

Summary
Oral leukoplakia is the most prevalent and potentially malignant disorder of the oral mucosa. Previous studies have demonstrated that molecular changes of the WWOX gene (WW-domain containing oxidoreductase), a candidate tumor suppressor gene located at 16q23.3–24.1 that spans FRA16D, the second most common fragile site, are present in several malignant neoplasias, including oral squamous cell carcinoma. In this report, the role of the WWOX gene was investigated in 23 cases of oral leukoplakias. Using nested RT-PCR and immunohistochemistry, altered mRNA transcription and/or reduced Wwox protein expression was observed in 35% of the lesions when compared with normal mucosa. The majority of lesions (4/6) with altered transcripts had a reduction in the expression of Wwox protein. Although normal WWOX expression was found in some lesions with dysplasia, all lesions with WWOX mRNA and/or protein expression showed histological evidence of dysplasia and none of the cases without dysplasia presented this alteration. These results show that the WWOX gene alteration is an early genetic alteration and may contribute to oral carcinogenesis.
Keywords: WWOX gene, Tumor suppressor gene, Oral leukoplakia, Oral premalignant lesion, Oral carcinogenesis
Introduction
Oral leukoplakia (OL) is defined as a “predominantly white patch or plaque that cannot be characterized clinically or pathologically as any other disease and is not associated with any physical or chemical causative agent except the use of tobacco”.1,2 The development of OL is strongly associated with the exogenous exposure to carcinogens, mainly smoking, chewing tobacco and betel nut. Of these, tobacco use is the most important etiological factor and is present in 80% of all cases.3,4 OL may show histologically the presence of dysplasia which often undergoes malignant transformation to oral squamous cell carcinoma (OSCC) ranging from 1% to 28% and approximately 80% of oral cancers progresses from precancerous lesions.5–7 According to the degree of dysplasia, the lesions are classified as mild, moderate or severe, based on histopathological findings.1 Although these designations are thought to be sequential phases of oral carcinogenesis, transformation is found in lesions with mild dysplasia.8 In addition, many studies have demonstrated that susceptibility to oral cancer is associated with phase I and phase II metabolic enzymes.9–11 It has also been suggested that the detection of genetic changes such as DNA aneuploidy and loss of heterozygosity could improve the possibilities for predicting malignant development from precursor lesions.12 However, there are no reliable biomarkers to predict which oral leukoplakias will be quiescent or will rapidly become invasive squamous cell carcinoma.12
WWOX (WW-domain containing oxidoreductase) is a candidate tumor suppressor gene that was identified in chromosome 16q23.3–24.1. It was determined that the WWOX gene spans the common fragile site region FRA16D.13–15 Chromosomal fragile sites are often hot spots for translocations, deletions, gene amplification and integration of oncogenic viruses. These fragile sites are chromosomal regions that frequently exhibit DNA strand breaks, often following exposure to chemicals that delay DNA replication.16 Of the common chromosomal fragile site loci, FRA3B and FRA16D are the most frequently expressed.17 The tumor suppressor gene fragile histidine triad (FHIT) spans the FRA3B fragile site,18 and abnormal FHIT transcription and low FHIT expression were detected in various human cancers, including oral squamous cell carcinomas.19,20 Studies of the two most frequently affected common fragile site loci, FRA3B and FRA16D, have provided compelling evidence that these regions are indeed prone to DNA instability in cancer cells.21 It has also been shown that exposure to environmental carcinogens such as smoking and alcohol consumption increases the potential for chromosome breakage at fragile sites FRA3B and FRA16D in esophageal, non-small-cell lung and cutaneous squamous cancer.21–25 Furthermore, we have recently described alterations on WWOX gene in 50% of oral squamous cell carcinomas.26 Therefore, given that OLs have been associated with squamous carcinomas, we investigated whether molecular changes of the WWOX gene were present in these potentially malignant disorders.
Materials and methods
Human tissue
Fresh tissue samples were consecutively obtained from 23 patients (14 males and nine females; age ranged from 29 to 67 years, mean 47 years; smoking status27: four non-smokers and 19 smokers) undergoing therapeutic surgical resection for OL at the Oral Diagnosis Clinic at School of Dentistry between March 2005 and June 2006. The cases were submitted to histological grade accord World Health Organization Classification of Tumors Pathology & Genetic of Head Neck Tumors, 20051 and are shown in Table 1. Normal oral mucosa was obtained from volunteers without leukoplakia during non-neoplastic or preprosthetic surgical procedures (control samples). Age, sex and smoking habits of the healthy volunteers matched that from patients. The present study was approved by the local Ethics Committee and a signed informed consent was obtained from all patients as well as healthy volunteers. In each case, a portion of the lesion was resected, immediately snap frozen and stored at −80 °C. For immunohistochemistry, a portion of the tissue was fixed in 10% buffered formalin and paraffin embedded.
Table 1.
Summary of the RT-PCR and Immunohistochemistry results
| Lesions | RT-PCRa | Aberrant product | IH scoreb | Histologyc |
|---|---|---|---|---|
| #OL1 | N | – | 3 | 1 |
| #OL2 | N and aberrant | Exons 2d – 7d deletion | 1 | 2 |
| #OL3 | N | – | 3 | 1 |
| #OL4 | N | – | 2 | 2 |
| #OL5 | N | – | 3 | 2 |
| #OL6 | N | – | 2 | 4 |
| #OL7 | N | – | 3 | 2 |
| #OL8 | N | – | 3 | 3 |
| #OL9 | N | – | 3 | 3 |
| #OL10 | N | – | 3 | 2 |
| #OL11 | N | – | 3 | 3 |
| #OL12 | N | – | 3 | 2 |
| #OL13 | N | – | 3 | 1 |
| #OL14 | N and aberrant | Exons 1d – 7d deletion | 2 | 2 |
| #OL15 | N | – | 3 | 4 |
| #OL16 | N and aberrant | NS | 2 | 4 |
| #OL17 | N | – | 3 | 3 |
| #OL18 | N and aberrant | Exons 2d – 8d deletion | 3 | 3 |
| #OL19 | N | – | 3 | 2 |
| #OL20 | N | – | 3 | 3 |
| #OL21 | N | – | 3 | 1 |
| #OL22 | A | – | 3 | 4 |
| #OL23 | N and aberrant | Exons 1d – 8d deletion | 2 | 3 |
| Exons 6 – 8 deletion |
RT-PCR: N: wild type transcript; A: absence of transcript; NS: not sequenced.
Immunohistochemistry scores; +1, 0–50% positive cells; +2, 51–75% positive cells; +3, >76% positive cells. Normal mucosas were used as positive controls and all were score +3.
Histological grading (1): 1 – without dysplasia; 2 – mild dysplasia; 3 – moderated dysplasia; 4 – severe dysplasia.
Partial exon deletion.
Reverse transcription-PCR analysis
Total RNA was extracted from cells of the OL lesions with Trizol reagent (Invitrogen Life Technologies, Carlsbad, CA), according to the manufacturer's recommendations and treated with DNAse (Invitrogen Life Technologies, Carlsbad, CA), after microdissection. First-strand cDNA was prepared from 1 μg of total RNA treated with DNAse, using the Superscript first-strand synthesis system (Invitrogen Life Technologies). After reverse transcription, the cDNA was used as a template for PCR amplification of the human WWOX cDNA. The first and second amplifications were performed with nested primers as previously described.28 Both reactions were carried out in a volume of 25 μl containing 10 pmol of each primer, 2.5 mM MgCl2, 1.5 mM dNTP mix, 1× PCR buffer, and 1.25 unit of Taq DNA Polymerase Recombinant (Invitrogen Life Technologies). Amplifications were carried out in a Mastercycler gradient thermocycler (Eppendorf AG) as follows: an initial denaturation for 8 min at 95 °C followed by 35 cycles of 94 °C for 30 s, 57 °C for 30 s, 72 °C for 1 min, and a final extension for 5 min at 72 °C. One microliter of the amplification product from the first reaction was used for the second reaction under the same conditions stated earlier. Glyceraldehyde-3-phosphate dehydrogenase cDNA was amplified as a control for cDNA quality.29 The amplified products were subjected to electrophoresis on a 6.5% polyacrylamide gel, followed by silver staining. DNA bands corresponding to the normal and abnormal size transcripts were purified using the GFX PCR DNA and Gel Band Purification Kit (Amersham Biosciences, Piscataway, NJ), and sequenced on the ABI PRISM 310 Genetic Analyzer (Applied Biosystems, Foster City, CA). The two primer sets of the second PCR amplification that amplify the whole open reading frame and two other primers designed to facilitate sequencing of the open reading frame (WW1FOR 5′-CGGCAAAGATACGACGGCAG-3′, exon 4 and WW2FOR 5′-ACTTTTGCTCTACCCTGG-3′, exon 7) were used. To each case, two different amplified products were sequenced.
WWOX immunohistochemistry
Tissue sections from OLs and oral normal mucosas were stained with Wwox antiserum.30 Briefly, 4 μm paraffin-embedded sections were dewaxed in xylene and hydrated with graded ethanol. Heat-induced epitope retrieval was performed with 1 mM EDTA buffer pH 8.0 for 30 min in a steamer at 94 °C. Endogenous peroxidase activity was blocked with 3% H2O2 in water for 10 min. Primary polyclonal rabbit antiWwox antiserum (140 μg/ml) was used at a 1:100 dilution (in BSA 0.5%) for 18 h at 4 °C. This was followed by incubation with the labeled streptavidin–biotin (LSAB) Kit (DakoCytomation California Inc., Carpinteria, CA). Peroxidase activity was developed with DAB (Sigma, St Louis, MI) with timed monitoring using a positive control sample. The sections were then counterstained with hematoxylin, dehydrated and mounted. Two experienced independent pathologists examined multiple fields and scored tissue sections for extent of staining, regardless of staining intensity (+1, 0–50% positive cells; +2, 51–75% positive cells; +3, greater than 76% positive cells). OL scored as +3 were considered as having normal expression to Wwox (oral normal mucosas were all +3).
Results
Aberrant WWOX gene transcripts
Aberrant WWOX gene transcript RT-PCR amplification was performed to analyze WWOX mRNA expression and six of 23 cases (26%) showed altered or absent expression of WWOX gene transcription. Of these six lesions, five showed transcripts with total or partial loss of exons. In one lesion (#OL22) no transcript was detected. This sample was positive for Cytokeratin 19 that was used as a marker of epithelial cells.31 All normal mucosa had normal size transcripts without aberrant transcripts (Fig. 1a). Sequence analyses of the RT-PCR products showed partial loss of exon 2, total loss of exons 3–6 and partial loss of exon 7 (#OL2) (Fig. 1b); partial loss of exon 1, total loss of exons 3–6 and partial loss of exon 7(#OL14) (Fig. 1c); partial loss of exon 2, total loss of exons 3–7 and partial loss of exon 8 (#OL18) (Fig. 1d). Interestingly, two transcript variants were present in lesion #OL23, one with partial loss of exon 1, total loss of exons 2–7 and partial loss of 8 in one (Fig. 1e) and the other with loss of exons 6–8 (Fig. 1f). In case #OL16 we detected a band of abnormal size and we sequenced the exons 1–6 and part of exon 7 but the aberrant transcript was not possible detected because of minute sample amount. The remaining 17 OLs samples showed only the normal size transcript. Representative results of nested RT-PCR analysis are shown in Figure 1.
Figure 1.

RT-PCR amplifications of the WWOX cDNA of oral leukoplasia (OL) cases. (a) Electrophoresis in polyacrylamide gel 6.5% was used to detect wild-type transcript (1284 bp) and alterations in the WWOX mRNA transcripts. Absence of transcript was seen in 1 lesion (#OL22). Representative sequences from the OLs showing five different types of aberrant transcripts: #OL2 (b); #OL14 (c); #OL18 (d); two in #OL23 – < (e) and > (f) bands. * represents a partial loss of the exon. In case #OL16 we detected a band of abnormal size and we sequenced the exons 1–6 and part of exon 7 but the aberrant transcript was not possible detected because of minute sample amount. WT – Normal WWOX transcription RNA with 9 exons.
Expression of Wwox protein
Wwox expression in normal oral mucosa and OL was also confirmed by immunohistochemistry. Epithelial cells in normal oral mucosa displayed a cytoplasmic staining for Wwox protein as shown in Figure 2a and b. All normal tissue showed a score +3 for Wwox (Fig. 2a). Of the 23 OLs, six lesions (26%) showed a reduced expression to Wwox. Five of these (#OL4, #OL6, #OL14, #OL16, #OL23) were scored +2 and one lesion (#OL2) scored +1 (Fig. 2c). Taken together, the results obtained by RT-PCR and immunohistochemistry showed that 35% of the lesions presented mRNA and/or protein alteration and the majority of lesions (4/6) with altered transcripts had a significant reduction in the expression of Wwox protein (Table 1).
Figure 2.
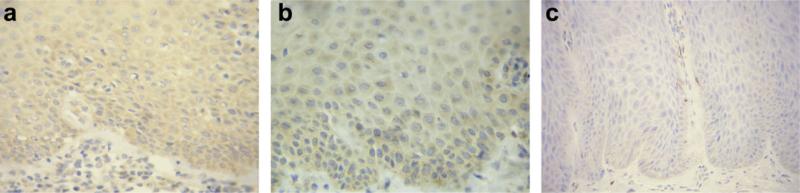
Immunohistochemical detection of Wwox protein in normal oral mucosa and oral leukoplakia (OL). (a) Normal oral mucosa showing (strong) cytoplasmic staining in all layers of the epithelium (score +3). (b) OL showing normal expression (score + 3) of Wwox protein (#OL10). (c) OL with reduced expression (score +1) of Wwox protein (#OL2) (streptavidin–biotin complex stain, 400×).
Correlation between the presence of dysplasia and WWOX alteration
All lesions with altered WWOX mRNA and/or protein expression showed histological evidence of dysplasia. While five out of the 11 samples with moderate or severe dysplasia showed WWOX alteration, three out of eight lesions with mild dysplasia and none of the four cases without dysplasia presented this alteration.
Discussion
Most of earlier studies have shown a risk of malignant transformation of OL in the range of 3.6–6%.6 However, this data has changed as recent studies have demonstrated an increase in malignant transformation rates now ranging from 8.9% to 17.5%.32,33 However, there are no genetic biomarkers to predict which OLs will be quiescent or will rapidly become invasive squamous cell carcinoma.12
In this study, we demonstrated an altered WWOX mRNA and/or protein expression in eight of 23 samples (35%) of OL. Among these samples, RT-PCR showed loss or aberrant transcripts in 26% of them. By immunohistochemistry, Wwox protein expression was reduced in 26%. We have recently investigated the WWOX gene in OSCCs and we showed molecular changes in 50% of primary tumors by either genomic DNA, RT-PCR or protein expression. The current findings suggest that WWOX changes are early events during OSCC development, a finding previously reported in breast carcinogenesis.34,35
WWOX gene underexpression in human cancers could result from distinct events such as allelic losses, point mutations, promoter hypermethylation, aberrant mRNA transcription, or a combination of two or more events resulting in a loss of tumor suppressor activity.36 Point mutations are rare in WWOX gene28 but loss of heterozygosity of WWOX allele is observed in a variety of cancers.15,28,37 The expression of some tumor suppressor genes is down-regulated in many cancers by epigenetic mechanisms. CpG rich areas of gene promoter regions are frequently methylated in cancer.38 WWOX hypermethylation was previously reported in breast and lung cancers.38,39
The WWOX aberrant forms can be generated by alternative splicing of WWOX mRNA as well as by exon deletions of the WWOX allele in cancer. Alternative splicing has been associated with various diseases.40 Splicing abnormalities occur frequently in cancer, perhaps due to increased cellular proliferation or other uncommon abnormalities in the splicing machinery. Furthermore, the splicing abnormalities would more often affect genes that have very long transcripts such as WWOX.30
The pattern of aberrations affecting WWOX is similar to that of FHIT gene in human tumors41 and could be caused by its location at a fragile site in the human genome.21 It has been known that a fragile site replicates late during the cell cycle and common fragile sites are susceptible to and preferentially targeted by the same carcinogenic agents.42 It is conceivable that breakage at WWOX/FRA16D and FHIT/FRA3B loci could be inflicted concomitantly.43
Four lesions with WWOX mRNA alteration had reduced expression of the protein. Interestingly, in two lesions the molecular changes of the mRNA were not followed by reduced protein expression. One them (#OL22) showed normal protein expression but no transcript was detected by RTPCR. The other sample (#OL18) had normal protein expression associated with partial loss of exon 2, total loss of exons 3–7 and partial loss of exon 8. As the molecular and immunohistochemical analysis were performed on separate areas of the lesion, this discrepancy may be due to the occurrence of heterogeneous expression. Furthermore, two lesions with reduced protein expression (#OL4 and #OL6) did not exhibit any abnormality at genetic level, thus the possibility of WWOX hypermethylation or deletion of one allele cannot also be excluded.36,39 In addition, despite we have performed microdissection, the possibility of contamination with non-epithelial cells should not be discarded.
The presence of dysplasia in OLs ranges from 15.6% to 39.2%.33 While lesions with severe dysplasia are prone to malignant transformation, this alteration is also found in OLs with mild dysplasia. Up to now, no molecular markers have been demonstrated as good prognostic predictors of potentially malignant oral lesions. Therefore, dysplasia recording continues to be the most reliable method for prognostic evaluation. Although we found WWOX expression in some lesions with dysplasia, all lesions with WWOX mRNA and/or protein expression showed histological evidence of dysplasia and none of the cases without dysplasia presented this alteration.
In summary, in this report, we demonstrate a reduction in the expression of WWOX gene in 35% of the OL lesions. These results show that the WWOX gene alteration is an early genetic alteration that could contribute to oral carcinogenesis and further studies on differential gene expression are required to help clarify the molecular changes associated with the transformation of OL to OSCC.
Acknowledgements
Grant sponsor: Milênio CNPq/MCT, PRONEX and FAPEMIG, Brazil; Grant sponsor: NCI RO1 CA102444. Dr. M.V. Gomez, L. De Marco and R.S. Gomez are research fellows of CNPq.
Footnotes
Conflict of Interest Statement
None declared.
References
- 1.Gale N, Pilch BZ, Sedransky D, El Nagzar A, Westra W, Califano J, et al. Precursor Lesions. In: Barnes L, Eveson JW, Reichart P, Sedransky D, editors. World Health Organization classification of tumors pathology & genetic of head neck tumors. Lyon: 2005. pp. 177–9. [Google Scholar]
- 2.Axéll T, Pindborg JJ, Smith CJ, van der Waal I. An International Collaborative Group on oral white lesions. Oral white lesions with special reference to precancerous and tabacco-related lesions: conclusions of an international symposium held in Uppsala, Sweden, May 18–21, 1994. J Oral Pathol Med. 1996;25(2):25–49. doi: 10.1111/j.1600-0714.1996.tb00191.x. [DOI] [PubMed] [Google Scholar]
- 3.Johnson NW, Warnakulasuriya S, Tavassoli M. Hereditary and environmental risk factors; clinical and laboratory risk matters for head and neck, especially oral, cancer and precancer. Eur J Cancer. 1996;5(1):5–17. [PubMed] [Google Scholar]
- 4.Neville BW, Day TA. Oral cancer and precancerous lesions. CA Cancer J Clin. 2002;52(4):195–215. doi: 10.3322/canjclin.52.4.195. [DOI] [PubMed] [Google Scholar]
- 5.Schepman KP, van der Meij EH, Smeele LE, van der Waal I. Malignant transformation of oral leukoplakia: a follow-up study of a hospital-based population of 166 patients with oral leuko plakia from The Netherlands. Oral Oncol. 1998;34(4):270–5. [PubMed] [Google Scholar]
- 6.Silverman S, Jr, Gorsky M, Lozada F. Oral leukoplakia and malignant transformation. A follow-up study of 257 patients. Cancer. 1984;53(3):563–8. doi: 10.1002/1097-0142(19840201)53:3<563::aid-cncr2820530332>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 7.Shiu MN, Chen TH. Impact of betel quid, tobacco and alcohol on three-stage disease natural history of oral leukoplakia and cancer: implication for prevention of oral cancer. Eur J Cancer Prev. 2004;13(1):39–45. doi: 10.1097/00008469-200402000-00007. [DOI] [PubMed] [Google Scholar]
- 8.Scully C, Sudbo J, Speight PM. Progress in determining the malignant potential of oral lesions. J Oral Pathol Med. 2003;32(5):251–6. doi: 10.1034/j.1600-0714.2003.00108.x. [DOI] [PubMed] [Google Scholar]
- 9.Drummond SN, De Marco L, Noronha JC, Gomez RS. GSTM1 polymorphism and oral squamous cell carcinoma. Oral Oncol. 2004;40(1):52–5. doi: 10.1016/s1368-8375(03)00132-5. [DOI] [PubMed] [Google Scholar]
- 10.Drummond SN, Gomez RS, Motta Noronha JC, Pordeus IA, Barbosa AA, De Marco L. Association between GSTT-1 gene deletion and the susceptibility to oral squamous cell carcinoma in cigarette-smoking subjects. Oral Oncol. 2005;41(5):515–9. doi: 10.1016/j.oraloncology.2004.12.014. [DOI] [PubMed] [Google Scholar]
- 11.Majumder M, Sikdar N, Paul RR, Roy B. Increased risk of oral leukoplakia and cancer among mixed tobacco users carrying XRCC1 variant Haplotypes and cancer among smokers carrying two risk genotypes: one on each of two loci, GSTM3 and XRCC1 (Codon 280). Cancer Epidem Biomar Prevent. 2005;14(9):2106–12. doi: 10.1158/1055-9965.EPI-05-0108. [DOI] [PubMed] [Google Scholar]
- 12.Reibel J. Prognosis of oral pre-malignant lesions: significance of clinical, histopathological, and molecular biological characteristics: review. Crit Rev Oral Biol Med. 2003;14(1):47–62. doi: 10.1177/154411130301400105. [DOI] [PubMed] [Google Scholar]
- 13.Thavathiru E, Ludes-Meyers JH, MacLeod MC, Aldaz CM. Expression of common chromosomal fragile site genes, WWOX/FRA16D and FHIT/FRA3B is downregulated by exposure to environmental carcinogens, UV, and BPDE but not by IR. Mol Carcinogen. 2005;44(3):174–82. doi: 10.1002/mc.20122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bednarek AK, Laflin KJ, Daniel RL, Liao Q, Hawkins KA, Aldaz CM. WWOX, a novel WW domain-containing protein mapping to human chromosome 16q23.3–24.1, a region frequently affected in breast cancer. Cancer Res. 2000;60(8):2140–5. [PubMed] [Google Scholar]
- 15.Bednarek AK, Keck-Waggoner CL, Daniel RL, Laflin KJ, Bergsagel PL, Kiguchi K, et al. WWOX, the FRA16D gene, behaves as a suppressor of tumor growth. Cancer Res. 2001;61(22):8068–73. [PubMed] [Google Scholar]
- 16.Popescu NC. Genetic alterations in cancer as a result of breakage at fragile sites. Cancer Lett. 2003;192(1):1–17. doi: 10.1016/s0304-3835(02)00596-7. [DOI] [PubMed] [Google Scholar]
- 17.Glover TW. Common fragile sites: review. Cancer Lett. 2006;232(1):4–12. doi: 10.1016/j.canlet.2005.08.032. [DOI] [PubMed] [Google Scholar]
- 18.Ohta M, Inoue H, Cotticelli MG, Kastury K, Baffa R, Palazzo J, et al. The FHIT gene, spanning the chromosome 3p14.2 fragile site and renal carcinoma-associated t(3;8) breakpoint, is abnormal in digestive tract cancers. Cell. 1996;84(4):587–97. doi: 10.1016/s0092-8674(00)81034-x. [DOI] [PubMed] [Google Scholar]
- 19.Mineta H, Miura K, Takebayashi S, Misawa K, Ueda Y, Suzuki I, et al. Low expression of fragile histidine triad gene correlates with high proliferation in head and neck squamous cell carcinoma. Oral Oncol. 2003;39(1):56–63. doi: 10.1016/s1368-8375(02)00022-2. [DOI] [PubMed] [Google Scholar]
- 20.Tai SK, Lee JI, Ang KK, El-Naggar AK, Hassan KA, Liu D, et al. Loss of Fhit expression in head and neck squamous cell carcinoma and its potential clinical implication. Clin Cancer Res. 2004;10(16):5554–7. doi: 10.1158/1078-0432.CCR-04-0208. [DOI] [PubMed] [Google Scholar]
- 21.O'Keefe LV, Richards RI. Common chromosomal fragile sites and cancer: focus on FRA16D. Cancer Lett. 2006;232(1):37–47. doi: 10.1016/j.canlet.2005.07.041. [DOI] [PubMed] [Google Scholar]
- 22.Sozzi G, Sard L, De Gregorio L, Marchetti A, Musso K, Buttitta F, et al. Association between cigarette smoking and FHIT gene alterations in lung cancer. Cancer Res. 1997;57(11):2121–3. [PubMed] [Google Scholar]
- 23.Mori M, Mimori K, Shiraishi T, Alder H, Inoue H, Tanaka Y, et al. Altered expression of FHIT in carcinoma and precarcinomatous lesions of the esophagus. Cancer Res. 2000;60(5):1177–82. [PubMed] [Google Scholar]
- 24.Stein CK, Glover TW, Palmer JL, Glisson BS. Direct correlation between FRA3B expression and cigarette smoking. Gene Chromosome Cancer. 2002;34(3):333–40. doi: 10.1002/gcc.10061. [DOI] [PubMed] [Google Scholar]
- 25.Lai FJ, Cheng CL, Chen ST, Wu CH, Hsu LJ, Lee JY, et al. WOX1 is essential for UVB irradiation-induced apoptosis and down-regulated via translational blockade in UVB-induced cutaneous squamous cell carcinoma in vivo. Clin Cancer Res. 2005;11(16):5769–77. doi: 10.1158/1078-0432.CCR-04-2274. [DOI] [PubMed] [Google Scholar]
- 26.Pimenta FJ, Gomes DA, Perdigao PF, Barbosa AA, Romano-Silva MV, Gomez MV, et al. Characterization of the tumor suppressor gene WWOX in primary human oral squamous cell carcinomas. Int J Cancer. 2006;118(5):1154–8. doi: 10.1002/ijc.21446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moreno-López LA, Esparza-Gómez GC, González-Navarro A, Cerero-Lapiedra R, González-Hernández MJ, Domínguez-Rojas V. Risk of oral câncer associatd with tobacco smoking, alcohol consumption and oral hygiene: a case-control study in Madrid, Spain. Oral Oncol. 2000;36(2):170–4. doi: 10.1016/s1368-8375(99)00084-6. [DOI] [PubMed] [Google Scholar]
- 28.Kuroki T, Trapasso F, Shiraishi T, Alder H, Mimori K, Mori M, et al. Genetic alterations of the tumor suppressor gene WWOX in esophageal squamous cell carcinoma. Cancer Res. 2002;62(8):2258–60. [PubMed] [Google Scholar]
- 29.Maier JA, Voulalas P, Roeder D, Maciag T. Extension of the lifespan of human endothelial cells by an interleukin-1 a antisense oligomer. Science. 1990;249(4976):1570–4. doi: 10.1126/science.2218499. [DOI] [PubMed] [Google Scholar]
- 30.Ludes-Meyers JH, Bednarek AK, Popescu NC, Bedford M, Aldaz CM. WWOX, the common chromosomal fragile site, FRA16D, cancer gene. Cytogenet Genome Res. 2003;100(1–4):101–10. doi: 10.1159/000072844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aihara T, Noguchi S, Ishikawa O, Furukawa H, Hiratsuka M, Ohigashi H, et al. Detection of pancreatic and gastric cancer cells in peripheral and portal blood by amplification of keratin 19 mRNA with reverse transcriptase-polymerase chain reaction. Int J Cancer. 1997;72(3):408–11. doi: 10.1002/(sici)1097-0215(19970729)72:3<408::aid-ijc6>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 32.Bouquot JE, Whitaker SB. Oral leukoplakia – rationale for diagnosis and prognosis of its clinical subtypes or “phases”. Quintessence Int. 1994;25(2):133–40. [PubMed] [Google Scholar]
- 33.Bouquot JE, Speight PM, Farthing PM. Epithelial dysplasia of the oral mucosa-Diagnostic problems and prognostic features. Current Diagn Pathol. 2006;12(1):11–21. [Google Scholar]
- 34.Chen T, Sahin A, Aldaz C. Deletion map of chromosome 16q in ductal carcinoma in situ of the breast: refining a putative tumor suppressor gene region. Cancer Res. 1996;56(24):5605–9. [PubMed] [Google Scholar]
- 35.Guler G, Uner A, Guler N, Han SY, Iliopoulos D, McCue P, et al. Concordant loss of fragile gene expression early in breast cancer development. Pathol Int. 2005;55(8):471–8. doi: 10.1111/j.1440-1827.2005.01855.x. [DOI] [PubMed] [Google Scholar]
- 36.Mahajan NP, Whang YE, Mohler JL, Earp HS. Activated tyrosine kinase Ack1 promotes prostate tumorigenesis: role of Ack1 in polyubiquitination of tumor suppressor Wwox. Cancer Res. 2005;65(22):10514–23. doi: 10.1158/0008-5472.CAN-05-1127. [DOI] [PubMed] [Google Scholar]
- 37.Yendamuri S, Kuroki T, Trapasso F, Henry AC, Dumon KR, Huebner K, et al. WW domain containing oxidoreductase gene expression is altered in non-small cell lung cancer. Cancer Res. 2003;63(4):878–81. [PubMed] [Google Scholar]
- 38.Iliopoulos D, Guler G, Han SY, Druck T, Ottey M, McCorkell KA, et al. Roles of FHIT and WWOX fragile genes in cancer. Cancer Lett. 2006;232(1):27–36. doi: 10.1016/j.canlet.2005.06.048. [DOI] [PubMed] [Google Scholar]
- 39.Iliopoulos D, Guler G, Han SY, Johnston D, Druck T, McCorkell KA, et al. Fragile genes as biomarkers: epigenetic control of WWOX and FHIT in lung, breast and bladder cancer. Oncogene. 2005;24(9):1625–33. doi: 10.1038/sj.onc.1208398. [DOI] [PubMed] [Google Scholar]
- 40.Venables JP. Aberrant and alternative splicing in cancer. Cancer Res. 2004;64(21):7647–54. doi: 10.1158/0008-5472.CAN-04-1910. [DOI] [PubMed] [Google Scholar]
- 41.Aqeilan RI, Kuroki T, Pekarsky Y, Albagha O, Trapasso F, Baffa R, et al. Loss of WWOX expression in gastric carcinoma. Clin Cancer Res. 2004;10(9):3053–8. doi: 10.1158/1078-0432.ccr-03-0594. [DOI] [PubMed] [Google Scholar]
- 42.Park SW, Ludes-Meyers J, Zimonjic DB, Durkin ME, Popescu NC, Aldaz CM. Frequent downregulation and loss of WWOX gene expression in human hepatocellular carcinoma. Brit J Cancer. 2004;91(4):753–9. doi: 10.1038/sj.bjc.6602023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guler G, Uner A, Guler N, Han S, Iliopoulos D, Hauck WW, et al. The fragile genes FHIT and WWOX are inactivated coordinately in invasive breast carcinoma. Cancer. 2004;100(8):1605–14. doi: 10.1002/cncr.20137. [DOI] [PubMed] [Google Scholar]