

Abstract
Nonribosomal peptides (NRPs) such as vancomycin and daptomycin are among the most effective antibiotics. While NRPs are biomedically important, the computational techniques for sequencing these peptides are still in their infancy. The recent emergence of mass spectrometry techniques for NRP analysis (capable of sequencing an NRP from small amounts of nonpurified material) revealed an enormous diversity of NRPs. However, as many NRPs have nonlinear structure (e.g., cyclic or branched-cyclic peptides), the standard de novo sequencing tools (developed for linear peptides) are not applicable to NRP analysis. Here, we introduce the first NRP identification algorithm, NRPquest, that performs mutation-tolerant and modification-tolerant searches of spectral data sets against a database of putative NRPs. In contrast to previous studies aimed at NRP discovery (that usually report very few NRPs), NRPquest revealed nearly a hundred NRPs (including unknown variants of previously known peptides) in a single study. This result indicates that NRPquest can potentially make MS-based NRP identification as robust as the identification of linear peptides in traditional proteomics.
About 70% of new chemical entities introduced as antibacterials over the last 25 years are natural product derivatives.1 Peptide natural products have diverse biological activities (such as cell signaling, immune response, and development) and are divided into nonribosomal peptides (NRPs)2 and ribosomally synthesized and post-translationally modified peptides (RiPPs).3 NRPs represent a widely distributed and biomedically important class of peptidic natural products that includes antibiotics, antitumor agents, immunosuppressors, and toxins. NRPs do not follow the central dogma “DNA produces RNA produces protein”. Instead, they are assembled by nonribosomal peptide synthetases (NRPSs) that represent both the mRNA-free template and the building machinery for the peptide biosynthesis.4 Thus, NRPs are not directly inscribed in the genomes and cannot be inferred with traditional DNA sequencing. Instead, they are coded by NRPSs using “nonribosomal code” (also called the Stachelhaus rule) discovered 15 years ago.5
Many NRPs are nonlinear peptides that contain nonstandard amino acids, increasing the number of possible building blocks from 20 to several hundred. The now dominant NMR-based methods for NRP characterization are time-consuming and error prone and require large amounts of highly purified material. Because NRPs are often produced by difficult-to-cultivate microorganisms, it may not be possible to obtain sufficient quantities for NMR-based NRP sequencing, calling for a new nanomolar scale NRP sequencing approach.6 Such methods based on mass spectrometry (MS) promise to greatly accelerate NRP screening and may provide a vast resource for the discovery of pharmaceutical agents.7 MS and NMR are complementary approaches to NRP discovery, with MS being a fast nanomolar scale technique able to identify multiple, diverse NRPs in a single MS study, while NMR studies are being aimed at a single NRP but are capable of differentiating between monomers with identical masses (that are impossible to distinguish by MS).
In this paper we develop algorithms for identification of both cyclic and branched-cyclic NRPs. Branched-cyclic NRPs have a cyclic peptide backbone with a side chain consisting of one or more amino acids or a linear peptide backbone and a side chain bond formed between two arbitrary residues. While these two categories of peptides are chemically different, we use the same fragmentation model for them since their fragmentation patterns are similar.
The first automated MS approach to sequencing cyclic peptides was proposed by Ng et al. in 20098 and was used in a number of follow-up NRP discovery efforts.9−12 This approach was further extended to NRP sequencing by multistage mass spectrometry13 and multiplex sequencing of NRP families.14 Ng et al., 2009,8 also described algorithms for dereplication of cyclic peptides, answering the question of whether a spectrum arises from a known peptide in a chemical database such as Norine.15 They further introduced a variable dereplication algorithm answering the question of whether a spectrum is derived from a peptide that is similar to a known peptide. Ibrahim et al., 2012,16 further extended the dereplication approach8 from cyclic to branched-cyclic peptides. However, all of these approaches represent either de novo sequencing algorithms or dereplication searches in chemical databases and do not utilize valuable information about biosynthetic genes in the genomes of the NRP-producing organisms.17
In contrast to the dereplication approaches (that use the same chemical database for different organisms), our new NRPquest tool, available at www.cyclo.ucsd.edu, first generates a database of putative NRPs extracted from the genome sequence of the organism using the nonribosomal code. Because the nonribosomal code, in contrast to the ribosomal genetic code, is not yet fully understood (and is not as specific as the ribosomal genetic code), it may result in a multitude of peptides predicted from a single NRPS domain, and these putative NRPs may miss the correct NRP variant. Thus, the NRP identification approach “from genome to NRPS gene prediction to NRP prediction to spectra matching” is more intricate than the classical proteomics approach “from genome to gene prediction to spectra matching”. NRPquest couples MS and genome mining and represents the first tool that transforms previous approaches for de novo sequencing/dereplication of NRPs into an MS/MS database search approach for the identification of NRPs. We demonstrate that, similarly to proteomics, peptide identification tools for NRP discovery are much more accurate than de novo sequencing tools.
NRPSs are organized in modules that are responsible for the incorporation (and, if necessary, modification) of each additional amino acid in the synthesized NRP. Each module consists of several domains with defined functions, separated by short spacer regions of about 15 amino acids. A minimum of three domains are required for each NRPS module, termed the adenylation domain (A-domain), the peptidyl carrier domain (PCP-domain), and the condensation domain (C-domain). The A-domain is responsible for picking the specific amino acid monomers that are to be incorporated into the final product. Hundreds of different A-domain specificities have been classified using the Stachelhaus code, each one recruiting a specific amino acid as a monomer. This allows one to determine the putative sequence of the NRP by looking at the sequential order of A-domains along the assembly line and assigning a specific amino acid to each one. Many genome mining tools have been introduced for identification of NRPS gene clusters and the determination of their adenylation specificities, such as NRPSpredictor,18 ClustScan,19 NP.searcher,20 antiSMASH,21 and NRPSpredictor2.22 As the nonribosomal code5 is not yet fully understood (particularly with respect to unusual nonstandard amino acids) and is promiscuous (adenylation domains can often load multiple amino acids), the accurate determination of the specificities of the adenylation domains remains difficult. Moreover, most NRPs go through postassembly line modifications such as backbone macrocyclization and the addition of fatty acid chains, and genome mining tools currently fail to predict most of these modifications. NRPquest recruits NRPSpredictor2 for the prediction of adenylation specificities and implements a mutation-tolerant and modification-tolerant (blind) MS/MS search that allows for multiple modifications and mutations. This is a difficult computational problem even in the case of linear peptides,23 let alone nonlinear peptides.
As the first tool integrating genomic and mass spectrometric evidence for identification of NRPs, NRPquest promises to greatly increase the number of known peptide–spectrum matches (PSMs) for NRPs; for example, this study alone identified nearly a hundred PSMs. Previous studies emphasized the importance of increasing the number of PSMs (formed by NRPs) for developing rigorous statistical approaches and more adequate scoring functions for cyclic peptide analysis.8,13
Results and Discussion
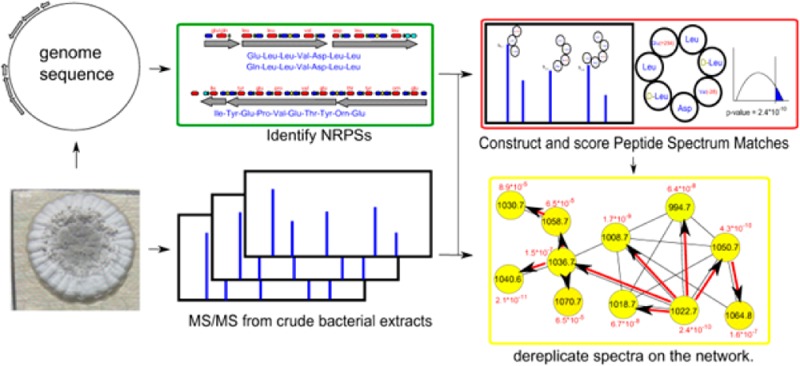
NRPquest uses a sequenced genome and a mass spectral data set as an input and includes the following steps: (i) uses NRP prediction tools for identifying NRPSs in the genome and constructing the database of putative NRPs, (ii) matches a spectral data set against the database of putative NRPs in a blind mode, (iii) computes statistical significance of the resulting cyclic, branched-cyclic, and linear PSMs and further ranks confident PSMs, (iv) constructs a spectral network29 to enlarge the set of identified PSMs and reveal families of related NRPs via spectral network dereplication (Figure 1).
Figure 1.

NRPquest pipeline starts with mining the microbial genome for putative NRPs using standard tools such as NRPSpredictor222 and constructing a database of putative NRPs. In the green rectangle, the results of NRPSpredictor2 are illustrated for Bacillus subtilis subsp. subtilis NCIMB 3610. This strain has two NRPS gene clusters, which according to NRPSpredictor2 produce two surfactins (7 amino acids each) and one plipastatin (10 amino acids). Adenylation domains are shown in red, condensation domains in blue, PCP domains in green, and thioesterase domains in light blue. Two blind modifications (with arbitrary offsets) are added to each NRP, and different possible structures (linear/cyclic/branched-cyclic) are considered (blue rectangle), resulting in ∼134 million modified peptides. The red rectangle illustrates PSMs formed between each spectrum and each putative modified NRP with feasible mass difference. PSMs are scored and their p-values are computed using MS-DPR.28 MS-DPR approximates the probability distribution of scores of PSMs formed by a random peptide and the spectrum and further derives the p-value as the area under the extreme tail of the distribution. Spectra are further analyzed by spectral networks to enlarge the set of identified statistically significant PSMs. The yellow rectangle illustrates a spectral network of surfactins. The red arrows in the network illustrate how annotations are propagated from a node with low p-value 2.4 × 10–10 (precursor m/z 1022.7 Da) to nodes with higher p-values (e.g., a node with precursor m/z 1030.7 Da), thus rescuing these nodes from being discarded as statistically insignificant.
NRP Database Construction
The NRPquest software pipeline starts with annotation of the genome of an organism of interest by NRPSpredictor2,22 which attempts to predict a set of all possible monomers for each adenylation domain in the genome (Figure 1). By considering all possible combinations of monomers, we construct a database of all putative NRPs (referred to as NRP database) that can be produced by the organism. NRPquest further searches the genome for methylation domains (PF08242) and accounts for the corresponding modification in the NRPs database. If NRPquest finds a methylation domain, it allows these modifications for the corresponding residues. If NRPquest finds a cytochrome P450 domain, it models side chain bonds between any two residues of the peptide, turning a linear peptide into a branched-cyclic peptide. Note that NRPquest also allows all peptides to have cyclic backbones with amino acid side chains. For each amino acid sequence in the database of putative NRPs, NRPquest considers linear, cyclic, and branched-cyclic structures representing these amino acid sequences.
Blind Search for Modifications
Each spectrum is matched against each putative peptide in the NRP database using a brute force algorithm that allows for blind modifications. The standard blind PTM search in traditional proteomics with tools (e.g., InsPecT23 and MODa24) typically limits the searches to at most two modifications, since further increase in the number of blind modifications makes the identified peptides less reliable. Since predictions provided by NRPSpredictor2 are typically within two mutations/modifications from the correct peptide, NRPquest also limits searches to at most two blind modifications whose total mass does not exceed 300 Da. This accounts for possible inaccurate adenylation specificity prediction of rare nonstandard amino acids (such as kynurenine from daptomycin), postassembly line modifications (such as modification of homoproline to 4-oxo-homoproline in pristinamycin), and addition of a fatty acid tail. Similarly to identification of linear peptides, MS-based methods for NRP identifications are limited to finding modification masses but do not provide insights into specific chemistry of modifications.
For example, in the case of Bacillus subtilis, the genome sequence has two NRPS clusters (surfactin and plipastatin). NRPSpredictor2 predicts a single NRP for plipastatin but two NRPs for surfactin (both Glu and Gln are predicted as first amino acid). As a result, we have only three possible NRPs initially predicted for B. subtilis. However, after considering different structures (linear, cyclic, branched-cyclic) there are 48 possible structures. If we consider two blind modifications with masses less than 300 Da for each peptide, there would be ∼134 million possibilities. However, many of these peptides have masses very different from the spectrum precursor mass, and on average, only ∼450 thousand of these putative modified peptides have a precursor mass matching each spectrum within 0.5 Da.
Statistical Significance
As there is still no large data set of PSMs formed by nonlinear peptides for automated learning of fragmentation propensities,25 previous studies of cyclic peptides8,16,26 used a somewhat primitive scoring based on the “shared peak” count. NRPquest scores cyclic PSMs using a previously proposed approach26 (Figure 1) and, to be consistent with the cyclic case, uses the same “shared peak count” approach for linear peptides. For branched-cyclic peptides, NRPquest simply adds up the scores of linear and cyclic parts (assuming that the linear branch is a modification on the cyclic part).
While methods for evaluating statistical significance of linear PSMs are well developed,27 until 2013 there were no methods for evaluating statistical significance of nonlinear PSMs. NRPquest calculates the statistical significance of each PSM using the recently introduced MS-DPR algorithm,28 which works for linear, cyclic, and branched-cyclic peptides (Figure 1). It further reports PSMs with low p-values (Table S1).
Spectral Networks
We use spectral networks29 to enlarge the set of identified NRPs. Spectral networks (also known as molecular networks12) analyze multiple spectra to simultaneously sequence related unknown peptides. The advantage of this approach (compared to de novo sequencing of individual spectra) is that finding peptides that simultaneously explain all spectra in a spectral network results in more accurate peptide reconstructions. Most NRPs form families of related peptides, and spectral network analysis can be used to reveal relationships between different spectra without knowing the amino acid sequences corresponding to these spectra.
Given a set of peptides P1 , ..., Pm, their peptide network is a graph with m nodes P1, ..., Pm and edges connecting two peptides if they differ by a single amino acid substitution or a single modification. Figure 2 shows the peptide network for nine variants of tyrocidine, a well-studied NRP from Bacilus brevis. For example, peptide 1 (tyrocidine B1) in this network (red node) is connected to four peptides differing from tyrocidine B1 by a single mutation or modification: tyrocidine A1 (peptide 2), tyrocidine B (peptide 5), tyrocidine C1 (peptide 8), and a previously unreported peptide with mass 1338.7 (peptide 9). However, it is not connected to peptides 3, 4, 6, and 7 since they differ from peptide 1 by multiple modifications. Six of these nine tyrocidines (1, 2, 3, 5, 7, 8) are contained in the database of putative NRPs generated by NRPSpredictor2 (without modifications), and three more differ from these variants by one or two modifications/mutations.
Figure 2.

Peptide network (a), spectral network (b), and annotations of nodes in the spectral networks (c) in the case of tyrocidines. The multitag algorithm13 for rescoring PSMs starts from a node with a known annotation in the spectral network and propagates annotations from known to unknown peptides through the edges in the network. The peptide network and spectral network of the tyrocidines are shown in parts (a) and (b). In part (c), annotations of each node in the spectral network are shown. Note that the nine nodes in the spectral network correspond to nine singly charged tyrocidines shown in Table S2. The spectral network revealed two novel tyrocidine variants at masses 1294.7 Da (node 6) and 1338.7 Da (node 9).
In reality, we are not given peptides P1 , ..., Pm but only their spectra S1, ..., Sm. Nevertheless, one can approximate the peptide network by constructing the spectral network on nodes S1, ..., Sm, where spectra Si and Sj are connected by an edge if they can be aligned against each other.29 For linear peptides, the spectral alignment algorithm23,30 reveals the lion’s share of spectra that correspond to peptides differing by a single mutation/modification, and similar approaches are proposed for nonlinear peptides.13 Figure 2 shows the spectral network of nine tyrocidines and illustrates that it captures all edges from the peptide network (shared edges between peptide and spectral networks are shown by thick lines). While the spectral network in Figure 2a is not identical to the peptide network in Figure 2b, their shared edges usually allow one to interpret the peptides corresponding to the nodes of the spectral network using the spectral network dereplication algorithm.13 NRPquest requires a p-value threshold to report reliable PSMs, and with a threshold of 10–10, four of nine peptides in this spectral network were identified (Figure 2c).
Below we illustrate how spectral networks allow us to confidently identify less reliable PSMs. The nine PSM peptides found by NRPquest have p-values ranging from 4.7 × 10–16 (extremely reliable identification) to 1.6 × 10–8 (less reliable identification). However, since NRPquest reports only PSMs with low p-values (otherwise, many statistically insignificant PSMs would be reported), some of these PSMs are deemed problematic and are not reported as identified peptides. For example, the spectrum corresponding to node 4 (with mass 1371.6 Da) was not identified by NRPquest (its p-value exceeds the threshold 10–10), but it is connected to spectrum 7, corresponding to the reliably identified peptide tyrocidine C (mass 1348.7 Da), with a low p-value of 2.7 × 10–12. Thus, peptide 4 differs from tyrocidine C by a modification with mass 23.1 Da. The variable dereplication algorithm13 further predicts the site of this modification at the tyrosine residue. This means the change is likely to be a mutation of tyrosine to tryptophan, and the peptide at node 4 is tyrocidine D.31
After constructing the spectral network, we extract their connected components (that correspond to families of related peptides) and further consider two possible scenarios: (i) no node (spectrum) in the connected component has been identified by NRPquest and (ii) at least one node in the connected component has been identified by NRPquest. In the former case, we use the multitag algorithm13 for multiplexed de novo sequencing of peptides represented by spectra forming this connected component. In the latter case (like in Figure 2), we use a variable dereplication version of the multitag algorithm.
Variable dereplication8 via peptide propagation through a spectral network29 allows one to identify peptides with more than two modifications that NRPquest missed during blind searches of individual spectra (as blind searches with more than two modifications become prohibitively time-consuming and increase the number of false PSMs). In the example in Figure 2a, four out of nine tyrocidines were identified (with p-values below 10–10) even before constructing the spectral network. After peptide propagation through the spectral network, all nine peptides were identified by the multitag algorithm with p-values below the 10–7 threshold.13 The case of the tyrocidines is somewhat special because many of them form statistically significant PSMs with exceptionally low p-values. For other spectral networks, the number of statistically significant PSMs is smaller; for example, in the case of arylomycins, no PSMs were identified as statistically significant before applying spectral networks.
Figure 3 shows the connected components of the spectral networks corresponding to six peptide families representing previously sequenced peptides. Daptomycin32 and arylomycin33 are identified from S. roseosporous, surfactin34 and plipastatin35 are identified from B. subtilis, pristinamycin36 is identified from S. pristinaespiralis, and tyrocidine37,38 is identified from B. brevis. For example, the spectral networks for the tyrocidines consist of nine peptides in the case of charge +1 spectra shown in red (eight peptides in the case of charge +2 spectra shown in green). Six out of these nine peptides have been previously sequenced in various studies,38 while others have not been reported in the literature yet (some of them may represent chemical adducts of known peptides). Similarly, only a small fraction of peptides in the spectral networks for daptomycin, arylomycin, surfactin, and plipastatin have been previously identified. The ability to identify known NRPs in a blind experiment and to discover previously unknown variants of known NRPs illustrates the power of NRPquest.
Figure 3.

Spectral networks of six NRP families identified by NRPquest: (a) daptomycin, (b) arylomycin, (c) pristinamycin, (d) plipastatin, (e) surfactin, and (f) tyrocidine (Table S1). Only spectra forming the most statistically significant PSMs are shown. Each node in these spectral networks may represent either a single spectrum or a group of very similar spectra (with similar precursor masses) compressed into a single node to simplify the network (in the latter case, the m/z of a cluster is the average of m/z of spectra in the cluster). The thickness of the edges indicates the level of similarity between the nodes in the spectral networks. Two connected components (in the case of plipastatin and tyrocidine) correspond to two different charge states (currently, the spectral alignment algorithm may fail to connect spectra from related peptides with different charges by an edge).
We have described how spectral networks help in identifying PSMs with high p-values (that would otherwise be discarded) that are adjacent to statistically significant PSMs with low p-values in the spectral networks. Another contribution of spectral networks is reranking and potentially reusing the entire spectral network of PSMs even if all PSMs in this spectral network have high p-values. Starting from a PSM PSM(P1,S1) and a spectral network with nodes S1, ..., Sm, the dereplication algorithm determines unknown peptides P1, ..., Pm that form PSMs with spectra S1, ..., Sm and computes the SpecNetScore:
Afterward, it reports all found PSMs (P1,S1), ..., (Pm,Sm) if the SpecNetScore exceeds a threshold.
Benchmarking NRPquest
We analyzed bacterial extracts from Streptomycesroseosporous NRRL 15998 (SR), Streptomyces pristinaespiralis ATCC 25486 (SP), Bacillus subtilis subsp. subtilis NCIMB 3610 (BS), and Bacillus brevis (BB). Table S1 illustrates that peptides with the lowest p-values that were identified by NRPquest in a blind experiment correspond to previously sequenced NRPs and provides confidence that some other PSMs that NRPquest finds may correspond to novel variants of known NRPs. While it is not possible to verify these low-abundance NRP variants by NMR, we were able to verify that these novel compounds are related to known NRPs using spectral networks (Figure 3). We further verified the significance of the resulting PSMs by computing their p-values (Table S1).
NRPquest constructs spectral networks (Figure 3) to improve the identification of individual spectra and to enlarge the set of identified PSMs (Table S2). After annotating spectra in the spectral network, we removed all nodes corresponding to PSMs with high p-values above 0.0001, as some large networks are difficult to visualize. Table S2 illustrates that spectral networks allow one to confidently sequence all spectra in a connected component of a spectral network via peptide propagation as long as even a single spectrum in this component is identified at the previous MS/MS database search step.
In addition to NRPs, peptide natural products include RiPPs. In a separate paper, Mohimani et al., 2013,40 described RiPPquest, an automated genome mining approach for RiPP identification. As NRPquest and RiPPquest are complementary tools searching completely different protein databases, the sets of peptides they identify do not overlap. Thus, the cases when NRPquest reports RiPPs (or RiPPquest reports NRPs) are not expected to occur in practice.
For example, in the case of S. roseosporous NRRL 15998, the spectral data set has 4355 spectra, out of which only six are assigned to NRPs with a p-value smaller than 10–10. In this particular data set, two RiPPs, called SRO-2212 and SRO-3108, were previously discovered.17 NRPquest correctly discards spectra of these RiPPs since they have a low score against the putative NRPs.
The identification of arylomycin via spectral networks illustrates the power of combining genome mining with spectral networks. Arylomycin is an antibiotic that selectively binds type I signal peptidases (SPases). Despite in vivo activity, and the fact that SPase is conserved and easily accessible as a drug target, arylomycin was dismissed as a drug candidate in the past, as it was perceived to have a narrow spectrum of activity. However, recent studies39 demonstrated that modified arylomycins are actually broad-spectrum antibiotics and that, if optimized to bind their targets with slightly more affinity, arylomycins would have a spectrum of activity that supports their progression as broad-spectrum therapeutics. Currently, RQx Pharmaceuticals is proceeding with clinical trials of arylomycin variants. Our study reveals a variety of arylomycins, opening the possibility to examine some of them as potential therapeutic agents. The limited number of fragmentation sites observed in some arylomycin MS/MS spectra makes it difficult to identify them individually. Table S2(A) illustrates that no arylomycin was identified with a p-value below the threshold. However, simultaneous analysis of different variants of arylomycin enables identification of seven arylomycin variants, as Table S2 and Figure 3b illustrate. Note that identification of arylomycins was based solely on their MS/MS and genomic information, and no prior information about known arylomycin peptides was used.
The effort involved in NRP discovery from MS/MS can be divided into three steps. The first step is determining which of the MS/MS spectra represent NRPs. This is a crucial step in natural product discovery because most of the time bacterial extracts also include endogenous peptides, RiPPs, PKs, and other chemical molecules in addition to NRPs. The second step is the correlation of MS/MS spectra to NRPSs in the genome. The third step is the annotation of spectra by assigning modifications to individual NRP residues. Our results in Table S1 show that while NRPquest has succeeded in the first two steps, the last step remains problematic and needs a better understanding of NRPS modification enzymes and NRP fragmentation. We hope that bioinformatics methods such as NRPquest will enable this progress by helping in the development of large data sets of annotated NRPS gene clusters.
Papers describing NRPs are usually limited to the analysis of a single peptide or a very few peptides. The first application of NRPquest revealed nearly a hundred NRPs (including unknown variants of previously known peptides) in a single study. This result provides hope that NRPquest can potentially make NRP identification as robust as peptide identification in traditional proteomics with popular spectral identification tools.
NRPquest utilizes NRPSpredictor2 for predicting NRPs from the bacterial genome. Because at most two blind modifications are considered, NRPquest would fail if NRPSpredictor2 prediction differs from the correct peptide by more than two modifications or mutations. Thus, users of NRPquest should be aware that it can work only within the limitations of genome mining tools (such as NRPSpredictor2 and antiSMASH), which in turn depend on accurate genome sequence information.
Experimental Section
Bacterial Metabolite Extraction
We analyzed bacterial strains of Streptomyces roseosporous NRRL 15998, Streptomyces pristinaespiralis ATCC 25486, Bacillus subtilis subsp. subtilis NCIMB 3610, and Bacillus brevis ATCC 8185. Each agar plate was inoculated with each bacterial strain by four parallel streaks. The plates were incubated for 10 days at 28 °C. The agar was sliced into small pieces, then put in a 50 mL centrifuge tube, covered with equal amounts of Milli-Q H2O and nBuOH/MeOH for 12 h at 28 °C, and shaken at 225 rpm. The nBuOH/MeOH layer was collected using a transfer pipet and dried with a rotary evaporator.
Genome Data Sets
Genomes of S. roseosporous and S. pristinaespiralis were recently sequenced at the Broad Institute and are available from the Actinomycetales database Web site.41 Genomes of B. subtilis and B. brevis are available from NCBI.
Spectral Data Sets
Collision-induced dissociation (CID) MS/MS data sets were collected with or without liquid chromatography (LC) separation in-line with mass spectrometry. For LC-MS, capillary columns were prepared by drawing a 360 μm o.d., 100 μm i.d. deactivated, fused silica tubing (Agilent) with a model P-2000 laser puller (Sutter Instruments) (Heat: 330, 325, 320; Vel, 45; Del, 125) and were packed at 600 psi to a length of about 10 cm with C18 reversed-phase resin suspended in MeOH. The column was equilibrated with 95% solvent A (H2O, 0.1% AcOH) and loaded with 10 μL (10 ng/μL in 10% CH3CN) of bacterial BuOH/MeOH extract by flowing 95% of solvent A and 5% of solvent B (CH3CN, 0.1% AcOH) at 200 μL/min for 15 min. A gradient was established with a time-varying solvent mixture [(min, % of solvent A): (20, 95), (30, 60), (75, 5)] and directly electrosprayed into the LTQ-FT MS inlet (source voltage, 1.8 kV; capillary temperature, 180 °C). The first scan was a high-resolution broadband scan. The subsequent six scans were low-resolution scans data-dependent on the first scan. In each data-dependent scan, the top intensity ions were selected to be fragmented by CID, which generated hundreds of fragmentation spectra collected as individual data events. The resulting .RAW files were converted to .mzXML using the program ReAdW (http://tools.proteomecenter.org).
Acknowledgments
We would like to thank M. Rottig and M. Medama for providing the source code and insightful suggestions on using NRPS specificity prediction software, and N. Bandeira for insightful suggestions on using spectral networks. This work was supported by U.S. National Institutes of Health grants 1-P41-RR024851-01, GM086283, and GM097509.
Supporting Information Available
A user manual on how to use the NRPquest Web server for discovering NRPs from genomic and mass spectral data and scoring tables are included in the Supporting Information. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Newman D. J.; Cragg G. M. J. Nat. Prod. 2007, 70, 461–477. [DOI] [PubMed] [Google Scholar]
- Strieker M.; Tanovi A.; Marahiel M. A. Curr. Opin. Struct. Biol. 2010, 20, 234–240. [DOI] [PubMed] [Google Scholar]
- Arnison P. G.; Bibb M. J.; Bierbaum G.; Bowers A. A.; Bugni T. S.; Bulaj G.; Camarero J. A.; Campopiano D. J.; Challis G. S.; Clardy J.; Cotter P. D.; Craik D. J.; Dawson M.; Dittmann E.; Donadio S.; Dorrestein P. C.; Entian K. D.; Fischbach M. A.; Garavelli J. S.; Gransson U.; Gruber C. W.; Haft D. H.; Hemscheidt T. K.; Hertweck C.; Hill C.; Horswill A. R.; Jaspars M.; Kelly W. L.; Klinman J. P.; Kuipers O. P.; Link A. J.; Liu W.; Marahiel M. A.; Mitchell D. A.; Moll G. L.; Moore B. S.; Muller R.; Nair S. K.; Nes I. F.; Norris G. E.; Olivera B. M.; Onaka H.; Patchett M. L.; Reaney M. J. T.; Rebuffat S.; Ross R. P.; Sahl H. G.; Schmidt E. W.; Selsted M. E.; Severinov K.; Shen B.; Sivonen K.; Smith L.; Stein T.; Sussmuth R. E.; Tagg J. R.; Tang G. L.; Truman A. W.; Vederas J. C.; Walsh C. T.; Walton J. D.; Wenzel S. C.; Willey J. M.; van der Donk W. A. Nat. Prod. Rep. 2013, 30, 108–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieber S. A.; Marahiel M. A. Chem. Rev. 2005, 105, 715–738. [DOI] [PubMed] [Google Scholar]
- Stachelhaus T.; Mootz H. D.; Marahiel M. A. Chem. Biol. 1999, 6, 493–505. [DOI] [PubMed] [Google Scholar]
- Molinski T. F. Curr. Opin. Biotechnol. 2010, 21, 819–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J. W.; Vederas J. C. Science 2009, 325, 161–165. [DOI] [PubMed] [Google Scholar]
- Ng J.; Bandeira N.; Liu W. T.; Ghassemian M.; Simmons T. L.; Gerwick W. H.; Linington R.; Dorrestein P. C.; Pevzner P. A. Nat. Methods 2009, 6, 596–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leao P. N.; Pereirab A. R.; Liu W. T.; Ng J.; Pevzner P. A.; Dorrestein P. C.; Konig G. M.; Teresa M.; Vasconcelos S. D.; Vasconcelos V. M.; Gerwick W. H. Int. J. Mass Spectrom. Ion Processes 2010, 107, 11183–11188. [Google Scholar]
- Liu W. T.; Yang Y. L.; Xu Y.; Lamsa A.; Haste N. M.; Yang J. Y.; Ng J.; Gonzalez D.; Ellermeier C. D.; Straight P. D.; Pevzner P. A.; Pogliano J.; Nizet V.; Pogliano K.; Dorrestein P. C. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 16286–16290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mevers E.; Liu W. T.; Engene N.; Mohimani H.; Byrum T.; Pevzner P. A.; Dorrestein P. C.; Spadafora C.; Gerwick W. H. J. Nat. Prod. 2011, 74, 928–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watrous J.; Roach P.; Alexandrov T.; Heath B.; Yang J.; Kersten R.; van der Voort M.; Pogliano K.; Gross H.; Raaijmakers J.; Moore B.; Laskin J.; Bandeira N.; Dorrestein P. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, E1743–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohimani H.; Liu W. T.; Liang Y.; Gaudenico S.; Fenical W.; Dorrestein P. C.; Pevzner P. J. Comput. Biol. 2011, 18, 1371–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohimani H.; Liang Y.; Liu W. T.; Hsieh P. W.; Dorrestein P. C.; Pevzner P. J. Proteomics 2011, 11, 3642–3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caboche S.; Pupin M.; LeclÃ̈re V.; Fontaine A.; Jacques P.; Kucherov G. Nucleic Acids Res. 2008, 36, D326–D331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim A.; Yang L.; Johnston C.; Liu X.; Ma B.; Magarveya N. A. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 19196–19201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kersten R.; Yang Y.; Cimermancic P.; Nam S.; Fenical W.; Fischbach M.; Moore B.; Dorrestein P. C. Nat. Chem. Biol. 2011, 7, 794–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rausch C.; Weber T.; Kohlbacher O.; Wohlleben W.; Huson D. H. Nucleic Acids Res. 2005, 33, 5799–4808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starcevic A.; Zucko J.; Simunkovic J.; Long P. F.; Cullum J.; Hranueli D. Nucleic Acids Res. 2008, 36, 6882–6892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M. H.; Ung P. M.; Zajkowski J.; Garneau-Tsodikova S.; Sherman D. H. Nucleic Acids Res. 2009, 10, 185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medema M. H.; Blin K.; Cimermancic P.; Jager V.; Zakrzewski P.; Fischbach M. A.; Weber T.; Takan E.; Breitling R. Nucleic Acids Res. 2011, 39, W339–W346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottig M.; Medema M. H.; Blin K.; Weber T.; Rausch C.; Kohlbacher O. Nucleic Acids Res. 2011, 39, W332–W367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsur D.; Tanner S.; Zandi E.; Bafna V.; Pevzner P. Nat. Biotechnol. 2005, 23, 1562–1567. [DOI] [PubMed] [Google Scholar]
- Na S.; Bandeira N.; Paek E.. Mol. Cell Proteomics 2012, 11, M111, DOI: 10.1074/mcp.M111.010199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dancik V.; Addona T. A.; Clauser K. R.; Vath J. E.; Pevzner P. A. J. Comput. Biol. 1999, 6, 327–342. [DOI] [PubMed] [Google Scholar]
- Mohimani H.; Liu W. T.; Mylne J. S.; Poth A. G.; Colgrave M. L.; Tran D.; Selsted M. E.; Dorrestein P. C.; Pevzner P. J. Proteome Res. 2011, 10, 4505–4512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta N.; Bandeira N.; Keich U.; Pevzner P. J. Am. Soc. Mass Spectrom. 2011, 22, 1111–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohimani H.; Kim S.; Pevzner P. A. J. Proteome Res. 2013, 12, 1560–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandeira N.; Tsur D.; Frank A.; Pevzner P. A. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 6140–6145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pevzner P.; Dancik V.; Tang C. J. Comput. Biol. 2000, 7, 777–787. [DOI] [PubMed] [Google Scholar]
- Tang X.; Thibault P.; Boyd R. Int. J. Mass Spectrom. Ion Processes 1992, 122, 153–179. [Google Scholar]
- Debono M.; Barnhart M.; Carrell C. B.; Hoffmann J. A.; Occolowitz J. L.; Abbott B. J.; Fukuda D. S.; Hamill R. L.; Biemann K.; Herlihy W. C. J. Antibiot. 1987, 40, 761–777. [DOI] [PubMed] [Google Scholar]
- Holtzel A.; Schmid D. G.; Nicholson G. J.; Stevanovic S.; Schimana J.; Gebhardt K.; Fiedler H. P.; Jung G. J. Antibiot. 2002, 55, 571–577. [DOI] [PubMed] [Google Scholar]
- Arima K.; Kakinuma A.; Tamura G. Biochem. Biophys. Res. Commun. 1968, 31, 488–494. [DOI] [PubMed] [Google Scholar]
- Umezawa H.; Aoyagi T.; Nishikiori T.; Okuyama A.; Yamagishi Y.; Hamada M.; Takeuchi T. J. Antibiot. 1986, 39, 737–744. [DOI] [PubMed] [Google Scholar]
- de Crecy-Lagard V.; Saurin W.; Thibaut D.; Gil P.; Naudin L.; Crouzet J.; Blanc V. Antimicrob. Agents Chemother. 1997, 41, 1904–1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootz H. D.; Marahiel M. A. J. Bacteriol. 1997, 197, 6843–6850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roskoski R.; Gevers W.; Kleinkauf H.; Lipmann F. Biochemistry 1970, 9, 4839–4845. [DOI] [PubMed] [Google Scholar]
- Smith P. A.; Romesberg F. E. Antimicrob. Agents Chemother. 2012, 56, 5054–5060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohimani H.; Kersten R.; Liu W. T.; Wang M.; Purvine S. O.; Wu S.; Brewer H. M.; Pasa-Tolic L.; Moore B. S.; Pevzner P. A.; Dorrestein P. C. ACS Chem. Biol. 2014, 9, 1545–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Actinomycetales group database, Broad Institute of Harvard and MIT. http://www.broadinstitute.org/.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.