

Abstract
Alkylation of 2-mercaptopyridine with 1,2-dibromoethane affords a cyclic dihydrothiazolopyridinium salt that can serve as a precursor of 2-aminopyridines. Its reaction with primary or secondary amines, either neat or in DMSO, under mild conditions gives the title compounds.
Keywords: aminopyridine, heteroaryl amine, thiazolopyridinium, N-alkyl pyridinium
Introduction
2-Aminopyridines serve as useful chelating ligands in a variety of inorganic and organometallic applications.1 The high incidence of pharmacological activity among heteroaromatic amines also has stimulated many research efforts to prepare compounds of this structural type.2 The most common syntheses of 2-aminopyridines involve either the N-alkylation of 2-aminopyridine (sidechain elaboration),3 or the substitution of a 2-halopyridine by an amine. The former approach often requires multiple steps to ensure that over-alkylation does not occur. The latter method has been applied to fluoro-,4 chloro-,5 bromo-6 and iodo-pyridines7 using either lithiated amines, high temperature, transition metal catalysis or combinations thereof.8 Indeed, a powerful synthetic approach to aminopyridines uses the Buchwald-Hartwig amination9 wherein the halide substitution is facilitated by palladium (e.g., cat. Pd2(dba)3)10 at an elevated temperature.11 Copper-catalyzed methods also have been developed for halide to amine substitutions in heteroaromatic systems.12
In an alternative approach, the formation of N-alkyl pyridinium salts has been used to improve the nucleophilic addition of amines to C(2).13 One drawback of this strategy, depending on substitution pattern, is competing addition to C(4) of the N-alkyl pyridinium salt. However, given that nucleophilic substitution reactions of activated aryl halides are highly regioselective, we surmised that a combination of pyridinium salt activation and a C(2)-leaving group might provide a mild, regioselective method for pyridyl C(2)–N bond formation. We tested this concept by preparing a pyridinium salt having a C(2)-thioether as the leaving group14 and report herein the results of its nucleophilic addition-elimination reactions.
Results and Discussion
Treatment of 2-mercaptopyridine (1) with 1,2-dibromoethane in DMF cleanly furnished dihydrothiazolopyridinium salt 215 as a tan solid suitable for use in subsequent reactions without further purification (Scheme 1).16 We next examined the reaction of 2 with morpholine as a test case. Although the reaction was sluggish and the yield low (ca. 23% at 48 h), we were gratified to find that the reaction of 2 with 2.0 equivalents of morpholine in DMSO at room temperature afforded a single regioadduct, 2-morpholinopyridine (3a). Doubling the equivalents of amine and warming the reaction to 50 °C improved the yield of 3a to 75%. We also found that for simple, liquid amines, such as morpholine, solvent-free conditions afforded the corresponding 2-aminopyridines regioselectively and in good yield (Table). Presumably the transformation proceeds via amine addition to C(2) of 2 leading to adduct [4]. We have not isolated the postulated intermediates [4] nor observed sulfur-containing by-products, such as ethylene sulfide or the corresponding amino adducts of ethylene sulfide; thus, the mechanism of the reaction remains unclear.
Scheme 1.
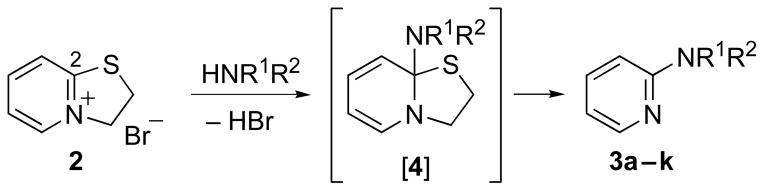
Synthesis of 2-aminopyridines.
Table.
Synthesis of 2-aminopyridines.
| Entry | Amine | Conditionsb | Yield 3c |
|---|---|---|---|
| a |

|
A B C |
75 78 94 |
| b |

|
A | 94 |
| c |
|
A B C |
78 77 75 |
| d |
|
A B C |
56 63 65 |
| e |
|
A B C |
65 57 79 |
| f |
|
A B C |
75 59 61 |
| g |
|
A - |
36 48d |
| h |
|
A | 73 |
| i |
|
A | 71 |
| j |
|
A | 86 |
| k |
|
A | 78 |
Reactions analyzed after 48h; the spectral data for products 3a-f and 3h-j are consistent with previous reports.
A: 4.0 eq. amine, DMSO, 50 °C; B: neat amine (ca. 0.4 – 0.5M), rt; C: neat amine (ca. 0.4–0.5M), 50 °C.
percent, after chromatography.
4.0 eq. amine, DMSO, rt.
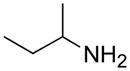
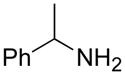
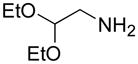
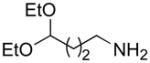
From the entries in the Table, optimal conversions of the pyridinium salt into 2-aminopyridines generally required warming to 50 °C. Reaction temperatures above 50 °C effected a gradual decomposition of 2. The only case not improved by warming was the reaction of 2 with benzyl glycine (entry g). We noted that considerable ester cleavage occurred on warming this reaction. A change to the t-butyl ester of glycine did not improve the yield (data not shown). The modest yield obtained for formation of glycine derivative 3g, however, does represent an improvement in synthesis of such N-pyridyl glycine analogs. Previous attempts to react glycine or the tert-butyl ester of glycine with 2-fluoropyridine under various conditions are reported to fail or give polymerized products.17 Other glycine equivalents, such as allylamine and ethanolamine, required reaction with 2-fluoropyridine under microwave irradiation at 190 °C and 210 °C, respectively, to give the corresponding substituted pyridines in 64% and 74% yield.17 In comparison, the reactions of allylamine and ethanolamine with 2 (entries h and i) proceeded smoothly under milder conditions. The mild conditions of the present method also tolerate acetal functionality. The reactions of aminoacetaldehyde diethyl acetal and 4-amino-butyraldehyde diethyl acetal with 2 (entries j and k) gave 3j18 and 3k in good yields.
In regards to the time dependence of the reaction, an nmr experiment (DMSO-d6 version of entry c at 50 °C)19 showed that the reaction was essentially 80% complete within 24 hours, but required more than one week to reach completion. The use of excess amine, albeit the principal drawback of the present method, improves the efficiency of the transformation. We also examined the reactions of 2 in water since the salt is freely soluble in water and to explore whether the carbon–heteroatom bond formation behaves analogously to other click chemistry examples.20 Treatment of 2 with either piperidine, N,N-dimethylamine, or N-methylbutylamine (4 equivalents of amine in water at 50 °C) gave the corresponding 2-aminopyridines in 41%, 22% and 19% yields, respectively. The lower yields in these reactions relative to the neat or DMSO reactions are due to a faster decomposition of 2 in warm water.
To determine the susceptibility of 2 toward base-induced elimination, we reacted 2 with the non-nucleophilic base DBU (Scheme 2). Heating 2 with DBU in DMSO resulted in formation of N-vinyl pyridine-2-thione (5) in good yield. We had not observed this elimination product in any of the aforementioned syntheses of 2-aminopyridines. The two-step synthesis of 5 represents a convenient and improved means for N-vinylation of pyridine-2-thione.21
Scheme 2.
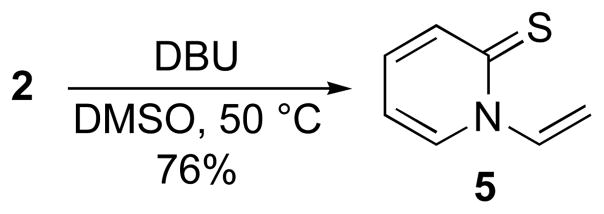
Synthesis of N-vinylpyridine-2-thione.
Finally, we briefly examined extension of this method to quinoline. In similar manner to the formation of pyridinium salt 2, 2-mercaptoquinoline (6, Scheme 3) was transformed into the dihydrothiazoloquinolinium salt 7.22 Reaction of 7 with either dimethylamine or piperidine under the conditions developed for 2 showed that amine addition occurred to give the 2-substituted quinolines. Again, the substitution reactions proceeded under milder conditions relative to previously reported halide displacement methods.23
Scheme 3.
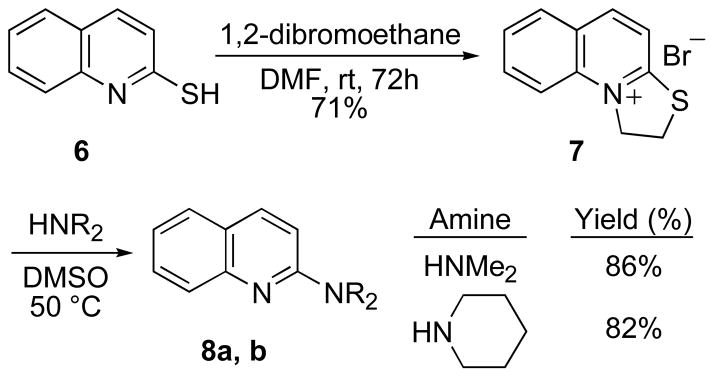
Synthesis of 2-aminoquinolines.
Conclusion
In summary, we have described a new, two-step synthesis of 2-aminopyridines from 2-mercaptopyridine. Conversion of the dihydrothiazolopyridinium salt 2 into the title compounds requires only stirring with excess amines.
Experimental Section
General
Prior to use, DMF and DMSO were distilled from CaH2 under reduced pressure. After reaction work-up, solutions were dried using Na2SO4 and solvents were subsequently removed by rotary evaporation. Nuclear magnetic resonance (NMR) spectra were recorded in CDCl3 unless otherwise indicated. Infrared spectra were recorded on a Mattson 5020 FTIR spectrometer. Melting points are uncorrected. Elemental analyses were performed by Midwest Microlabs (Indianapolis, IN).
2,3-Dihydrothiazolo[3,2-a]pyridinium bromide (2)
To a solution of 2-mercaptopyridine (5.0 g, 45 mmol) in DMF (0.5 L) at room temperature was added 1,2-dibromoethane (19.4 mL, 225 mmol). The reaction mixture was stirred 72 h whereupon the precipitate, pyridinium bromide 2, was collected by filtration. The filtrate was washed with a small portion of dichloromethane and dried under vacuum to afford 2 (7.2 g, 73%) in essentially pure form; mp 235–236 °C (lit.15 222 °C). A second crop of 2 was obtained (1.7 g) by concentrating the mother liquor (combined yield: 8.9 g, 91%); 1H NMR (300 MHz) δ 8.94 (d, J = 6.3 Hz, 1H), 8.31 (m, 1H), 8.14 (d, J = 6.3 Hz, 1H), 7.71 (m, 1H), 5.10 (t, J = 6.3 Hz, 2H), 3.82 (t, J = 6.3 Hz, 2H); 13C NMR (75 MHz) δ 159.4, 144.4, 142.7, 122.9, 122.2, 60.0, 30.0; Anal. Calcd for C7H8BrNS: C, 38.55; H, 3.70; N, 6.42. Found: C, 38.59; H, 3.65; N, 6.42.
General procedure for 2-aminopyridine formation; Method A
To a solution of salt 2 (300 mg, 1.38 mmol) in DMSO (5 mL) at room temperature was added the amine (4.8 mmol) in one portion. The reaction was warmed to 50 °C and stirred 48 h. After cooling to room temperature, the reaction mixture was diluted with water (20 mL) and 0.5M aq. NaOH (5 mL). The resultant solution was extracted with diethyl ether (5X) and the combined organic extract was washed with brine and dried (anhydr. Na2SO4). The solvent was removed and the residue purified by column chromatography (SiO2).
Synthesis of N-(2-pyridyl)-glycine benzyl ester (3g)
To a solution of pyridinium bromide 2 (300 mg, 1.38 mmol) and the p-toluenesulfonate salt of glycine benzyl ester (1.85 g, 5.5 mmol) in dry DMSO (7 mL) at room temperature was added triethylamine (1.16 mL, 8.25 mmol). The reaction was stirred 48 h at room temperature and then quenched by addition of water (20 mL) and saturated aq. NaHCO3 (50 mL). The resultant solution was extracted with Et2O (5×50 mL), and the combined organic extract was dried (Na2SO4). The solvents were removed and the residue was purified by column chromatography (SiO2) eluting with mixture of EtOAc:hexane (35:65) to obtain ester 3g (160 mg, 48%) as a colorless oil; IR (neat) 3404, 3029, 2931, 1740, 1604 cm−1; 1H NMR (500 MHz) δ 8.08 (d, J = 4.5 Hz, 1H), 7.42–7.25 (m, 6H), 6.61 (m, 1H), 6.45 (d, J = 8.5 Hz, 1H), 5.20 (s, 2H), 5.01 (br. s, NH), 4.19 (d, J = 5.0 Hz, 2H); 13C NMR (125 MHz) δ 171.4, 157.7, 147.9, 137.7, 135.7, 128.8, 128.6, 128.4, 113.9, 108.7, 67.1, 44.0; HRMS, m/z [M+H]+ calcd for C14H14N2O2 243.1133, found: 243.1133.
4-N-(2-Pyridyl)amino-butyraldehyde diethyl acetal (3k)
Following method A outlined above, salt 2 (0.20 g, 0.92 mmol) was transformed into aminopyridine 3k (0.17 g, 78%); IR (neat) 3376, 3261, 2972, 1602 cm−1; 1H NMR (500 MHz) δ 8.0–7.98 (m, 1H), 7.31 (t, J = 8 Hz, 1H), 6.46 (t, J = 6 Hz, 1H), 6.29 (d, J = 8.5 Hz, 1H), 4.70 (br s, 1H), 4.45–4.43 (m, 1H), 3.60–3.50 (m, 2H), 3.45–3.40 (m, 2H), 3.24–3.21 (m, 2H), 1.65–1.64 (m, 4H), 1.15–1.10 (m, 6H); 13C NMR (125 MHz) δ 158.7, 147.9, 137.2, 112.4, 106.4, 102.5, 61.0, 41.8, 31.0, 24.6, 15.2; HRMS, m/z [M+H]+ calcd for C13H22N2O2 239.1759, found 239.1753.
N-Vinylpyridine-2-thione (5)
Following method A, 100 mg (0.46 mmol) of salt 2 was treated with DBU (0.28 mL, 1.83 mmol) to afford 5 (47 mg, 76%) as yellow solid, mp 67–68 °C (lit.2172 °C); 1H NMR (500 MHz) δ 7.86 (dd, J = 15.5, 8.5 Hz, 1H), 7.75 (d, J = 7.0 Hz, 1H), 7.60 (d, J = 9.0 Hz, 1H), 7.19 (t, J = 8.5 Hz, 1H), 6.66 (t, J = 6.5 Hz, 1H), 5.38 (dd, J = 15.5, 1.5 Hz, 1H), 5.31 (dd, J = 8.0, 1.5 Hz, 1H); 13C NMR (125 MHz) δ 180.1, 138.8, 137.3, 136.2, 134.1, 113.4, 109.2.
1,2-Dihydrothiazolo[3,2-a]quinolinium bromide (7)
To a solution of 2-quinolinethiol (6) (1.0 g, 6.20 mmol) in DMF (50 mL) at room temperature was added 1,2-dibromoethane (2.67 mL, 31 mmol). The reaction mixture was stirred 72 h. Et2O (200 mL) was added to the reaction mixture to cause precipitation of the product. The solids were collected by filtration and washed with a small portion of Et2O and dried under vacuum to afford 7 (1.17 g, 71%); mp 246–247 °C (lit.21 234 °C); 1H NMR (500 MHz, DMSO-d6) δ 8.97–8.85 (m, 1H), 8.38–8.28 (m, 1H), 8.25–8.05 (m, 3H), 7.92–7.80 (m, 1H), 5.41–5.30 (m, 2H), 4.05–3.90 (m, 2H); 13C NMR (125 MHz, DMSO-d6) δ 165.3, 144.3, 137.6, 134.7, 130.0, 128.0, 126.0, 118.5, 56.8, 29.2; Anal. Calcd for C11H10BrNS·1/2 H2O: C, 47.62; H, 3.96; N, 5.05. Found: C, 47.85; H, 3.84; N, 5.09.
Supplementary Material
Acknowledgments
Funding from the NIH (NS-040489) is gratefully acknowledged.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Footnotes
- 1.(a) Kawasaki M, Suzuki Y, Terashima S. Chem Lett. 1984:239. [Google Scholar]; (b) Kempte R, Brenner S, Arndt P. Organometallics. 1996;15:1071. [Google Scholar]; (c) Fuhrmann H, Brenner S, Arndt P. Inorg Chem. 1996;35:6742. doi: 10.1021/ic960182r. [DOI] [PubMed] [Google Scholar]; (d) Scott NM, Schareina T, Tok O, Kempe R. Eur J Inorg Chem. 2004:3297. [Google Scholar]
- 2.(a) Lechat P, Tesleff S, Bownan WC. Aminopyridines and Similarly Acting Drugs. Pergamon Press; Oxford: 1982. [Google Scholar]; (b) Zhang J, Pettersson HI, Huitema C, Niu C, Yin J, James MNG, Eltis LD, Vederas JC. J Med Chem. 2007;50:1850. doi: 10.1021/jm061425k. [DOI] [PubMed] [Google Scholar]; (c) Queiroz MJRP, Ferreira ICFR, Calhelha RC, Estevinho LM. Biorg Med Chem. 2007;15:1788. doi: 10.1016/j.bmc.2006.11.035. [DOI] [PubMed] [Google Scholar]
- 3.(a) Alcaide B, Plumet J, Sierra MA. J Org Chem. 1990;55:3143. [Google Scholar]; (b) Abarghaz M, Kerbal A, Bourguignon JJ. Tetrahedron Lett. 1995;36:6463. [Google Scholar]; (c) Watanabe Y, Morisaki Y, Kondo T, Mitsudo T. J Org Chem. 1996;61:4214. doi: 10.1021/jo9516289. [DOI] [PubMed] [Google Scholar]; (d) Krein DM, Lowary TL. J Org Chem. 2002;67:4965. doi: 10.1021/jo020057z. [DOI] [PubMed] [Google Scholar]; (e) Sato S, Sakamoto T, Miyazawa E, Kikugawa Y. Tetrahedron. 2004;60:7899. [Google Scholar]; (f) Singh OM, Singh SJ, Kim SN, Lee SG. Bull Korean Chem Soc. 2007;28:115. [Google Scholar]
- 4.Pasumansky L, Hernandez AR, Gamsey S, Goralski CT, Singaram B. Tetrahedron Lett. 2004;45:6417. [Google Scholar]
- 5.(a) Grasa GA, Viciu MS, Huang J, Nolan SP. J Org Chem. 2001;66:7729. doi: 10.1021/jo010613+. [DOI] [PubMed] [Google Scholar]; (b) Rataboul F, Zapf A, Jackstell R, Harkal S, Riermeier T, Monsees A, Dingerdissen U, Beller M. Chem Eur J. 2004;10:2983. doi: 10.1002/chem.200306026. [DOI] [PubMed] [Google Scholar]
- 6.(a) Strekowski L, Dworniczak M, Kowalewski A. Polish J Chem. 1980;54:1557. [Google Scholar]; (b) Crust EJ, Clarke AJ, Deeth RJ, Morton C, Scott P. J Chem Soc, Dalton Trans. 2004;42:4050. doi: 10.1039/b413023e. [DOI] [PubMed] [Google Scholar]
- 7.Okano K, Tokuyama H, Fukuyama T. Org Lett. 2003;5:4987. doi: 10.1021/ol035942y. [DOI] [PubMed] [Google Scholar]
- 8.For other 2-aminopyridine syntheses, see: Heller B, Sundermann B, Buschmann H, Drexler HJ, You J, Holzgrabe U, Heller E, Oehme G. J Org Chem. 2002;67:4414. doi: 10.1021/jo011032n.Penney JM. Tetrahedron Lett. 2004;45:2667.
- 9.(a) Yang BH, Buchwald SL. J Organomet Chem. 1999;576:125. [Google Scholar]; (b) Hartwig JF. In: In Modern Amination Methods. Ricci A, editor. Wiley–VCH; Weinheim, Germany: 2000. [Google Scholar]; (c) Muci AR, Buchwald SL. Top Curr Chem. 2002;219:131. [Google Scholar]; (d) Zim D, Buchwald SL. Org Lett. 2003;5:2413. doi: 10.1021/ol034561h. [DOI] [PubMed] [Google Scholar]
- 10.Urgaonkar S, Verkade JG. J Org Chem. 2004;69:9135. doi: 10.1021/jo048716q. [DOI] [PubMed] [Google Scholar]
- 11.(a) Wagaw S, Buchwald SL. J Org Chem. 1996;61:7240. doi: 10.1021/jo9612739. [DOI] [PubMed] [Google Scholar]; (b) Munson PM, Thompson WJ. Synth Commun. 2004;34:759. [Google Scholar]; (c) Loones KTJ, Maes BUW, Dommisse RA, Lemiere GLF. Chem Commun. 2004;21:2466. doi: 10.1039/b409161b. [DOI] [PubMed] [Google Scholar]; (d) Navarro O, Marion N, Mei J, Nolan SP. Chem Eur J. 2006;12:5142. doi: 10.1002/chem.200600283. [DOI] [PubMed] [Google Scholar]; (e) Begouin A, Hesse S, Queiroz MJRP, Kirsch G. Eur J Org Chem. 2007;10:1678–1682. [Google Scholar]; (f) Li JJ, Wang Z, Mitchell LH. J Org Chem. 2007;72:3606. doi: 10.1021/jo070366v. [DOI] [PubMed] [Google Scholar]; (g) Schön U, Messinger J, Buckendahl M, Prabhu MS, Konda A. Tetrahedron Lett. 2007;48:2519. [Google Scholar]
- 12.(a) Lang F, Zewge D, Houpis IN, Volante RP. Tetrahedron Lett. 2001;42:3251–3254. [Google Scholar]; (b) Antilla JC, Baskin JM, Barder TE, Buchwald SL. J Org Chem. 2004;69:5578. doi: 10.1021/jo049658b. [DOI] [PubMed] [Google Scholar]
- 13.(a) Angelino SAGF, van Veldhuizen A, Buurman DJ, Van Der Plas HC. Tetrahedron. 1984;40:433. [Google Scholar]; (b) Lavilla R, Gotsens T, Guerrero M, Masdeu C, Santano MC, Minguillon C, Bosch J. Tetrahedron. 1997;53:13959. [Google Scholar]
- 14.Amine substitution of aryl sulfoxide and sulfonate groups is known; for example, see: Kawai T, Kodera Y, Furukawa N, Oae S, Ishida M, Takeda T, Wakabaysashi S. Phosphorus and Sulfur and the Related Elements. 1987;34:139.Andreassen EJ, Bakke JM, Sletvold I, Svenson H. Org Biomol Chem. 2004;2:2671. doi: 10.1039/B408840A.
- 15.Singh H, Malhotra SC. Indian J Chem. 1981;20B:213. [Google Scholar]
- 16.Although unnecessary for subsequent reactions with amines, pyridinium bromide 2 is readily recrystallized from ethanol.
- 17.Blomberg D, Brickmann K, Kihlberg J. Tetrahedron. 2006;62:10937. [Google Scholar]
- 18.Previously prepared in 47% yield from 2-aminopyridine; see: Ziegler R, Ho B, Castagnoli N., Jr J Med Chem. 1981;24:1133. doi: 10.1021/jm00142a004.
- 19.The reaction progress is conveniently monitored by observing the pyridyl-H resonances in 1H nmr between δ 6–9 ppm.
- 20.Kolb HC, Finn MG, Sharpless KB. Angew Chem Intl Ed. 2001;40:2004. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 21.Ulsaker GA, Undheim K. Acta Chem Scand. 1977;B31:917. [Google Scholar]
- 22.Sugiyama M, Ohi R, Shishido T, Omura M. DE 71–2133054 Ger Offen. 1972
- 23.(a) Gupton JT, Idoux JP, Baker G, Colon C, Crews AD, Jurss CD, Rampi RC. J Org Chem. 1983;48:2933. [Google Scholar]; (b) Cho YH, Park JC. Tetrahedron Lett. 1997;38:8331. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.