

Abstract
Atypical invariant chain (Ii)3 CLIP fragments (CLIP2) have been found in association with HLA-DQ2 (DQ2) purified from cell lysates. We mapped the binding register of CLIP2 (Ii 96-104) to DQ2 and found proline at the P1 position, in contrast to the canonical CLIP1 (Ii 83-101) register with methionine at P1. CLIP1/2 peptides are the predominant peptide species, even for DQ2 from HLA-DM (DM)-expressing cells. We hypothesized that DQ2-CLIP1/2 might be poor substrates for DM. We measured DM-mediated exchange of CLIP peptides for high affinity indicator peptides and found it is inefficient for DQ2 compared to HLA-DR3 (DR3). DM-DQ binding and DM chaperone effects on conformation and levels of DQ are also reduced for DQ2, compared to DQ1. We suggest that the unusual interaction of DQ2 with Ii and DM may provide a basis for the known disease associations of DQ2.
Keywords: Antigen Presentation/Processing MHC class II, invariant chain, HLA-DM
Introduction
MHC class II molecules are αβ dimeric membrane glycoproteins expressed on the surface of antigen presenting cells of the immune system. Their function is to present peptide antigens derived from endosomal proteins to CD4+ T cells. To ensure that newly synthesized class II molecules intersect with and bind endocytosed peptides, the αβ dimer associates with the chaperone molecule invariant chain (Ii), which both prevents premature binding of ligands and promotes the entrance of the class II-Ii complexes into the endocytic pathway (1).
Ii is progressively degraded by proteases in the endocytic pathway, leaving CLIP (class II-associated Ii peptides) bound to the binding groove of all previously studied MHC class II molecules. The subsequent release of CLIP in late endosomes is required for antigenic peptide binding, a process known to be accelerated by HLA-DM (DM). The MHC II-peptide complex is then transported to the cell surface for inspection by CD4+ T cells (2).
Previous data on peptides eluted from HLA-DQ2 (DQ2) molecules (DQA1*0501, DQB1*0201) that were affinity purified from B lymphoblastoid cell lines (B-LCL), revealed the presence of large amounts of Ii-derived peptides (54% of total) (3,4). Intriguingly, the population of Ii peptides was dominated by a unique peptide species, here termed CLIP2, which had lower IC50 values/higher affinity for DQ2 when compared to the conventional CLIP (CLIP1) peptides (3,4). DQ2 is part of the “autoimmune” HLA-DR3/-DQ2 haplotype associated with multiple autoimmune diseases (5,6). Motivated by this fact, we investigated the binding of CLIP2 to DQ2 and asked whether the presence of substantial amounts of CLIP peptides reflected altered interaction of DQ2 with DM, in comparison to DM interaction with HLA-DR3 (DR3) and HLA-DQ1 (DQ1).
Material and Methods
Cell lines
The following Epstein Barr-virus transformed B lymphoblastoid cell lines (B-LCL) were used in this study: CD114 (from a celiac disease patient) expressing DQ2, DR3; 2.2.93 expressing DP4, DQ1, DR1, DR3, and no DM (7); 8.1.6 expressing DP4, DQ2, DR3 (8); 9.5.3 expressing DP4, DQ2, DR3 and no DM (8); 3.1.3 expressing DP4 and DQ1 (9); 9.22.3 expressing DP4 and DQ2 (8). The DR3 expressing cells also express DR52a. The DM transfectants of 2.2.93 and 9.5.3 (2.2.93-DM and 9.5.3-DM, respectively) were generated by retroviral transduction as described (7).
Antibodies
Antibodies used in this study were B8.11 (IgG2b, anti-DR, kind gift of B.Malissen), L243 (IgG2a, anti-DR), ISCR3 (IgG2b, anti-DR), SPV-L3 (IgG2a, anti-DQ, kind gift of H.Spits), Ia3 (IgG2a, anti-DQ, Biodesign), 2.12.E11 (IgG1, anti-DQ2), B7/21.2 (IgG3, anti-DP), XD5.A11 (IgG1, anti-class II), 5C1 (IgG1, anti-DM), 16.23 (IgG3, anti-DR3), anti-HLA-DM conjugated with PE (IgG1, BD Pharmingen), CerCLIP (IgG1, anti-human CLIP, kind gift of P.Cresswell)
Flow cytometry
For cell surface staining, cells were incubated (40 min, on ice) with primary antibodies and washed. Bound antibody was detected by incubation (40 min, on ice) with goat F(ab')2 anti-mouse IgG (H+L) conjugated with FITC or PE (CalTag). For intracellular staining, cells were fixed and permeabilized using the Cytofix/Cytoperm kit (BD Pharmingen) and then stained using PE-conjugated antibody. Cells were analyzed using a FACScan flow cytometer (Becton Dickinson) and data were analyzed using FlowJo software (Tree Star, Inc).
Pulse-chase immunoprecipitation
Cells were washed three times and starved for 1-2 hours in Met/Cys-free RPMI containing 10% dialyzed FBS (Invitrogen). Cells were pulsed with 0.1 μCi/ml ExpreSS [35S] labeling mix (Perkin Elmer) for the indicated times, then washed and chased in complete RPMI containing 10% FBS and 2mM L-glutamine (37°C, 5% CO2). Aliquots of cells were collected and washed at the indicated time points and lysed in buffer (Tris-HCl, pH 8.0 with MgCl2, 1% NP-40 and complete protease inhibitors (Roche Diagnostics)) at 4°C for 1–2 hrs. Lysates were pre-cleared three times with normal mouse serum and Pansorbin® (Calbiochem) and once with protein A sepharose beads (Amersham), then normalized based on total radioactivity, measured by beta-counter (Wallac). Immunoprecipitations were performed by incubating the normalized lysates with protein A sepharose beads conjugated with class II-specific antibodies overnight at 4°C. Proteins were eluted by boiling the precipitates in reducing SDS sample buffer containing 62.5 mM Tris-HCl (pH 6.8), 1% SDS, 3% glycerol, 0.007% bromophenolblue, 1% 2-mercaptoethanol, and then separated by SDS-PAGE. Bands were visualized by exposing dried gels to radiography films (Kodak).
Generation of soluble DQ2- and DR3-peptide complexes
Water soluble DQ2 (sDQ2), engineered to have a peptide ligand tethered to β-chain via a thrombin cleavable linker and a Fos-Jun leucine zipper pair replacing the transmembrane domains was expressed in insects cells using a baculovirus expression vector system (10). The sDQ2 molecules with deamidated αI gliadin peptide (QLQPFPQPELPY) and deamindated γI gliadin peptide (PEQPQQSFPEQERP) have been previously described (10). The insertion of CLIP1 and CLIP2 sequences (PVSKMRMATPLLMQA and MATPLLMQALPMGAL respectively), into the DQ2 β-chain construct were completed by ligation of hybridized oligonucleotides (corresponding to the peptide sequences) with single strand overhangs complementary to those of the MroI/NarI restriction sites of the original β-chain construct. The sDQ2-CLIP1 M91A and sDQ2-CLIP M91P complexes were constructed by performing site-directed mutagenesis of the sDQ2-CLIP1 plasmid using primers containing the desired nucleotide change (Quikchange Multi Site-Directed Mutagenesis Kit, Stratagene) according to manufacturer's instructions. The water soluble DR3-CLIP1 (sDR3-CLIP1) construct was created in a similar fashion as the original soluble DQ2 construct. cDNA was synthesized by RT-PCR (Superscript II, Invitrogen) from mRNA (RNeasy, QIAGEN) isolated from the 8.1.6 cell line, according to the manufacturers' instructions. The DRA1*0101 and DRB1*0301 sequences were PCR-amplified from cDNA, where the DRA1*0101 primer contained an N-terminal BglII restriction site. The Jun-Fos leucine-zippers from the soluble DQ2-CLIP1 construct containing C-terminal BglII (DQα-chain) or BamHI (DQβ-chain) restriction sites were PCR-amplified with primers containing DRα or DRβ complimentary tails, allowing for megaprimer PCR attachment to the C-terminal ends of the DRα (Fos) and DRβ (Jun) constructs. The N-terminal end of the sDQ2-CLIP1 molecule containing the BamHI restriction site, leader sequence, CLIP1, and linkers was PCR-amplified with primers containing DRβ-complimentary tails, allowing for megaprimer PCR attachment to the N-terminal end of the DRβ-chain. The constructs were cloned into the pAcAB3 vector by means of BamHI (DRβ) and BglII (DRα) restriction enzymes. The oligonucleotides used for the construction of the molecules can be found in Table I Constructs were verified by DNA sequencing. The sDQ2 and sDR3 molecules were affinity purified using the mAbs 2.12.E11 or B8.11, respectively.
TABLE I.
Oligonucleotides used in the construction of sDQ2-peptide and sDR3-CLIP1 complexes.
| CLIP1 oligonucleotides | Fwd | 5'-CCGGACCTGTGAGCAAGATGAGAATGGCCACCCCCCTGCTCATGCAGGCTGG-3' |
| Rev | 5'-CCGGAATGGCCACCCCCCTCCTTATGCAGGCACTGCCTATGGGCGCTCTGGG-3' | |
| CLIP2 oligonucleotides | Fwd | 5'-CGCCAGCCTGCATGAGCAGGGGGGTGGCCATTCTCATCTTGCTCACAGGT-3' |
| Rev | 5'-CGCCCAGAGCGCCCATAGGCAGTGCCTGCATAAGGAGGGGGGTGGCCATT-3' | |
| CLIP1 M91A mutagenesis primer | Fwd | 5'-CCTGTGAGCAAGGCGAGAATGGCCACCCC-3' |
| CLIP1 M91P mutagenesis primer | Fwd | 5'-CCTGTGAGCAAGCCGAGAATGGCCACCCC-3' |
| DRα amplification primers | Fwd | 5'-GAAGATCTATGGCCATAAGTGGAGTC-3' |
| Rev | 5'-ATAATGATGCCCACCAGACC-3' | |
| DRβ amplification primers | Fwd | 5'-CGGCGGAGGCGGTAGTGGGGACACCAGACCACGTTT-3' |
| Rev | 5'-CCGCGACCTTCAATGTCTACCTTGCTCTGTGCAGATTCAG-3' | |
| Fos amplification primers | Fwd | 5'-TCTCCCAGAGACTACAGAGAACGTGGACATCGAAGGACGTGG-3' |
| Rev | 5'-GAAGATCTTCAGGCGGCCAGGATG-3' | |
| Jun amplification primers | Fwd | 5'-GTAGACATTGAAGGTCGCGG-3' |
| Rev | 5'-GAGGATCCTTAGTTGTGCCATTCT-3' | |
| DQB1*0201-CLIP1+LS amplification primers | Fwd | 5'-GAGGATCCATGTCCTGGAAAAAGG-3' |
| Rev | 5'-ACTACCGCCTCCGCCG-3' |
Generation of soluble DM
Soluble DM molecules were produced in stably transfected S2 cells and purified by immunoaffinity chromatography and size exclusion chromatography (11).
Fluid-phase peptide binding assay
Detergent-solubilized DQ2 and DR3 molecules were purified from the CD114 cell line and used in a fluid-phase competitive inhibition assay with 125I labeled indicator peptides as previously described (12). Briefly, labeled indicator peptides (KPLLIIAEDVEGEY; MB 65-kDa Hsp 243–255Y or EPRAPWIEQEGPEYW; HLA class I α 46–60) for DQ2 and KTIAYDEEARR; MT 65kDa 3–13 for DR3) and unlabeled peptides were incubated with DQ2 or DR3 molecules overnight at 37°C in a pH 4.9 citrate phosphate buffer. Complexes of peptide and DQ2/DR3 molecules were separated from unbound peptides by spin column chromatography. Radioactivity was determined, and the concentrations of the competing peptides required for half-maximal inhibition of the binding of the indicator peptide (IC50) were calculated.
Peptide binding assays measuring the DM effect using water soluble peptide-linked molecules were done in a similar manner. Molecules were treated with 1/16 u thrombin (Novagen) for 90 minutes at RT followed by inhibition with 1 mM AEBSF (Sigma). The cleaved molecules were incubated overnight at 37°C in a pH 4.3 citrate phosphate buffer in the presence or absence of various amounts of soluble DM (as indicated in figures) and, in some experiments, non-cleaved molecules (0-7 μM) as well, along with the 125I-labeled indicator peptides and protease inhibitors.
Solid-phase peptide exchange assay
HLA class II molecules captured from cell lysates were used for a peptide exchange assay (13). Briefly, clarified cell lysates of 0.5×106 cells of B-LCL 8.1.6 and 9.5.3 were added to wells coated with either mAb 2.12.E11 (anti-DQ2), or mAb L243 (anti-DR). After incubation at 4°C overnight, the wells were washed, and 2.5 μM biotinylated indicator peptides were added (identical peptide sequences as in fluid-phase binding assay) in a pH 4.3 citrate phosphate buffer containing protease inhibitors. To adjust for the amount of captured DQ2 and DR3, biotinylated mAb B8.11 (anti-DR) or SPV-L3 (anti-DQ) in pH 7.4 citrate phosphate buffer was added to the relevant wells and incubated for 48 hrs at 37°C. The wells were washed and streptavidineuropium diluted 1:2000 in assay buffer (Wallac) was added, incubating for 60 min at 37°C. The wells were washed and enhancement solution (Wallac) was added, incubating for 20 min before measurement in a time-resolved fluorometer (1234, Wallac).
Dissociation experiments of peptides from water soluble DQ2 and DR3
Water soluble DQ2-CLIP1, DQ2-CLIP2, DQ2-CLIP1 M91A, DQ2-CLIP1 M91P, DQ2-αI gliadin, DQ2-γI gliadin and DR3-CLIP1 molecules (1.6 μM) were treated with thrombin (1 u per 32 μg) for 90 min at RT, followed by AEBSF treatment (1 mM). The cleaved molecules were incubated at 37°C in the presence or absence of 6 μM soluble DM in a pH 5.0 citrate phosphate buffer for the required length of time. High affinity competitor peptides were added in excess (35 μM) (MB 65-kDa Hsp 243–255Y for DQ2, and MT 65kDa 3-13 for DR3). The dissociation was stopped by neutralizing the sample with cold citrate phosphate buffer pH 7.2. Released peptides (CLIP1: RDSGPVSKMRMATPLLMQAGAGSLVPR, CLIP2: RDSGMATPLLMQALPM GALGAGSLVPR, CLIP1 M91A: RDSGPVSKARMATPLLMQAGAGSLVPR, CLIP1 M91P: RDSGPVSKPRMATPLLMQAGAGSLVPR αI-gliadin: RDSGQLQPFPQPELPYGAGSLVPR, and γI-gliadin: RDSGPEQPQQSFPEQERPGAGSLVPR) were removed by filtration using 10,000MWCO microcon centrifugal filters (Millipore), and the bound peptides were eluted with 0.1% TFA/2 mM DTT in the presence of indicator peptides (16.1 pmol of each) for 30 min at 37°C. The indicator peptides have identical sequence to the released peptides except for a Ser-Thr substitution in position 3. Peptides were isolated using microcon filters, lyophilized, and treated with 0.1 M TMSBr in 100% acetonitrile (ACN) for 60 min at room temperature. Following lyophilization, the peptides were reconstituted in 0.1% TFA and purified over Poros 20 R2 reversed phase particles (Applied Biosystems) packed in GELoader tips (Eppendorf), and eluted directly onto a stainless steel target plate (Bruker Daltonics) using ACN/0.1% TFA (70/30, v/v) containing 5 mg/ml α-cyano-4-hydroxycinnamic acid (α-CHCA). After crystallization, the samples were analyzed on an MALDI-TOF/TOF mass spectrometer (Ultraflex II, Bruker Daltonics). The relative quantification of the eluted peptides was performed by comparing the first two peaks of the isotopic distribution of the eluted peptides with the corresponding peaks of the indicator peptide, which has a +14 Da mass increase due to the Ser-Thr substitution. The dissociation kinetics were fit into the single exponential decay function (Y = As × exp(−ksx)) using GraphPad Prism (Version 3.02).
Isolation of peptides bound to DQ2 and DR3 molecules from 8.1.6 and 9.5.3 cells
DQ2 and DR3 molecules were affinity purified from the 8.1.6 and 9.5.3 B-LCLs using anti-DQ2 mAb 2.12.E11 and anti-DR mAb B8.11 essentially as described (12). Purified molecules (50 μg) were washed with Milli-Q water using 10.000 MWCO microcon filters, followed by acid-elution (0.1% TFA) of the peptides. The peptides were isolated using microcon filters and lyophilized. Sequencing of the peptides was performed by reconstituting the peptides in 10 mM formic acid and separating on a series 1100 nanoLC instrument (Agilent) coupled to an ion trap mass spectrometer (Esquire 3000+, Bruker Daltonics). Quantification of the eluted peptides was executed by LC-MS using the Isotope Coded Protein Labeling (ICPL) Kit (Serva), according to the manufacturer's instructions. Briefly, lyophilized peptides and 75 pmol of each of 6 synthesized Ii peptides (Ii 81-104, Ii 81-103, Ii 82-103, Ii 81-101, Ii 93-109, Ii 93-108, all peptides synthesized by EZ-Biolabs) were dissolved in 4 μl 50 mM NaHCO3 pH 8.5 buffer and incubated 120 min at 25°C with 0.6 μl of 12C-Nic-reagent, or 13C-Nic-reagent respectively. Subsequently, 0.7 μl stop solution was added, and the samples were incubated for 20 min at 25°C prior to combining. The samples were treated with pH 11.9 phosphate buffer for 20 min at 25°C, neutralized with a citrate phosphate buffer to pH 7.2, and pre-purified by HPLC (Agilent, Column: Zorbax 300SB-C18 3.5Micron), collecting the Ii peptides. The collected peptides were then separated on a series 1100 nanoLC instrument coupled to a Q-TOF mass spectrometer (microTOF-Q, Bruker Daltonics). Quantification of Ii peptides was completed by comparing the peak intensities observed in the mass spectrometer of the light and heavy ICPL-labeled peptides.
In vitro DM-association assay
Cell pellets were lysed in buffer (50 mM Tris-HCl, pH 8.0 with 150 mM NaCl, 2 mM EDTA, 1% CHAPS and complete protease inhibitors; Roche Diagnostics). Protein concentration was measured by Bradford assay, and class II molecules were immunoprecipitated from titrated amounts of lysate, by overnight incubation with anti-DQ antibody (SPV-L3)-conjugated protein G sepharose beads. After washing with D-PBS containing 0.01% CHAPS, 1 ml of reaction buffer (1x PBS, 0.2% CHAPS) and 1–4 μg of soluble DM were added and then rotated for 3 hours at 37°C. After washing with D-PBS containing 0.01% CHAPS, samples were boiled for 10 minutes with non-reducing sample buffer, cooled, and separated by SDS-PAGE and then transferred onto Immobilon PVDF membrane (Millipore). The amount of DQ1/DQ2 and the associated sDM was detected using anti-class II β chain antibody (XD5.A11) and anti DMα-specific antibody (5C1), respectively. Antibody binding was detected with horseradish peroxidase-conjugated goat anti-mouse followed by an enhanced chemiluminescence (ECL) substrate (Amersham).
Results
In cells, CLIP is released from DR3 more efficiently than from DQ2
Based on previous work showing the presence of significant amounts of CLIP peptides in elution profiles from DQ2-expressing cells (3,4), we examined the relative stability of DR3-CLIP and DQ2-CLIP in cells by pulse/chase immunoprecipitation, with antibodies that bind to DR3 or DQ2 both in the presence and absence of CLIP peptides. To evaluate the influence of the level of DM, we used a panel of related cells expressing variable amounts of DM. Intracellular FACS staining revealed a hierarchy in DM levels, with 9.5.3 cells < 8.1.6 cells < 9.5.3-DM transfectant cells (Fig. 1A). In the absence of DM (9.5.3), CLIP is detected in association with both DQ2 and DR3 molecules at the 8 hour post-synthesis time point. In the presence of DM (8.1.6 cells), the CLIP:DR3 ratio is substantially lower at 8 hours of chase (23% of 9.5.3 ratio), whereas the change in the CLIP:DQ2 ratio is more modest (80% of 9.5.3 ratio) (Fig. 1B and densitometric analysis in 1C). Thus, DQ2-CLIP complexes appear more resistant than DR3-CLIP to DM-mediated peptide exchange. This resistance can be overcome, as increased DM (9.5.3-DM) reduces DQ2-CLIP levels at 8 hours (31% of 9.5.3 ratio). At all levels of DM, CLIP removal from DR3 is more efficient than from DQ2, however (Figs. 1B and 1C).
FIGURE 1.

The efficiency of Ii peptide release from DQ2 is lower than from DR3 and related to the level of HLA-DM. (A) Intracellular DM staining of the indicated cell lines using anti-HLA-DM dimer antibody. (B) 9.5.3, 8.1.6, and DM transfectant of 9.5.3 (9.5.3-DM) cells were pulsed for 1 hr with S35-Met/Cys and then chased in label-free media. Cell lysates (3 × 106 cell equivalents/lane) were immunoprecipitated with anti-DQ mAb, SPV-L3, (top) or anti-DR mAb, L243, (bottom) at the indicated times (hours) of chase and then analyzed by SDS-PAGE. Representative images from one of two independent experiments are shown. (C) Densitometry analysis calculating ratios of CLIP to class II band intensity at 8 hours from images shown in (B). (D) Assessment of cell surface CLIP-class II accumulation by FACS analysis. Indicated cell lines (9.5.3: DR3, DQ2, DP4, DM-null; 9.22.3: DQ2, DP4, DM+; 3.1.3: DQ1, DP4, DM+; 5.2.4: DP4, DM-null) were stained with anti-CLIP (CerCLIP.1; left) and anti-DQ (SPVL-3; right panel) followed by 2nd antibody (FITC conjugated goat anti-mouse IgG). Control staining (without primary antibody: dotted lines) is comparable on all cell lines and histogram for one representative cell line is shown in each panel. CLIP:DQ ratios were calculated as MFICerCLIP − MFIisotype ÷ MFISPVL# − MFIisotype: CLIP:DQ1=1:108; CLIP:DQ2=1:108 p<0.04. Representative data from one of >3 experiments are shown.
DQ2-CLIP complexes are surface-expressed in DM-expressing cells
The finding of DQ2-CLIP complexes persisting during pulse-chase experiments leaves open the possibility that we are detecting mostly intracellular (predominantly pre-DM) complexes Therefore, we asked whether DQ2-CLIP complexes are also found at the cell surface in DM-expressing cells. We used DR-negative, DM-positive B-LCL expressing either DQ1, DP4 (3.1.3) or DQ2, DP4 (9.22.3), both derived from the same parental B-LCL in several steps of mutagenesis and selection [(8) and Mellins, unpublished]. We carried out staining and FACS analyses using CerCLIP, an antibody that recognizes CLIP peptides, bound to class II (14). DP4-CLIP complexes are not surface-expressed, even in the absence of DM (see B-LCL 5.2.4, Fig. 1D). Thus, CerCLIP staining of B-LCLs 3.1.3 and 9.22.3 reflects DQ-CLIP complexes. DQ2-CLIP complexes are present at substantially higher levels than DQ1-CLIP complexes (with normalization for differences in levels of DQ) at the surface of DM-expressing cells, (Fig. 1D). This result also implies DQ2-CLIP resistance to DM action.
DQ2 binds two CLIP sequences in different registers
To identify the specific CLIP peptides associated with DQ2, we eluted peptides from DQ2 molecules purified from the DM-expressing B-LCL 8.1.6 and analyzed them by MALDI-TOF and LC-coupled ESI mass spectrometry (MS). We confirmed the previous observation that Ii-derived peptides are associated with DQ2 expressed in B-LCL, even in the presence of DM (data not shown) (3,4). Traces of CLIP are found among the peptides eluted from many, though not all, class II alleles expressed in B-LCL (15). The proportion of CLIP peptides in the total eluate from DQ2 is notably high, however. We also confirmed the prior finding that two families of CLIP peptides are associated with DQ2 from B-LCL: conventional CLIP peptides, here termed CLIP1, and unusual CLIP peptides, CLIP2 (see Table IIA).
TABLE II.
Identification and quantification of the Ii-chain peptides bound to DQ2 and DR3. (A) Peptides acid-eluted from 50 μg of affinity-purified DQ2 and DR3 from 8.1.6 and 9.5.3 B-LCLs were sequenced using LC-coupled ESI-MS/MS to identify all Ii-chain peptide truncations present in at least one of the elutions. The peptides were present in multiple charged states (2–5 fold) and the observed molecular weight (MW) was calculated by deconvolution. (B) The quantities of the individual Ii-chain peptides as listed above are given, and the data have been summarized in Fig. 3. ND= not detected, na=not applicable.
| A | |||||||
|---|---|---|---|---|---|---|---|
| Residues | Sequence | Detected | Length | MW | |||
| DQ2DM+ | DQ2DM− | DR3DM+ | DR3DM− | ||||
| Ii81-104 | LPKPPKPVSKMRMATPLLMQALPM | − | − | − | + | 24 | 2674.5 |
| Ii81-103 | LPKPPKPVSKMRMATPLLMQALP | + | + | + | + | 23 | 2543.4 |
| Ii82-103 | PKPPKPVSKMRMATPLLMQALP | + | + | − | + | 22 | 2430.4 |
| Ii81-101 | LPKPPKPVSKMRMATPLLMQA | + | + | − | + | 21 | 2333.3 |
| Ii93-109 | MATPLLMQALPMGALPQ | + | + | − | − | 17 | 1781.9 |
| Ii93-108 | MATPLLMQALPMGALP | + | + | − | − | 16 | 1653.9 |
| B | |||
|---|---|---|---|
| MHC II | Peptide | pmol | SD |
| DQ2 (8.1.6) | Ii81-104 | ND | na |
| Ii81-103 | 79,5 | 31,8 | |
| Ii82-103 | 16,5 | 9,5 | |
| Ii81-101 | 31,5 | 3,2 | |
| Ii93-109 | ND | na | |
| Ii93-108 | 159,0 | 1,1 | |
| DQ2 (9.5.3) | Ii81-104 | ND | na |
| Ii81-103 | 71,1* | 29,2 | |
| Ii82-103 | 36,8* | 14,5 | |
| Ii81-101 | 88,4* | 35,6 | |
| Ii93-109 | 3,8 | 6,5 | |
| Ii93-108 | 156,3 | 62,6 | |
| DR3 (8.1.6) | Ii81-104 | ND | na |
| Ii81-103 | 14,9 | 19,7 | |
| Ii82-103 | ND | na | |
| Ii81-101 | ND | na | |
| Ii93-109 | ND | na | |
| Ii93-108 | ND | na | |
| DR3 (9.5.3) | Ii81-104 | 42,4 | 3,7 |
| Ii81-103 | 128,3* | 23,3 | |
| Ii82-103 | 113,3 | 22,3 | |
| Ii81-101 | 106,1 | 8,0 | |
| Ii93-109 | ND | na | |
| Ii93-108 | ND | na | |
Underestimated due to suboptimal ICPL labeling of eluted peptides from 9.5.3 cells. A rough estimation indicate that about 3% and 8% percent of total CLIP peptides remained unlabeled when eluted from DQ2 and DR3, respectively.
CLIP1 peptides are derived from a sequence around Ii 83-101 and contain the Ii 91-99 core binding motif. CLIP2 sequences, from around Ii 92-107, overlap with, but are C-terminal to, the sequences of CLIP1 peptides. The sequence differences in CLIP1 versus CLIP2 suggested that the peptides bound DQ2 in different frames. CLIP1 has been found to bind in the same frame to all alleles where this has been examined, with M91 in the P1 pocket (16). To identify the binding register of CLIP2 peptides to DQ2, we examined the binding of five 11-mers (Ii 97-107, Ii 96-106, Ii 95-105, Ii 94-104, Ii 93-103) that scan through Ii in positions 93-107 in comparison to a naturally processed form of CLIP2 peptide (Ii 92-107: RMATPLLMQALPMGAL) (4). We used a peptide binding assay that measures the IC50 value of the test peptide, which is the amount of peptide needed to give 50% inhibition of the binding of a high affinity indicator peptide. This analysis demonstrated that Ii 94-104 and Ii 92-107 had similarly low IC50 values, whereas Ii 95-105 and Ii 93-103 had increased IC50 values, consistent with weaker binding (Fig. 2A). This pattern implied that the core binding region of CLIP2 was Ii 96-104 (PLLMQALPM), with P96 at the P1 position. This conclusion is compatible with the observation that N-terminally extended peptides bind with increased affinity due to hydrogen bond formation between the MHC II main chain and P-1 and P-2 (17).
FIGURE 2.

The binding frame of CLIP2 for binding to DQ2 is determined to be PLLMQALPM. (A) The IC50 values of truncated and mutated versions of the CLIP2 peptide were measured. IC50 is the peptide concentration required to give 50% inhibition of the indicator peptide KPLLIIAEDVEGEY. The underlined residues P and M denote positions P1 and P9 in the DQ2 peptide binding frame. Dotted bars indicate IC50 values higher than 167 μM. Data are from one of at least three independent experiments. (B) Depiction of the CLIP1 and CLIP2 binding frames.
To confirm the binding frame, we carried out a competitive inhibition binding assay with peptide variants of Ii 94-104, in which a lysine (K) was introduced in the putative P4, P5, or P6 positions (Ii 94-104;M99K, Ii 94-104;Q100K, and Ii 94-104;A101K, respectively). A positively-charged lysine would be expected to prohibit binding to DQ2 when introduced at P4 and P6, but not at P5, as the P5 side chain is predicted to face the solvent. As seen in Fig. 2A, the IC50 value of Ii 94-104;Q100K is low, while the Ii 94-104;M99K and Ii 94-104;A101K values are high, consistent with our conclusion that PLLMQALPM (Ii 96-104) represents the core binding region of CLIP2. The overlapping, but distinct, binding frames of CLIP1 and CLIP2 are shown schematically in Fig. 2B.
High levels of DQ2-Ii complexes in presence or absence of DM
The finding of abundant CLIP peptides associated with DQ2 in cell lysates of DM-expressing cells led us to ask whether the DQ2-CLIP complexes found in DM-expressing cells represent a novel repertoire. We compared the peptide elution profile of DQ2 purified from 8.1.6 cells (DM-expressing) with the elution profile of DQ2 purified from the DM-deficient mutant, 9.5.3, derived from 8.1.6. MS analysis revealed that, like DQ2 from 8.1.6 cells, DQ2 from DM-deficient 9.5.3 cells also bound predominantly Ii-derived peptides, representing both CLIP1 and CLIP2 species. In contrast, and as previously reported (18), peptides eluted from DR3 (and DR52a) were predominantly Ii-peptides in the absence of DM, whereas a heterogeneous mixture of self-peptides was associated with DR3 (and DR52a) from DM-expressing cells (data not shown). As DR52a has very low affinity for CLIP (19), the observed CLIP peptides most probably derive from DR3. An overview of the Ii variants associated with DQ2 and DR3 is presented in Table IIA.
To more precisely quantify the CLIP peptide pools associated with DR3 and DQ2 in the presence and absence of DM, both the eluted peptides and synthetic CLIP peptides of a known amount were ICPL-labeled and analyzed by nano-LC-coupled Q-TOF-MS. The peak heights for the light-labeled eluted peptides were compared to those obtained for the heavy-labeled synthetic peptides. The results demonstrated that DR3 from DM-deficient cells had a 25-fold higher level of CLIP peptides compared to the DM-expressing parental line, 8.1.6, while DQ2 from the DM-deficient cells showed only a 1.2-fold increase in total CLIP level compared to DQ2 from 8.1.6 (Fig. 3). This finding corroborated that DQ2-CLIP was significantly less affected by DM than DR3-CLIP.
FIGURE 3.
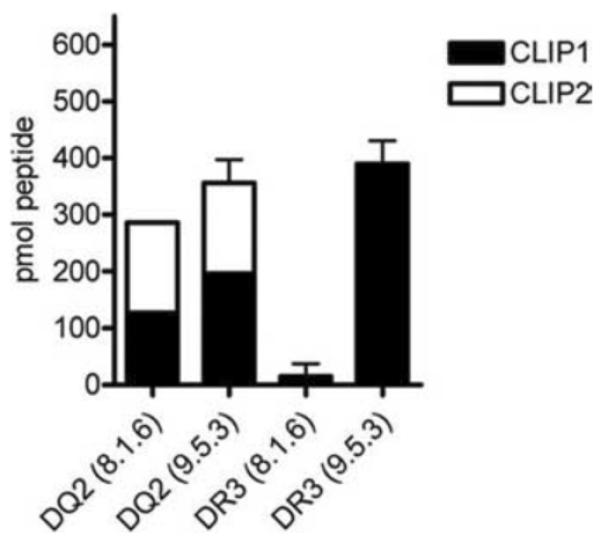
Peptide exchange of DQ2 is relatively DM-insensitive in vivo and in vitro. (A) Quantification of eluted CLIP peptides from 50 μg of affinity purified DQ2 and DR3 from 8.1.6 and 9.5.3 B-LCLs using LC-coupled Q-TOF-MS and ICPL labeling of eluted and indicator peptides. The mean and range of at least two sets of independent experiments are shown. The quantities of the different Ii chain peptides and the ESI-MSMS data justifying the selected peptides can be found in Table IIB.
Spontaneous peptide exchange is equivalent on DQ2 from DM+ or DM− cells
We next investigated the effect of the naturally processed peptide repertoires associated with DR3 and DQ2 on peptide exchange. We performed solid phase peptide binding assays, in which DQ2 and DR3 molecules purified from lysates of 8.1.6 (DM+) and 9.5.3 (DM−) cells were captured by monoclonal antibodies 2.12.E11 (anti-DQ2) or L243 (anti-DR dimer) and binding of biotinylated peptides to DQ2 or DR3 was measured. The spontaneous peptide exchange on DR3 originating from DM-null cells was increased more than 2-fold compared to DR3 from DM-expressing cells, reflecting the effects of DM editing of the peptide repertoire for less-exchangeable peptides. In contrast, peptide exchange on DQ2 from DM-deficient cells was similar to that observed with DQ2 from the DM-sufficient counterpart (Fig. 4A). These results are consistent with the similarity in the peptide profiles of DQ2 from 8.1.6 and 9.5.3 cells.
FIGURE 4.
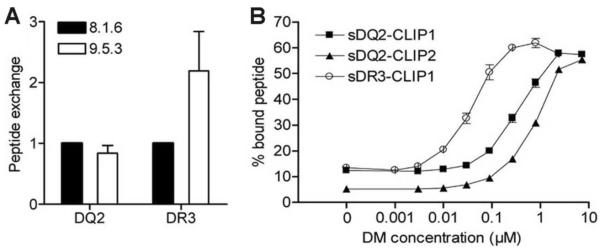
DQ2 displays reduced DM mediated peptide exchange compared to DR3 (A) Lysates of 8.1.6 and 9.5.3 B-LCLs were added to anti-DQ2 (2.12.E11) or anti-DR (L243) coated plates, followed by 48 h incubation with high affinity biotinylated indicator peptides (EPRAPWIEQEGPEYW for DQ2 and KTIAYDEEARR for DR3). The peptide exchange of DQ2 or DR3 from 8.1.6 (wt) is assigned a value of 1. Mean and SD from four independent experiments are shown. (B) Peptide exchange of cleaved sDQ2-CLIP1, sDQ2-CLIP2 and sDR3-CLIP1 (1.5 μM) with high affinity indicator peptides (1–5 nM of KPLLIIAEDVEGEY for DQ2 and KTIAYDEEARR for DR3) were measured in the absence (0 DM) or presence of an increasing amount of DM (0.001–7.2 μM) using the fluid-phase peptide binding assay. The complexes of DQ2 or DR3 with indicator peptides were used as a measure of peptide exchange. The negative control (without DQ2/DR3) did not give any signal (not shown). The mean and range of two independent experiments is shown.
DQ2-CLIP1 and DQ2-CLIP2 display high resistance to DM action
To more precisely quantify the extent of DM resistance of DQ2-CLIP compared to DR3-CLIP for peptide exchange, we measured DM-mediated peptide exchange, using a fluid-phase peptide exchange assay. Recombinant complexes (sDR3-CLIP1, sDQ2-CLIP1, sDQ2-CLIP2) were initially generated with tethered peptides, attached to the N-terminus of the class II beta chain with a linker including a thrombin cleavage site (see materials and methods). Thrombin-treated, soluble HLA molecules were incubated with high affinity, allele-specific indicator peptides and increasing DM concentrations before measurement of binding of the indicator peptides. This in vitro assay allowed us to increase the DM:DQ2 ratio beyond that achieved in cells (including the DM transfectants). sDR3-CLIP1 was significantly more sensitive to DM, showing 50% of maximum peptide exchange at 10-fold and 20-fold lower concentrations of DM compared to sDQ2-CLIP1 and sDQ2-CLIP2, respectively (Fig. 4B).
DQ2 shows reduced binding interaction with DM compared to DQ1
To further explore the interaction of DQ2 and DM, we compared DQ2 and DQ1 molecules, using an in vitro association assay in which bead-bound, immunoprecipitated DQ is incubated with soluble recombinant DM. DQ/DM binding is determined by isolating bead-associated DQ/DM complexes, and detecting DM by immunoblotting of boiled complexes. With similar input amounts of DQ1 and DQ2 by silver stain (not shown) and anti-class II β chain blotting (Fig. 5A, upper panels), reduced amounts of soluble recombinant DM were co-precipitated with DQ2 (Fig. 5A, lower panels). The observed binding is specific, as DM binding to the antibody-conjugated beads alone is negligible (Fig. 5A, lower panels). DM/class II co-precipitation assays are known to be influenced by the peptide cargo of class II. Peptide-free class II appears to be the best ligand for DM, so that binding in this assay does not provide information that is independent of the ability of DM to release peptide from the class II/peptide complex (20). Accordingly, DQ1 from DM-null cells binds better than DQ1 from DM-positive cells, a portion of which is peptide-edited (Fig. 5B). Notably, however, DM/DQ2 interaction is even more inefficient (Fig. 5B). We did not compare DR3 and DQ2 here, because of the confounding variable introduced by immunoprecipitating class II with different antibodies.
FIGURE 5.

DQ2 shows reduced binding interaction with DM compared to DQ1. DQ2 and DQ1 were immunoprecipitated with SPV-L3 from DM-positive (DR-null) B-LCLs 9.22.3 (DQ2) or 3.1.3 (DQ1), and DM-null cells 9.5.3 (DQ2) and 2.2.93 (DQ1), respectively, and then incubated with soluble recombinant DM to allow molecular interaction. (A) Input class II amount was measured by immunoblotting with anti-class IIβ chain Ab XD5.A11 (top) followed by measuring the amount of interacting DM by immunoblotting with anti-DMα mAb 5C1. Shown is one representative image of at least three experiments. (B) Ratio of sDM:class II band intensity was calculated by densitometric analysis of the images in 5A: DM+ cell lines: 3.1.3 (DQ1) and 9.22.3 (DQ2) and DM-null cells: 2.2.93 (DQ1) and 9.5.3 (DQ2).
DQ2-CLIP1/2 complexes show high intrinsic stability and reduced DM effect
Effects of DM on various class II-peptide complexes and changes in the peptide repertoires of cells expressing or lacking DM have led to the conclusion that intrinsically more stable complexes are, in general, more resistant to DM (21,22). The differential effect of DM on DQ2-CLIP and DR3-CLIP could reflect differences in the intrinsic stability of these complexes. Alternatively, the differences in peptide exchange could reflect reduced efficiency of DM interaction with the DQ2 allele. These two possibilities are not mutually exclusive. To assess the intrinsic stability of the DR3 and DQ2 complexes with CLIP peptides, we measured the spontaneous dissociation rate of the CLIP1 and CLIP2 peptides from sDQ2 and CLIP1 from sDR3 at pH 5 without DM, using thrombin-treated, soluble molecules. The dissociation was measured by eluting the bound peptides and quantifying them by MALDI-TOF MS analysis, comparing the intensity of the isotopic peaks to an added indicator peptide. The CLIP1 and CLIP2 peptides from sDQ2 in the absence of DM displayed nearly identical dissociation rates (t1/2 = ~140 h), compared to the 2-fold faster release of CLIP1 from DR3 (t1/2 = ~61.5 h) (Table III). Thus, both CLIP complexes with sDQ2 are more stable than sDR3-CLIP1, raising the possibility that increased intrinsic stability makes a contribution to the reduced efficacy of DM-mediated peptide exchange of the DQ2-CLIP complexes compared to DR3-CLIP1. In the presence of DM, the dissociation of CLIP1 from sDR3 increased 246X (t1/2 = 0.25 hr). DM also accelerated the release of CLIP1 and CLIP2 from sDQ2 (t1/2 = 9.0 h and t1/2 = 5.0 h, respectively); however, the DM-mediated enhancement of dissociation was substantially more modest (16–28X). To consider this issue further, we assessed four additional sDQ2-peptide complexes; sDQ2-γI-gliadin, sDQ2-αI-gliadin, and two mutated forms of CLIP1 where the residue buried in MHC class II pocket 1 is mutated from methionine to either alanine (M91A) or proline (M91P). Compared to sDR3-CLIP, the CLIP1 M91P peptide had a slightly longer spontaneous dissociation time (t1/2 = 93.6 h), the γI-gliadin peptide had a similar dissociation rate (t1/2 = 55.2 h), while the intrinsic stability of the CLIP1 M91A and αI-gliadin peptides were both lower than sDR3-CLIP1 (t1/2 = 13.1 h and 8.3 h, respectively) (Table III). Using a class II:DM molar ratio of 1:3.8 we then measured the effect of DM on the half-lives of the seven complexes (Table III). Only moderate enhancement in the presence of DM was also seen for the additional sDQ2-peptide complexes tested (5–19X) (Table III). These findings indicate that the abundance of CLIP-bound DQ2 in DM-containing cells may be a combined effect of the high intrinsic stability of CLIP and the limited effect of DM on DQ2.
TABLE III.
The DM effect on dissociation of Ii derived peptides is greater for DR3 than for DQ2. Dissociation of peptides from thrombin-treated water soluble molecules (1.6 μM) in the presence of excess competitive high affinity peptides (35 μM) and in the absence or presence of DM (6 μM). Peptide release was determined by MALDI-TOF MS analysis comparing the intensity of the isotopic peaks of added indicator peptides with those of the released peptides. Data from the mean of at least two independent experiments were fitted to a single exponential decay function (Y = As x exp(−ksx)) and values for t1/2 and the coefficient of determination (r2) were calculated.
| Complex | Spontaneous dissociation, t1/2 in hours (r2) | DM-mediated dissociation, t1/2 in hours (r2) | DM-mediated increase in dissociation | ||
|---|---|---|---|---|---|
| DR3-CLIP1 | 61.5 | (0.98) | 0.25 | (0.98) | 246x |
| DQ2-CLIP1 | 144.2 | (0.97) | 9.0 | (0.91) | 16x |
| DQ2-CLIP2 | 141.7 | (0.99) | 5.0 | (0.89) | 28x |
| DQ2-CLIP1 M91A | 13.1 | (0.99) | 0.7 | (0.92) | 19x |
| DQ2-CLIP1 M91P | 93.6 | (0.92) | 8.9 | (0.95) | 11x |
| DQ2-αI | 8.3 | (0.94) | 1.6 | (0.92) | 5x |
| DQ2-γI | 55.2 | (0.96) | 6.9 | (0.96) | 8x |
Low DM effect on DQ2-CLIP1/2 is not due to intrinsic stability
To further evaluate the DM susceptibility of these seven complexes in the context of their intrinsic stability, we compared our current results to previous findings where we measured peptide dissociation in the presence and absence of DM for a panel of complexes of varying intrinsic stability. In this prior work, we observed a consistent quantitative relationship between intrinsic stability and DM effect with a correlation of r=0.69 (21). Strikingly, 6 of 6 sDQ2 complexes in the current study did not conform to this quantitative relationship whereas the sDR3-CLIP1 complex resembled the predicted behavior (Fig. 6). These findings argue that the stability of the sDQ2-CLIP1/2 complexes in the presence of DM is not solely the result of their intrinsic stability.
FIGURE 6.

Relationship between intrinsic and DM-catalyzed dissociation. Dissociation rate constants, k, calculated from the t½, by the equation t½ =ln2/k, are plotted on a log10 scale in units of hours−1. The x-axis shows dissociation in the absence of DM; the y-axis shows dissociation in the presence of soluble DM. The solid best-fit straight line through the data has a slope of 0.47 (95% confidence interval, 0.3–0.64; thin dashed curves). The solid line of slope 1 through the origin indicates no DM effect; the vertical distance of each data point from this line is a measure of DM susceptibility. These data have been published with the exception of the data from the DQ2 and DR3 complexes (21). In order to compare current results to previous work on 36 complexes, it was necessary to correct values for the differences in concentration of DM used; previous work indicated that effects of DM in the DM concentration range of both these assays are linear (Mellins, unpublished). An outlier DR4 complex was excluded from the original correlation analysis (21) and is not shown.
Cell surface expression of DQ2 is not enhanced by co-expression of DM
In addition to its peptide exchange function, DM acts as a chaperone and influences the steady state and cell surface abundance of certain alleles ((23–27), Roh et al, submitted). To see whether DQ2 is susceptible to these other effects of DM, we measured cell surface binding of a DQ2 specific antibody (2.12.E11) and of two monomorphic anti-DQ antibodies to the DM-null cell, 9.5.3, and its DM-transfectant, 9.5.3-DM. At saturating amounts of antibody, the binding of each of these antibodies was comparable in the DM-expressing and non-expressing cells, suggesting that cell surface abundance of DQ2 was not influenced by DM. In contrast, the level of DQ1 expression in the DM-null B-LCL 2.2.93 was increased by expression of DM in the 2.2.93 transfectant. The difference in effects on DQ1 and DQ2 was not due to cell line differences, because the behavior of DP4 and DR3 molecules was consistent across DM-transfectants of both cell lines (Fig. 7).
FIGURE 7.

Cell surface expression level of DQ2 is not enhanced by co-expression of DM. Fold change in mean fluorescence intensities (MFI) of staining of 2.2.93-DM compared to 2.2.93 (anti-DR (L243), anti-DP (B7/21.2), anti-DQ (SPV-L3 and aIa3) staining) or of 9.5.3-DM compared to 9.5.3 (anti-DR (L243), anti-DP (B7/21.2), anti-DQ (SPV-L3 and aIa3), anti-DQ2 (b2.12.E11), and anti-DR3 (c16.23) staining). Mean values from multiple experiments are shown as horizontal bars. Dotted line indicates no change (MFI ratio= 1). Analyses using t-tests showed that all the changes except DQ2a, DQ2b, and DR3 are significant (p < 0.05).
Conformation of DQ1 but not DQ2 is altered by DM expression
DM interaction with nascent class II molecules influences their conformation, probably in both a peptide-dependent and independent manner (28). We assessed whether DQ2 was susceptible to DM effects on molecular maturation, in comparison to DQ1. After pulse/chase metabolic labeling of the same cell lines used in Fig. 7, we immunoprecipitated DQ1 and DQ2 with the monomorphic anti-DQ antibody SPV-L3. We observed increased binding of SPV-L3 to DQ1 at ~2.5 hours of chase in 2.2.93-DM, but not 2.2.93 cells, consistent with a DM-dependent conformational change in DQ1 (Fig. 8). In contrast, DQ2 (expressed in 9.5.3 ± DM) did not show the DM-dependent conformational change detected by SPV-L3 (Fig. 8) or the DQ2-specific antibody, 2.12.E11 (not shown). This result does not depend on the cell lines tested as DP4 undergoes DM-dependent increases in precipitable molecules in 9.5.3-DM and 2.2.93-DM (not shown; Roh et al, submitted). We conclude that DQ2 resists DM-mediated chaperoning as well as peptide exchange.
FIGURE 8.
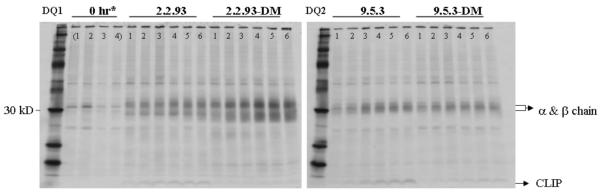
DQ2 does not show DM dependent conformational change during biosynthesis. 2.2.93, 9.5.3, and their DM transfectants (2.2.93-DM and 9.5.3-DM, respectively) were pulsed for 15 min with 35S-Met/Cys and then chased for 2, 2.5, 3, 3.5, 4, 4.5 hrs (lanes labeled as 1, 2, 3, 4, 5, 6, respectively), as described in Materials and Methods. Cell lysates (5 × 106 cell equivalents/lane) were immunoprecipitated with anti-DQ mAb, SPV-L3, and then analyzed by SDS-PAGE. Representative images from one of five independent experiments are shown. *(1. 2.2.93 2. 2.2.93-DM 3. 9.5.3 4. 9.5.3-DM) at 0 hr.
The interaction of DM with DQ2 is competitively inhibited by DR3
Given that DR3 and DQ2 are tightly linked alleles, they are generally expressed concurrently, and they likely are competing ligands for DM within peptide-loading compartments. To model this situation in vitro, we examined whether DR3 could competitively inhibit DQ2/DM interaction. We used soluble DR3 or DQ2 with tethered CLIP1 as a competitor for DQ2 or DR3, respectively, with thrombin-cleaved CLIP1, in a DM-mediated peptide exchange reaction. We varied the ratios of thrombin-treated to thrombin-untreated complexes and measured binding of labeled, high affinity peptides. At equimolar, 2-fold and 10-fold molar excess concentrations of DR3, the peptide exchange on DQ2 fell by 15%, 27% and 61%, respectively. DQ2 was a less effective inhibitor, reducing the peptide exchange on DR3 by 2%, 6% and 38% at the same concentrations (Fig. 9). These results argue that DR3 is a more potent inhibitor of DM-mediated peptide exchange of DQ2 than vice versa. At 10-fold molar excess of DR3 compared to DQ2, which roughly approximates physiological intracellular conditions (29), the insufficient DQ2/DM interaction is further hampered by the presence of a successful, competitive ligand for DM.
FIGURE 9.
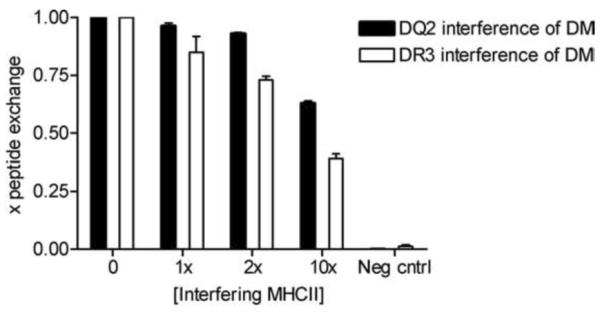
DQ2/DM interaction is subject to competitive inhibition by DR3. The level of peptide exchange of cleaved sDQ2-CLIP1 or sDR3-CLIP1 (0.7 μM) with indicator peptides (EPRAPWIEQEGPEYW for DQ2 and KTIAYDEEARR for DR3) was measured in the presence of DM (0.3 μM) and varying concentrations of non-cleaved sDR3-CLIP1 or sDQ2-CLIP1 (0–7 μM). Mean and SD of three independent experiments are shown.
Discussion
For most MHC class II alleles, biosynthesis and peptide loading is regulated by the actions of two co-factors: Ii and DM. Full length Ii typically associates with class II with its core CLIP region (CLIP1; Ii 91–99) bound to the class II peptide binding groove, and nested peptides from this region are left in the groove after Ii proteolysis. DM typically binds to class II to catalyze CLIP release, edit the repertoire of bound peptides and regulate class II conformation. The class II molecule DQ2 is associated with unusually high amounts of two CLIP variants (CLIP1 and CLIP2), and we find that the “CLIP-rich” phenotype of DQ2 in cells appears nearly identical in the presence and absence of DM. This unusual phenotype of DQ2 is apparently explained by a combination of the relatively high intrinsic stability of the DQ2-CLIP1/2 complexes and the inefficient interaction of DQ2 with DM.
Formally, DM resistance of DQ2-CLIP could result from a thermodynamic block (reduced DM binding to DQ2-CLIP) or a kinetic block (reduced ability of DM to release CLIP from DQ2). We find that increased concentrations of DM result in more effective CLIP release from DQ2, as shown by the reduction of co-precipitated CLIP in the presence of DM and by peptide exchange assays, arguing that the block is thermodynamic. However, much higher concentrations of DM are needed to obtain a peptide editing effect equivalent to that observed with DR3-CLIP1. The higher DM concentrations or prolonged exposure required for activity with DQ2 substrates may rarely be achieved in vivo for several reasons. DM levels are substoichiometric to class II in peptide loading compartments, with DM:DR molar ratio ≈ 1:5 ((30) and Mellins, unpublished). Endosomal residence time for class II-CLIP complexes is seemingly kinetically controlled, as DR3-CLIP complexes arrive at the cell surface at a rate that is similar to DR3-peptide complexes; prolonged retention of either class II-CLIP or of more DM-resistant complexes does not appear to occur (31). Third, with linkage disequilibrium in MHC class II haplotypes and co-regulated expression of class II isotypes, DQ2 is almost always expressed concurrently with DR3, which is capable of out-competing DQ2 for limiting amounts of DM, as evident in Fig. 9.
The structural basis of the unusual interaction of DQ2 with DM is likely to involve polymorphic residues of DQ2. DM is thought to disrupt H-bonds between the peptide backbone and residues around pocket 1 of the class II molecule (α53, β81) or stabilize a class II conformation in which these hydrogen bonds are disrupted, along with more global changes effecting pocket structure (7,32–37). Mutational analysis has demonstrated the critical role of the protruding α51F in DR3 for interaction with DM (34,35). This residue is predicted to participate in hydrophobic interactions with DM and may serve as a lever to alter the location of the extended strand including α51–53 (34,35) and its distance from the bound peptide. In DQ2, the α44–53 stretch (numbering as in (38)) contains several polymorphic residues and deletion of α53. Further work should identify if this region indeed is of significance and which residues are involved.
It is striking that CLIP2 is a prominent component of the DQ2 peptide repertoire. We show that CLIP2 binds to DQ2 by placing a proline at the P1 position. As part of a polypeptide chain, the proline side chain is unable to engage in amide hydrogen bonding because of its ring structure. In most MHC class II molecules, there is a hydrogen bond from residue 53 of the α-chain to the amide nitrogen of the P1 residue. Not only does DQ2 have a deletion of residue α53, but there is evidence that DQ2 is unable to form the P1-related hydrogen bond (39). Thus, proline can be accommodated by DQ2 at P1 without energetic penalty, perhaps explaining the unusual association of DQ2 with CLIP2 peptides. An interesting question is how these CLIP2 fragments come to be associated with DQ2. The CLIP region is unstructured within the full length Ii and may allow an alternate register of association in the ER. Alternatively, CLIP2 peptide associate with DQ2 after some proteolysis of Ii in endosomes, either through reptation or re-binding.
On first glance it may appear that our data on the DM susceptibility of DQ2-CLIP1 compared to DQ2-CLIP2 complexes are inconsistent. Among naturally processed peptides, the proportion of DQ2-CLIP2 is modestly increased in DM-expressing compared to non-expressing cells, suggesting increased resistance to DM editing. In the peptide exchange assay (Fig. 4B), the dose/response curve with DM is shifted to the right for sDQ2-CLIP2 compared to sDQ2-CLIP1. However, in Table III, off-rate measurements of the two forms of CLIP demonstrate comparable values in the absence of DM and a longer half-life of the sDQ2-CLIP1 complex in the presence of DM. These findings can be rationalized, because the data in Fig. 4B reflect the net effect of off-rate and on-rate of the CLIP variants in the presence of DM, while the assay in Table III includes excess competitor peptide, precluding any re-binding of the CLIP peptides and therefore any influence of peptide on-rates.
The DR3-DQ2 haplotype is associated with multiple autoimmune type disorders, including celiac disease, type 1 diabetes, systemic lupus erythematosus, Graves' disease and Addison's disease (5,6). Unusual interactions between DQ2 and Ii and DM could be one reason for this widespread predisposition to autoimmunity. Reduced interaction with DM may result in escape from DM editing. The residual CLIP1/2 peptides, though of relatively higher affinity for DQ2 than CLIP1 peptides for many other alleles, nonetheless may be less ideal than a DM-edited peptide repertoire for stabilizing class II surface expression and inhibiting exchange with extracellular peptides, including potential autoantigens (9,40). Our previous work on the half-life of complexes that survive DM editing shows a 9–70 fold increase in intrinsic half-life compared to DR3-CLIP, whereas DQ2-CLIP has only a 2.3-fold increase in t½ compared to DR3-CLIP (41). This difference suggests DQ2-CLIP1/2 would remain exchangeable on the surface of DM+cells, especially under conditions (e.g., peptide availability and pH) that favor exchange. In addition to these characteristics of DQ2, which may create a general predisposition to a breakdown in self-tolerance, unique features of DQ2 peptide binding may play a role in particular diseases. For example, we have hypothesized that DQ2 allows binding of proline-rich gluten epitopes of celiac disease, because it tolerates registers with proline residues at P1 (38,39).
Why has DQ2 evolved to be less DM sensitive? One possibility is that, under certain circumstances, this characteristic will lead to presentation of numerous antigenic epitopes instead of a few immunodominant epitopes (42), and this may provide a selective advantage in combating some infectious diseases. However, a deleterious side effect could be an increased susceptibility to autoimmune diseases. Studies in humanized mice models should shed light on the in vivo relevance of the phenomena described here.
Acknowledgements
We thank Peter Cresswell for the CerCLIP antibody, Hergen Spits for the SPV-L3 antibody, and Bernard Malissen for the B8.11 antibody. We would also like to thank Khoa Nguyen for assistance with figures.
Footnotes
Disclosures The authors have no conflicts to disclose.
This work was supported by grants from the Research Council of Norway, the National Institutes of Health (U19 AI02042 to EM), the Wasie Foundation (to EM) and the Norwegian Foundation for Health and Rehabilitation.
Abbreviations used in this paper: ACN, acetonitrile; B-LCL, B lymphoblastoid cell line; Ii, invariant chain; MFI, mean fluorescence intensity; MS, mass spectrometry.
Reference List
- 1.Cresswell P. Assembly, transport, and function of MHC class II molecules. Annu. Rev. Immunol. 1994;12:259. doi: 10.1146/annurev.iy.12.040194.001355. [DOI] [PubMed] [Google Scholar]
- 2.Busch R, Rinderknecht CH, Roh S, Lee AW, Harding JJ, Burster T, Hornell TM, Mellins ED. Achieving stability through editing and chaperoning: regulation of MHC class II peptide binding and expression. Immunol Rev. 2005;207:242. doi: 10.1111/j.0105-2896.2005.00306.x. [DOI] [PubMed] [Google Scholar]
- 3.van de Wal Y, Kooy YM, Drijfhout JW, Amons R, Koning F. Peptide binding characteristics of the coeliac disease-associated DQ(α1*0501, β1*0201) molecule. Immunogenetics. 1996;44:246. doi: 10.1007/BF02602553. [DOI] [PubMed] [Google Scholar]
- 4.Vartdal F, Johansen BH, Friede T, Thorpe CJ, Stevanovic S, Eriksen JE, Sletten K, Thorsby E, Rammensee HG, Sollid LM. The peptide binding motif of the disease associated HLA-DQ (α1* 0501, β1* 0201) molecule. Eur. J Immunol. 1996;26:2764. doi: 10.1002/eji.1830261132. [DOI] [PubMed] [Google Scholar]
- 5.Lechler R, Warrens A. HLA in health and disease. Academic; London: 2000. [Google Scholar]
- 6.Price P, Witt C, Allcock R, Sayer D, Garlepp M, Kok CC, French M, Mallal S, Christiansen F. The genetic basis for the association of the 8.1 ancestral haplotype (A1, B8, DR3) with multiple immunopathological diseases. Immunol Rev. 1999;167:257. doi: 10.1111/j.1600-065x.1999.tb01398.x. [DOI] [PubMed] [Google Scholar]
- 7.Pashine A, Busch R, Belmares MP, Munning JN, Doebele RC, Buckingham M, Nolan GP, Mellins ED. Interaction of HLA-DR with an acidic face of HLA-DM disrupts sequence-dependent interactions with peptides. Immunity. 2003;19:183. doi: 10.1016/s1074-7613(03)00200-0. [DOI] [PubMed] [Google Scholar]
- 8.Pious D, Dixon L, Levine F, Cotner T, Johnson R. HLA class II regulation and structure. Analysis with HLA-DR3 and HLA-DP point mutants. J Exp. Med. 1985;162:1193. doi: 10.1084/jem.162.4.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mellins E, Kempin S, Smith L, Monji T, Pious D. A gene required for class II-restricted antigen presentation maps to the major histocompatibility complex. J Exp. Med. 1991;174:1607. doi: 10.1084/jem.174.6.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Quarsten H, McAdam SN, Jensen T, Arentz-Hansen H, Molberg O, Lundin KE, Sollid LM. Staining of celiac disease-relevant T cells by peptide-DQ2 multimers. J Immunol. 2001;167:4861. doi: 10.4049/jimmunol.167.9.4861. [DOI] [PubMed] [Google Scholar]
- 11.Sloan VS, Cameron P, Porter G, Gammon M, Amaya M, Mellins E, Zaller DM. Mediation by HLA-DM of dissociation of peptides from HLA-DR. Nature. 1995;375:802. doi: 10.1038/375802a0. [DOI] [PubMed] [Google Scholar]
- 12.Johansen BH, Buus S, Vartdal F, Viken H, Eriksen JA, Thorsby E, Sollid LM. Binding of peptides to HLA-DQ molecules: peptide binding properties of the disease-associated HLA-DQ(α1*0501, β1*0201) molecule. Int. Immunol. 1994;6:453. doi: 10.1093/intimm/6.3.453. [DOI] [PubMed] [Google Scholar]
- 13.Vader W, Stepniak D, Kooy Y, Mearin L, Thompson A, van Rood JJ, Spaenij L, Koning F. The HLA-DQ2 gene dose effect in celiac disease is directly related to the magnitude and breadth of gluten-specific T cell responses. Proc. Natl. Acad. Sci. USA. 2003;100:12390. doi: 10.1073/pnas.2135229100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Avva RR, Cresswell P. In vivo and in vitro formation and dissociation of HLA-DR complexes with invariant chain-derived peptides. Immunity. 1994;1:763. doi: 10.1016/s1074-7613(94)80018-9. [DOI] [PubMed] [Google Scholar]
- 15.Chicz RM, Urban RG, Gorga JC, Vignali DA, Lane WS, Strominger JL. Specificity and promiscuity among naturally processed peptides bound to HLA-DR alleles. J Exp. Med. 1993;178:27. doi: 10.1084/jem.178.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ghosh P, Amaya M, Mellins E, Wiley DC. The structure of an intermediate in class II MHC maturation: CLIP bound to HLA-DR3. Nature. 1995;378:457. doi: 10.1038/378457a0. [DOI] [PubMed] [Google Scholar]
- 17.Sant AJ, Beeson C, McFarland B, Cao J, Ceman S, Bryant PW, Wu S. Individual hydrogen bonds play a critical role in MHC class II: peptide interactions: implications for the dynamic aspects of class II trafficking and DM-mediated peptide exchange. Immunol Rev. 1999;172:239. doi: 10.1111/j.1600-065x.1999.tb01369.x. [DOI] [PubMed] [Google Scholar]
- 18.Mellins E, Cameron P, Amaya M, Goodman S, Pious D, Smith L, Arp B. A mutant human histocompatibility leukocyte antigen DR molecule associated with invariant chain peptides. J Exp. Med. 1994;179:541. doi: 10.1084/jem.179.2.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sette A, Southwood S, Miller J, Appella E. Binding of major histocompatibility complex class II to the invariant chain-derived peptide, CLIP, is regulated by allelic polymorphism in class II. J Exp. Med. 1995;181:677. doi: 10.1084/jem.181.2.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Denzin LK, Hammond C, Cresswell P. HLA-DM interactions with intermediates in HLA-DR maturation and a role for HLA-DM in stabilizing empty HLA-DR molecules. J Exp. Med. 1996;184:2153. doi: 10.1084/jem.184.6.2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Belmares MP, Busch R, Wucherpfennig KW, McConnell HM, Mellins ED. Structural factors contributing to DM susceptibility of MHC class II/peptide complexes. J Immunol. 2002;169:5109. doi: 10.4049/jimmunol.169.9.5109. [DOI] [PubMed] [Google Scholar]
- 22.Weber DA, Evavold BD, Jensen PE. Enhanced dissociation of HLA-DR-bound peptides in the presence of HLA-DM. Science. 1996;274:618. doi: 10.1126/science.274.5287.618. [DOI] [PubMed] [Google Scholar]
- 23.Albert LJ, Ghumman B, Watts TH. Effect of HLA-DM transfection on hen egg lysozyme presentation by T2.Ak cells. J Immunol. 1996;157:2247. [PubMed] [Google Scholar]
- 24.Fallas JL, Tobin HM, Lou O, Guo D, Sant'Angelo DB, Denzin LK. Ectopic expression of HLA-DO in mouse dendritic cells diminishes MHC class II antigen presentation. J Immunol. 2004;173:1549. doi: 10.4049/jimmunol.173.3.1549. [DOI] [PubMed] [Google Scholar]
- 25.Honey K, Forbush K, Jensen PE, Rudensky AY. Effect of decreasing the affinity of the class II-associated invariant chain peptide on the MHC class II peptide repertoire in the presence or absence of H-2M. J Immunol. 2004;172:4142. doi: 10.4049/jimmunol.172.7.4142. [DOI] [PubMed] [Google Scholar]
- 26.Koonce CH, Wutz G, Robertson EJ, Vogt AB, Kropshofer H, Bikoff EK. DM loss in k haplotype mice reveals isotype-specific chaperone requirements. J Immunol. 2003;170:3751. doi: 10.4049/jimmunol.170.7.3751. [DOI] [PubMed] [Google Scholar]
- 27.Lich JD, Jayne JA, Zhou D, Elliott JF, Blum JS. Editing of an immunodominant epitope of glutamate decarboxylase by HLA-DM. J Immunol. 2003;171:853. doi: 10.4049/jimmunol.171.2.853. [DOI] [PubMed] [Google Scholar]
- 28.Lovitch SB, Pu Z, Unanue ER. Amino-terminal flanking residues determine the conformation of a peptide-class II MHC complex. J Immunol. 2006;176:2958. doi: 10.4049/jimmunol.176.5.2958. [DOI] [PubMed] [Google Scholar]
- 29.Gorga JC, Horejsi V, Johnson DR, Raghupathy R, Strominger JL. Purification and characterization of class II histocompatibility antigens from a homozygous human B cell line. J Biol. Chem. 1987;262:16087. [PubMed] [Google Scholar]
- 30.Schafer PH, Green JM, Malapati S, Gu L, Pierce SK. HLA-DM is present in one-fifth the amount of HLA-DR in the class II peptide-loading compartment where it associates with leupeptin-induced peptide (LIP)-HLA-DR complexes. J Immunol. 1996;157:5487. [PubMed] [Google Scholar]
- 31.Guerra CB, Busch R, Doebele RC, Liu W, Sawada T, Kwok WW, Chang M. d., Mellins ED. Novel glycosylation of HLA-DRa disrupts antigen presentation without altering endosomal localization. J Immunol. 1998;160:4289. [PubMed] [Google Scholar]
- 32.Busch R, Pashine A, Garcia KC, Mellins ED. Stabilization of soluble, low-affinity HLA-DM/HLA-DR1 complexes by leucine zippers. J Immunol Methods. 2002;263:111. doi: 10.1016/s0022-1759(02)00034-0. [DOI] [PubMed] [Google Scholar]
- 33.Chou CL, Sadegh-Nasseri S. HLA-DM recognizes the flexible conformation of major histocompatibility complex class II. J Exp. Med. 2000;192:1697. doi: 10.1084/jem.192.12.1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Doebele RC, Busch R, Scott HM, Pashine A, Mellins ED. Determination of the HLA-DM interaction site on HLA-DR molecules. Immunity. 2000;13:517. doi: 10.1016/s1074-7613(00)00051-0. [DOI] [PubMed] [Google Scholar]
- 35.Mosyak L, Zaller DM, Wiley DC. The structure of HLA-DM, the peptide exchange catalyst that loads antigen onto class II MHC molecules during antigen presentation. Immunity. 1998;9:377. doi: 10.1016/s1074-7613(00)80620-2. [DOI] [PubMed] [Google Scholar]
- 36.Narayan K, Chou CL, Kim A, Hartman IZ, Dalai S, Khoruzhenko S, Sadegh-Nasseri S. HLA-DM targets the hydrogen bond between the histidine at position β81 and peptide to dissociate HLA-DR-peptide complexes. Nat Immunol. 2007;8:92. doi: 10.1038/ni1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stratikos E, Wiley DC, Stern LJ. Enhanced catalytic action of HLA-DM on the exchange of peptides lacking backbone hydrogen bonds between their N-terminal region and the MHC class II alpha-chain. J Immunol. 2004;172:1109. doi: 10.4049/jimmunol.172.2.1109. [DOI] [PubMed] [Google Scholar]
- 38.Kim CY, Quarsten H, Bergseng E, Khosla C, Sollid LM. Structural basis for HLA-DQ2-mediated presentation of gluten epitopes in celiac disease. Proc. Natl. Acad. Sci. USA. 2004;101:4175. doi: 10.1073/pnas.0306885101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bergseng E, Xia J, Kim CY, Khosla C, Sollid LM. Main chain hydrogen bond interactions in the binding of proline-rich gluten peptides to the celiac disease associated HLA-DQ2 molecule. J Biol. Chem. 2005 doi: 10.1074/jbc.M501558200. [DOI] [PubMed] [Google Scholar]
- 40.Mellins E, Smith L, Arp B, Cotner T, Celis E, Pious D. Defective processing and presentation of exogenous antigens in mutants with normal HLA class II genes. Nature. 1990;343:71. doi: 10.1038/343071a0. [DOI] [PubMed] [Google Scholar]
- 41.Doebele RC, Pashine A, Liu W, Zaller DM, Belmares M, Busch R, Mellins ED. Point mutations in or near the antigen-binding groove of HLA-DR3 implicate class II-associated invariant chain peptide affinity as a constraint on MHC class II polymorphism. J Immunol. 2003;170:4683. doi: 10.4049/jimmunol.170.9.4683. [DOI] [PubMed] [Google Scholar]
- 42.Nanda NK, Bikoff EK. DM peptide-editing function leads to immunodominance in CD4 T cell responses in vivo. J Immunol. 2005;175:6473. doi: 10.4049/jimmunol.175.10.6473. [DOI] [PubMed] [Google Scholar]