhodopsin gene mutation responsible for autosomal dominant retinitis pigmentosa results in a protein that is defective in localization to the photoreceptor outer segment. J Neurosci 14: 5818–5833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan E, Wang Q, Quiambao AB, Xu X, Qtaishat NM, Peachey NS, Lem J, Fliesler SJ, Pepperberg DR, Naash MI, et al. 2001. The relationship between opsin overexpression and photoreceptor degeneration. Invest Ophthalmol Vis Sci 42: 589–600 [PubMed] [Google Scholar]
- Tso MO, Li WW, Zhang C, Lam TT, Hao Y, Petters RM, Wong F 1997. A pathologic study of degeneration of the rod and cone populations of the rhodopsin Pro347Leu transgenic pigs. Trans Am Ophthalmol Soc 95: 467–479 [PMC free article] [PubMed] [Google Scholar]
- Usui S, Komeima K, Lee SY, Jo YJ, Ueno S, Rogers BS, Wu Z, Shen J, Lu L, Oveson BC, et al. 2009. Increased expression of catalase and superoxide dismutase 2 reduces cone cell death in retinitis pigmentosa. Mol Ther 17: 778–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberghe LH, Bell P, Maguire AM, Cearley CN, Xiao R, Calcedo R, Wang L, Castle MJ, Maguire AC, Grant R, et al. 2011. Dosage thresholds for AAV2 and AAV8 photoreceptor gene therapy in monkey. Sci Transl Med 3: 88ra54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen XH, Shen L, Brush RS, Michaud N, Al-Ubaidi MR, Gurevich VV, Hamm HE, Lem J, DiBenedetto E, Anderson RE, et al. 2009. Overexpression of rhodopsin alters the structure and photoresponse of rod photoreceptors. Biophys J 96: 939–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson JH, Wensel TG 2003. The nature of dominant mutations of rhodopsin and implications for gene therapy. Mol Neurobiol 28: 149–158 [DOI] [PubMed] [Google Scholar]
- Wu TH, Ting TD, Okajima TI, Pepperberg DR, Ho YK, Ripps H, Naash MI 1998. Opsin localization and rhodopsin photochemistry in a transgenic mouse model of retinitis pigmentosa. Neuroscience 87: 709–717 [DOI] [PubMed] [Google Scholar]
- Yang Y, Mohand-Said S, Danan A, Simonutti M, Fontaine V, Clerin E, Picaud S, Leveillard T, Sahel JA 2009. Functional cone rescue by RdCVF protein in a dominant model of retinitis pigmentosa. Mol Ther 17: 787–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Y, Cai X, Cullen BR 2005. Use of RNA polymerase II to transcribe artificial microRNAs. Methods Enzymol 392: 371–380 [DOI] [PubMed] [Google Scholar]