

Abstract
Hematopoiesis is a complex process that relies on various cell types, signaling pathways, transcription factors and a specific niche. The integration of these various components is of critical importance to normal blood development, as deregulation of these may lead to bone marrow failure or malignancy. Tle4, a transcriptional corepressor, acts as a tumor suppressor gene in a subset of acute myeloid leukemia, yet little is known about its function in normal and malignant hematopoiesis or in mammalian development. We report here that Tle4 knockout mice are runted and die at around four weeks with defects in bone development and BM aplasia. By two weeks of age, Tle4 knockout mice exhibit leukocytopenia, B cell lymphopenia, and significant reductions in hematopoietic stem and progenitor cells. Tle4 deficient hematopoietic stem cells are intrinsically defective in B lymphopoiesis and exhaust upon stress, such as serial transplantation. In the absence of Tle4 there is a profound decrease in bone mineralization. In addition, Tle4 knockout stromal cells are defective at maintaining wild-type hematopoietic stem cell function in vitro. In summary, we illustrate a novel and essential role for Tle4 in the extrinsic and intrinsic regulation of hematopoiesis and in bone development.
Introduction
The bone marrow (BM) is a heterotypic organ that dynamically integrates a variety of signals to modulate both quantitative and qualitative output of hematopoiesis to meet specific needs such as oxygen transport, immunity, and clotting. It is increasingly recognized that the differentiation of blood cells is affected not only by factors intrinsic to hematopoietic stem and progenitor cells (HSPC) but also a variety of cell types in the HSPC niche, including endothelial cells, osteolineage cells, sympathetic neurons, Cxcl12-activated reticular (CAR) cells, and nestin expressing stromal cells [1]. Together, these external and internal regulators work in concert to dynamically respond to physiological demand.
Genetic aberrations in these HSPC can lead to clonal expansions of progenitor cells as in leukemia. One such recurrent genetic mutation occurring in approximately 2% of acute myeloid leukemia (AML) patients is del(9q), which is enriched in patients with the t(8∶21) fusion protein AML1-ETO [2]. Previously, we showed that loss of two genes, Tle1 and Tle4, within the commonly deleted region of 9q can cooperate specifically with the AML1-ETO fusion product in a zebrafish model of myeloblastic expansion. Moreover, modulation of these genes in cell lines harboring t(8; 21) can influence the proliferative and apoptotic rate of these cells [3]. Additionally, Tle1 has been shown to be silenced by methylation in a broader set of AML samples, as well as in non-Hodgkin’s lymphoma and diffuse large B-cell lymphomas [4].
The TLE family of genes is a group of highly conserved transcriptional corepressors. The TLE homologue in Drosophila, Groucho (Gro), plays a crucial role in multiple developmental processes including neurogenesis, segmentation, and sex determination [5]. Gro also has instructive roles in many signaling pathways including receptor tyrosine kinase/Ras/MAPK, Notch, Wingless (Wg)/Wnt, and Decapentaplegic (Dpp) [6]. Proteins in the TLE family can influence transcription by either direct binding to a variety of transcription factors essential to both hematopoiesis and leukemogenesis, including members of the Hes, Runx, LEF1/Tcf, Pax, and Myc families, as well as recruitment of histone deacetylases and methylases, leading to chromatin silencing via condensation over large domains [7]. A combination of these effects likely underlies the ability of this protein family to influence cell fate and malignant transformation. Depending on context, these proteins may behave as either tumor suppressor genes or as facilitators of oncogenesis as in invasive breast cancer and synovial cell sarcoma, respectively [8], [9].
To better understand the role of Tle4 in development and oncogenesis, we developed a Tle4 knockout (KO) mouse. Tle4 KO mice have significant postnatal growth abnormalities, including skeletal and hematological defects. By three weeks of age, KO mice are leukopenic and display specific deficiencies of B cells and HSPC. We show that these defects arise from a combination of both intrinsic and extrinsic defects.
Materials and Methods
Generation of Tle4 null Mice
A conditional Tle4 null mouse was constructed by targeting LoxP sites to flank exon 2 via homologous recombination using the 129S6/SvEvTac ES cell line (Figure 1a). Resultant mice were crossed with β-actin: Cre mice (gift of Susan Dymecki) to delete exon 2 in all tissues. Heterozygote mice were backcrossed to C57BL/6 background for over 6 generations and interbred to generate Tle4 null mice.
Figure 1. The development of a novel Tle4 null mouse model.

(a) Targeting schema for generation of Tle4 null animals. Conditional Tle4 null mice were created by homologous recombination with an original construct containing a pgk-neo positive selection cassette and a Diptheria toxin (Dt) negative selection sequence, The pgk-neo selection cassette was excised between flanking Frt sites (white arrow head) by breeding mice to beta actin-Flp mice, leaving loxp sites flanking exon 2. These conditionally Tle4 null mice were bred to beta-actin cre mice to generate Tle4 null mice with a deleted exon 2 between flanking Loxp sites (black arrowheads). (b) RT-PCR showed exon 2 was cleanly excised. Loss of exon 2 creates a frameshift in the cDNA and a truncated non-functional Tle4 peptide. (c) Loss of Tle4 does not significantly affect the expression of other Tle family members as shown by RT-PCR of cDNA from the bone marrow of 2 week old mice. (d) Loss of Tle4 expression in Tle4 null mice was demonstrated by Western blot with protein from brain and lung.
Whole Mount Staining Of Skeletons to Visualize Cartilage and Calcified Bone
Embryos at embryonic age day 19.5 (E19.5) and one day old newborn pups were euthanized and skeletons subsequently cleared of skin and viscera. Specimens were fixed in 95% ETOH for at least five days, followed by at least two days in 100% acetone to remove adipose tissue. Specimens were then stained in 0.3% Alcian blue, 0.1% Alizarin red in 70% ETOH, and 5% acetic acid for three days. After rinsing in water, specimens were cleared in 2% KOH for 24 hours, 1% KOH/20% glycerol for 5–7 days, 1% KOH/50% glycerol 5–7 days, 1% KOH/80% glycerol 5–7 days, and finally stored in 100% glycerol.
Terminal Deoxynucleotidyl transferase dUTP nick end labeling Stain (TUNEL)
Paraffin-embedded humeri harvested from two week old Tle4 WT and KO mice were sectioned for TUNEL staining using the Apoptag kit per manufacturer’s protocol (EMD Millipore, Billerica, MA). Briefly, sections were bathed in Tris buffer with Tween X, followed by proteinase K, peroxidase block, and TdT enzyme treatments. All antibodies used for TUNEL staining were included in the kit and sections were counterstained using Methyl Green.
BM Transplantation and Homing Assays
BM transplantation was performed by injecting bulk two week old BM or E13.5 fetal liver cells via tail vein into lethally irradiated six to eight week old CD45.1 expressing C57BL/6 recipient mice. Serial transplantation was conducted similarly with pooled BM of primary or secondary recipients for secondary or tertiary transplantation, respectively. For secondary fetal liver transplantation, 1×105 whole BM cells from companion WT CD45.1+ mice were included to aid in the post engraftment phase during secondary transplantation. Mice were followed for at least 16 weeks prior to BM and blood analysis by flow cytometry and complete blood count analysis (VetScan HM5, Abaxis, Union City, CA).
To assess homing potential, total BM from three week old KO and WT littermates were pooled by genotype and stained with 1∶500 dilutions of DiD or DiI (Molecular Probes, Inc., Eugene, OR) and 1×107 cells were injected into lethally irradiated WT CD45.1 expressing C57BL/6 mice. After 18 hours post-transplantation, total BM was isolated from recipient mice and analyzed using a FACS Calibur flow cytometer (Becton Dickinson, San Jose, CA). The homing index was calculated as a quotient of the Input Ratio (%KO/%WT) over the Recovered Ratio (%KO/%WT), as described previously [10].
Flow Cytometry and Cell Sorting
Cells were stained and analyzed on either a FACSCalibur II or LSR II (Becton Dickinson, San Jose, CA). The following anti-mouse antibodies were used for flow cytometry analysis: B220-PE, CD3e-FITC, CD11b-APC, Gr-1-APC-Cy7, CD48-Pacific Blue, CD150-PE-Cy7, CD34, c-Kit-APC, Sca-1-PE, FcγR-PE, CD45.1-Brilliant Violet 570, and CD45.2-Alexa Fluor 700 (all from eBiosciences, San Diego, CA). Cell sorting was performed using a FACS Aria (Becton Dickinson, San Jose, CA). Antibodies for Annexin V and Ki-67 assays were used as per manufacturer’s protocol (Becton Dickinson, San Jose, CA). Gating strategy for HSPC fractions are as follows: (1) LSK: LineageNegCkit+Sca1+; (2) CMP: LineageNegCkit+Sca1Neg CD34+FcγRIIIlow; (3) GMP: LineageNegCkit+Sca1Neg CD34+FcγRIIIhigh; (4) MEP: LineageNegCkit+Sca1Neg CD34NegFcγRIIINeg; (5) CLP: LineageNeg IL7RαhighCkit+Sca1low; ProB Frac A: B220lowCD43HighBP1NegHSANeg; ProB Frac B: B220lowCD43HighBP1NegHSAPos; ProB Frac C: B220lowCD43HighBP1HighHSAPos; ProB Frac D: B220HighCD43NegIgMNeg; ProB Frac E: B220HighCD43NegIgMMid; ProB Frac F: B220High++CD43NegIgMHigh/Mid.
Stromal co-culture and colony forming unit assays (CFU)
For the methylcellulose colonies, whole BM from 2 week old WT and KO mice were lineage depleted using Biotin lineage antibody cocktail (B220, CD3, CD4, CD8, Mac-1, Gr-1 and Ter119) and Streptavidin magnetic microbeads (Miltenyi Biotec, Cambridge, MA). Cells were subsequently sorted by flow cytometry using the following fluorescent antibodies: PE-Cy7 lineage cocktail, c-Kit-APC, Sca-1-PE (eBiosciences, San Diego, CA). 1×103 sorted LKS cells from each cohort were plated in M3434 Methocult (Stem Cell Technologies, Vancouver, CA) according to the manufacturer’s protocol. Colonies were manually counted 7 days and 14 days after plating for serial replating and colony differentiation assays, respectively.
Stromal co-culture was performed as previously described [11]. Briefly, cells from crushed bones of three day old KO or WT littermates were grown for two weeks in 20% FBS in α-MEM supplemented with Penicillin/Streptomycin. After reaching confluence, media was changed to osteogenic media containing 100 µM β-glycerophosphate, 2.84 µM ascorbic acid, and 10 nM dexamethasone. Three weeks after osteogenic induction, 100 Lin− c-Kit+ Sca-1+ CD34− CD48− CD150+ cells were sorted directly onto osteoblastic stromal cells, and allowed to expand for 4 weeks. Flow analysis for c-Kit and Sca-1 expression was performed two weeks after initiation of co-culture. After four weeks of co-culture, 1×103 cells from each co-culture assay sample were seeded in Methocult 3434 (Stem Cell Technologies, Vancouver, Canada) and assessed for colony forming units by manual count one week later.
Western Blot, qRT-PCR
Protein lysates of bone and osteoblast-like cells from WT and KO samples of above co-culture experiments, as well as from brain and lung from WT and KO animals were prepared for western blot as previously described [12]. Western blots were probed with an antibody to TLE4 (ab64833, Abcam, Cambridge, MA), GAPDH (Abcam), murine stem cell factor (Scf) (Santa Cruz Biotechnology Inc., Santa Cruz, CA) and β-actin (Santa Cruz Biotechnology Inc.), followed by horseradish peroxidase conjugated secondary antibodies (Santa Cruz Biotechnology, Inc.) and visualization by chemoluminescence (Pierce ECL, Thermo Scientific, Rockford, Ill).
qRT-PCR for Tle1, Tle2, Tle3, Tle6, and Aes were performed using TaqMan Assays (Life Technologies, Grand Island, NY).
Statistics
The Student’s unpaired t-test was used for most analyses. A paired Student’s t-test was used for the homing index. Significance was set at P<0.05.
Ethics Statement
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved the Massachusetts General Hospital Institutional Animal Care and Use Committee (IACUC, permit #2004N000277). Animals were sacrificed according to institutional guidelines by use of CO2 asphyxiation. All efforts were made to minimize suffering.
Results
Verification of Tle4 knockout
Exon 2 of Tle4 was deleted by crossing β-actin: Cre mice with conditional Tle4 null mice containing LoxP sites flanking the exon (Figure 1a). Excision of exon 2 removes a lysine zipper motif critical to the function of the Q-oligomerization domain [13] and results in a frameshift leading to a truncated protein of 13 amino acids. Correct targeting and exon 2 deletion was verified by Southern blot (not shown). Successful recombination and deletion of exon 2 was verified by cDNA sequencing (Figure 1b). Levels of other Tle protein family members did not significantly differ between WT and KO animals, as verified by qPCR (Figure 1c). Western blot of both brain and lung (Figure 1d) confirmed that genetic ablation of Tle4 resulted in the absence of Tle4 protein expression.
Tle4 null mice have growth defects and skeletal abnormalities
Tle4 null mice appear grossly normal at birth without obvious organ abnormalities. However, their growth plateaus by the second week of life compared to littermates that are wild-type (WT) or heterozygous (HET) for Tle4 (Figure 2a). Tle4 null mice become moribund and perish by four weeks of life. No phenotypic differences were observed between WT and HET mice. WT littermate mice served as controls for all following experiments.
Figure 2. Tle4 null mice exhibit growth retardation and under-mineralization of bone.

(a) Tle4 null mice are born of similar size to wild-type littermates, but exhibit severe growth retardation after birth. (b) One day old Tle4 null mice (KO) have decreased mineralization of the trabeculae and cortical bone in the tibiae compared to wild-type littermates (WT) as shown by Von Kossa staining. (c) Alizarin Red/Alcian Blue staining for ossified bone (red) and cartilage (blue) of 1 day old wild-type (WT) and Tle4 null (KO) skeletons shows decreased mineralization in Tle4 null mice of both membranous bone (skull) and endochondral bone (vertebrae and long bones).
Decreased calcification, as well as thinning of the cortical bone and decreased trabecular bone, was evident by Von Kossa staining of tibiae in one day old KO as compared to WT mice (Figure 2b). Alizarin Red and Alcian Blue staining of calcified bone and cartilage confirmed a significant reduction in the degree of calcification throughout the skeleton of one day old KO mice compared to WT littermates despite no significant differences in overall body size or weight (Figure 2a). By 21–28 days of age, trabecular bone was almost absent in KO tibiae and femurs and abnormal growth plates were present. Though similar at 2 weeks, the growth plates of Tle4 null mice were much thinner with shorter columns of cells in both the proliferating and hypertrophic zones by 3 weeks of age (Figure 3a–h). At three weeks of age, tartrate-resistant acid phosphatase (TRAP) staining [17] demonstrated osteoclasts underneath the growth plate in KO mice that might be contributing to trabecular resorption (Figure 3i–l) [14]. These data indicate that Tle4 null animals have developmental defects in the skeletal system, fail to thrive, and subsequently die prematurely by 4 weeks of life.
Figure 3. Progressive bone marrow hypoplasia with defective ossification, loss of trabecular bone and thinning of cortical bone is seen by 3 and 4 weeks in Tle4 null mice.

A–D. Hematoxylin and Eosin (H&E) staining of tibiae of 3 and 4 week old WT and Tle4 null (KO) mice demonstrate multiple abnormalities in Tle4 null mice including progressive pancytopenia of the bone marrow (BM), loss of trabecular bone (T), and thinning of the cortical bone layer (C). A higher power view shows a thinner proximal tibial growth plate in Tle4 null mice with a decrease in thickness of the resting (R), proliferative (P), and hypertrophic (H) zones and near complete loss of the trabeculae. E–H. Tartrate-resistant acid phosphatase (TRAP) staining (pink) demonstrates osteoclasts clustering under the hypertrophic zone at the boundary of the bone marrow cavity in Tle4 null mice at 3 and 4 weeks of age.
Tle4 null mice have hematological abnormalities by two weeks of age
At two weeks of age, there were no significant differences in absolute numbers of all blood cell lineages between KO and WT mice as determined by complete blood count (CBC) (Figure 4a). However, immunophenotypic analysis of peripheral blood illustrated a significant reduction in the frequency of B220+ cells, but no significant difference in CD3+ or CD11b+ cells (Figure 4a). BM of two-week-old KO mice exhibited a two-fold reduction in total cellularity (Figure 4b), significant reductions in the frequency of B220+ cells, and an increase in the frequency of CD3+ cells (Figures 4b). By four weeks of age, KO animals had progressed to severe leukopenia, and more specifically lymphopenia, in the blood (Figure 4b).
Figure 4. Peripheral blood counts are normal in Tle4 null mice at 2 weeks of age, but significant abnormalities are seen in peripheral blood and bone marrow at 4 weeks of age.

(a) At 2 weeks of age there was no significant difference in any of the cell compartments in the peripheral blood of Tle4 null mice (KO) as compared with normal control littermates (WT). However, by 4 weeks of age Tle4 null mice exhibit a marked leukopenia (decreased WBC) and lymphopenia in the peripheral blood, that primarily affects the B-cells (B220+ cells), and not T-cell (CD3+) or myeloid cells (CD11b+). (b) Tle4 null mice exhibit severe bone marrow aplasia with a 2-fold decrease in bone marrow cellularity. Within the remaining population of lymphoid cells, B-cell development appears particularly affected with a significant decrease in the percentage of B-cells (B220+) and relative increase in the percentage of T-cells (CD3+).
By three weeks of age, KO mice also developed abnormalities in the spleen and thymus. Splenic cellularity was significantly reduced in KO mice (Figure 5a), most likely due to significant reductions of total B220+ lymphocytes (Figures 5a). Furthermore, the splenic architecture was disrupted in KO mice with an almost complete absence of splenic follicular zones (Figure 5b). The total cellularity of the thymus and T cell subsets in three week old mice were significantly reduced (Figure 6a). The KO animals also had significant thymic atrophy compared to WT mice without a clear demarcation between the cortex and medulla (Figures 6b). TUNEL staining of the thymus harvested from three week old KO mice demonstrated increased thymic apoptosis (Figure 6c). In summary, deletion of Tle4 resulted in B-cell lymphopenia in peripheral blood and decreased cellularity in hematopoietic organs.
Figure 5. Tle4 null mice develop splenic atrophy with abnormal splenic architecture.

(a) At three weeks of age there is marked splenic atrophy and decreased cellularity especially of the major B-cell (B220+) compartment. (b) H&E staining of the spleen reveals an absence of splenic follicles (dashed oval) in two week old Tle4 null mice (KO). (n = 3–4 per genotype; mean +/−SEM; *: P<.05).
Figure 6. Tle4 null mice develop thymic atrophy with a block in T-cell differentiation.

(a) There is a dramatic decrease in total thymocytes in 3 week old Tle4 null mice as compared to wild-type littermates. The majority of this decrease is due to loss of double positive CD4+CD8+ cells. Within the double negative (DN) T progenitor populations there appears to be a block between DN1 (CD44+CD25−) and DN2 (CD44+CD25+) with a significant decrement in DN2 cells and an insignificant decrease in DN3 (CD44−, CD25+) cells. (n = 3–4 per genotype; mean +/−SEM; *: P<.05). (b) The thymus of 3 week old Tle4 null mice is atrophied with a loss in the demarcation between cortex and medulla as seen by H&E. (c) TUNEL staining demonstrates thymic apoptosis in Tle4 mice.
Loss of Tle4 has deleterious effects on HSPC
Tle4 null mice were found to have an almost four-fold reduction in the total number of Lin− c-Kit+ Sca-1+ (LKS) cells and a three-fold reduction in CD34+ LKS cells (Figure 7a). Interestingly, the total number of long-term HSC, defined by surface expression of SLAM markers CD150+ and CD48−, were not reduced in KO mice despite the five-fold reduction in total BM leukocytes at this age (Figure 7a). However, total numbers of common myeloid (CMP) and more so common lymphoid progenitors (CLP) were significantly reduced in Tle4 null mice (Figure 7b). Finally, Tle4 null mice also have significant reductions in Pre/ProB fractions A through C (Figure 7c). Pax5, which derives its repressive activity via interaction with Tle4, is necessary for progression through the Pre/ProB checkpoints. Deletion of Pax5 results specifically in a block in B cell development between Fractions B (early pro-B) and C (late pro-B) [15], [16]. Therefore, while some component of the Tle4 null ProB phenotype may be derived from deregulated Pax5 signaling, it also appears that loss of Tle4 led to more deleterious effects on B cell development than mere phenocopy of Pax5 deficiency. Taken together, loss of Tle4 appears to decrease the frequency of LKS cells and especially B lymphoid progenitors, but not of LKS SLAM cells.
Figure 7. Tle4 null mice have significant aberrations in hematopoietic stem and progenitor cells (HSPC).

Bar graphs with representative flow cytometry plots in two week old littermates (a) show significant loss of LSK and LKS CD34+ cells, though the most immature long-term HSC population (LKS CD34+CD48–CD150+ HSC) is relatively preserved. (b) Examination of CMP, GMP, MEP, and CLP progenitor fractions demonstrated significant decreases in CMP and CLP populations. (c) Amongst the Pre/Pro B cell progenitors the decrement is most prominent in the early Fractions A through C. (n = 3–8 per genotype; mean +/− SEM; *: P<.05, **: P<.001, ***: P<.0001). See methods for gating strategy of stem and progenitor cell compartments.
To further typify the above-described difference in LKS frequency between WT and KO mice, additional studies were done to examine cell proliferation and apoptosis within LKS populations. There is no difference in cell proliferation and cell cycling between two-week old WT and KO LKS, as indicated by flow cytometry analysis of Ki-67 (Figure 8a). Interestingly, however, Annexin V analysis showed LKS cells from two-week old KO mice exhibiting significantly reduced viability and increased apoptotic and dead cells (Figure 8b). This suggests the decrease in LKS frequency in KO mice may be due to Tle4-dependent increased apoptosis rather than changes in cell proliferation or cycling.
Figure 8. The decrease in LKS cells in Tle4 null mice is due to an increase in apoptosis and cell death rather than a decrease in proliferation and is accompanied by abnormalities of the bone marrow stroma.

(a) LKS cells isolated from the bone marrow of two week old mice show no difference in cell cycle distribution. (b) There is however an increase in apoptotic and dead LKS cells in the Tle4 null mice. (c) TUNEL staining of the growth plate of the femur in two week old mice marks the normal zone of cell death between the hypertrophic (H) layer and forming trabecular (T) bone (A, B) with an increase in staining in Tle4 null mice (F, G). Lacunae in the epiphysis are lined with periosteal cells undergoing apoptosis in Tle4 null mice (H), but was not seen in wild type (WT) littermates. Similar periosteal cells undergoing apoptosis and stained by TUNEL are seen under the cortex of diaphyseal bone in Tle4 null mice (I) but absent in wild-type mice (D). An increase in TUNEL staining is also observed in cells of the bone marrow in Tle4 null mice (J) as compared to wild-type bone marrow (E).
Co-culture of wild-type HSC on Tle4 null stromal cells impairs long term colony forming ability
Given the critical importance of the BM niche on hematopoiesis, we hypothesized that the abnormalities in the skeletal or stromal compartments of the BM were contributing to defective hematopoiesis [17], [18]. This was confirmed by TUNEL staining of paraffin-embedded humeri harvested from two-week old WT and KO mice (Figure 8c). Compared to WT, various compartments of KO bone exhibited increased TUNEL staining, including the periosteal cells lining lacunae in the epiphysis (Figure 8c, panel C vs H) and periosteal cells under the cortex of the diaphyseal portion of the humerus (Figure 8c, panel D vs I). Moreover, TUNEL staining was more extensively pervasive throughout trabeculae and bone marrow in KO compared to WT mice (Figure 8c, panel B vs G, E vs J). This further suggested the possibility that the hematopoietic abnormalities seen in KO mice might be at least in part due to the absence of support from the osteoblastic niche.
To further elucidate whether impaired HSC maintenance by KO BM was dependent on stromal cell irregularities, western blots using whole bone lysates from two week old WT and KO mouse were performed using antibodies against Scf, a known factor involved in the maintenance of HSC [1] [27]. Though some KO mice displayed a complete lack of Scf expression, others showed modest protein levels (results not shown). The considerable animal to animal variation suggests that down-regulation of Scf expression is not a direct effect of Tle4 loss, but might reflect a loss of an Scf producing cell type in Tle4 null mice. Other factors may account for the impairment of HSC maintenance.
Since Tle4 null animals do not survive long enough to serve as bone marrow recipients of wild-type HSPC, we adapted a co-culturing assay to determine whether HSC-extrinsic factors may have influenced the observed hematological phenotype [11]. Wild-type LKS cells from two week old mice were co-cultured on WT or KO stroma cells derived from 3 day old mice for two weeks (Figure 9a). Non-adherent cells were harvested and analyzed for expression of c-Kit and Sca-1. In WT co-cultures, 6–15% of recovered cells were positive for both markers. In stark contrast, less than 1% of cells recovered from WT LKS cells plated on KO stromal co-cultures were C-kit+ Sca-1+ (Figure 9b). Long-term co-culture experiments plated in methylcellulose revealed an even more pronounced effect. Cells recovered from WT co-cultures exhibited an average of 10-fold more colonies than their KO counterparts (Figure 9c). Some KO co-cultures failed to exhibit any colony forming ability. Thus, Tle4 null stromal cells cannot maintain and support HSPC growth as efficiently as WT stromal cells.
Figure 9. Co-culture of wild-type HSC on Tle4 null stromal cells impairs long term colony forming ability.

(a) Schematic of co-culture assay. Briefly, stromal cells were isolated from bones of 2 week old WT and KO animals and plated in aMEM with dexamethasone, ascorbic acid, and vitamin D3 as described in Methods. WT LKS were plated and used in LTC-IC experiments (b) Bar graph showing a dramatic decreased frequency of Sca-1+c-Kit+ cells after two weeks of co-culture on the stroma from Tle4 null mice with representative flow plots (n = 2 biological replicates per genotype, in triplicate *: P<.01). (c) Long term culture-initiating cell assay (LTC-IC) after four weeks of co-culture with WT or KO stroma showed relatively few colonies obtained after culturing on Tle4 null bone marrow stroma (n = 2 biological replicates, each in 9-plicate, seeded in duplicates for CFU; *: P<.0001).
Tle4 null HSPC are intrinsically impaired in B cell differentiation and exhaust after serial transplantation
Due to the progressive leukopenia in primary Tle4 null mice and the extrinsic effects noted above, we tested the colony-forming ability of sorted LKS cells from 2 week old WT or TLE4 null mice. After seven days in methylcellulose, TLE4 null LKS were significantly less efficient at forming colonies than WT LKS cells, suggesting intrinsic defects in HSC and HSPC and impaired HSC-self renewal in Tle4 null mice (Figure 10a). These observations were further reinforced by a continued decrease in the ability of KO LKS cells to produce colonies when serially replated in methylcellulose at 14 days (Figure 10a). Additionally, these colonies were also typified as granulocyte/macrophage-forming (CFU-GM), erythroid-forming (BFU-E), or granulocyte/erythrocyte/monocyte/megakaryocyte-forming (CFU-GEMM) colony forming units to identify any Tle4-dependent HSC-intrinsic effects on progenitor differentiation. After 13 days in methylcellulose culture, sorted LKS cells from KO were less efficient in forming CFU-GM and CFU-GEMM compared to their WT counterparts (Figure 10b).
Figure 10. LKS cells from TLE4 null mice are impaired in colony forming unit ability.

(a) The number of colonies in methyl cellulose originating from 1000 LKS cells from TLE4 null mice were fewer compared to wild-type LKS cells on Day 7 and again after replating on Day 14. (n = 4 biological replicates per genotype, in triplicate) (b) LKS cells from Tle4 null mice form fewer CFU-GM and CFU-GEMM colonies but similar BFU-E colonies compared to wild-type. (n = 5 biological replicates per genotype, in triplicate) (**: P<.01, ***: P<.0001).
To further test this effect, we performed in vivo transplantation assays using BM from 2 week old animal or fetal liver hematopoietic cells to determine whether loss of Tle4 intrinsically affected HSC self-renewal and repopulation efficiency. Whole BM from two-week-old CD45.2+ KO and WT animals was isolated and transplanted into lethally irradiated allogeneic WT mice expressing CD45.1 (Figure 11a). Given our observation of non-significant differences in LKS SLAM populations, representing long-term HSC, between WT and KO BM, equal numbers of cells from donor KO and WT BM were transplanted into recipient mice. Sixteen weeks after BM transplant (BMT), recipients of KO and WT BM showed non-significant differences in donor chimerism in blood (Figure 11b). However, FACS analysis of peripheral blood showed that recipient mice of KO BM had significant reductions in the frequency of B cells, with concomitant increases in both T and myeloid cell frequencies (Figure 11b). At 32 weeks after transplant, CBC analysis of KO BM recipients demonstrated the development of significant leukopenia, especially a decrease in myeloid and lymphoid lineages (Figure 11c, left panel). Immunophenotypic analysis showed that the leukopenia was mostly due to a decreased frequency of B cells (Figure 11c, right panel). Fractionation of B cell precursors in recipients 32 weeks post-transplant showed a reduction in the absolute number of Fraction D and E cells and an increase in Fraction C before progression to fraction D (Figure 11d). This suggests that transplanted Tle4 null BM cells have a block in B cell development at the late pro-B to pre-B-cell stage, more restricted than seen in situ with Tle4 null mice, but still distinct from the early pro-B to late pro-B block reported with Pax5 deficiency [15], [16], [19].
Figure 11. Tle4 null mice have cell-intrinsic defects in HSPC self-renewal and B-cell development.

Bone marrow transplants of wild-type (WT) or Tle4 null (KO) bone marrow from 2 week old mice into normal recipients were performed to evaluate cell intrinsic defects. (a) Schematic of BMT experimental design. (b) Analysis of peripheral blood at 16 after transplant by flow cytometry and CBC demonstrated insignificant differences in engraftment as measured by the percent CD45.2+ cells, but impaired B-cell numbers (B220+) with relatively increased T-cells (CD3+) and myeloid cells (CD11b+). (c) 32 weeks after transplant mice receiving Tle4 null bone marrow developed leukopenia (decreased WBC) primarily accounted for by a decrease in lymphocytes (c, left panel), which by immunophenotyping represented a decrease in B-cells (B220+). (n = 9–10 per transplant group, *: P<.005 **: P<.0001). (d) Analysis of B-cell differentiation in the BM of recipient mice 32 weeks after transplant by quantitation of ProB Fractions shows an increase in Fraction C, but decreased Fractions D and E indicating a relative block in differentiation between Fractions C and D (n = 5 per genotype, *: P<.05). (e) Analysis of bone marrow 32 weeks after transplant showed no significant differences between KO and WT recipients in the absolute number of LKS, CLP, CMP, or GMP compartments, but did show a decrease in MEPs in KO recipient mice. (f) Competitive homing 18 hours after transplantation using whole BM from two week old WT and KO littermates showed no defect in homing ability (n = 3). The homing index is represented as: Input/Output or 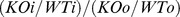 .
.
Analysis of HSPC in recipients 32 weeks after transplant, which has been shown to reflect long term repopulating activity of HSC [32], revealed no significant differences between KO and WT recipients in the absolute number of LKS, CLP, CMP, or GMP compartments. However, MEPs were significantly reduced in KO recipient mice (Figure 11e). A competitive homing experiment revealed that at 18 hours after transplant, KO HSC were able to home to the recipient niche as efficiently as WT cells (Figure 11f), indicating the observed abnormalities were not due to homing defects of these cells. These findings suggest that BMT with Tle4 null HSC results in leukopenia, specifically B lymphopenia arising from partial blocks in ProB development. However, KO donor BM retained normal frequencies of most HSPC fractions after transplantation into a normal BM niche.
To test self-renewal capacity of KO versus WT HSC while excluding confounding effects possibly due to prior exposure to a defective Tle4 null BM niche, we performed serial transplantations of fetal liver hematopoietic cells (FL) of E13.5 WT and KO CD45.2+ fetuses (Figure 12a). Recipients of KO and WT FL primary transplants exhibited peripheral leukopenia and specific B cell lymphopenia (Figure 7b, left and middle panels), recapitulating the phenotype observed in recipients of KO BM in prior BMT experiments (Figure 11c and 11d). Additionally, there were no differences in the frequency of HSC between primary recipients of KO or WT FL (Figure 12b, right). Upon serial transplantation of FL into secondary transplant recipients, we again saw highly significant leukopenia and lymphopenia 16 weeks after transplant in KO recipient mice (Figure 12c, left). Interestingly, secondary KO FL recipients also exhibited a significant reduction in Tle4 null-derived LKS (Figure 12c, right). Moreover, these LKS cells present at secondary transplant had high expression of CD34, with few CD34− LKS cells, while WT recipients had relatively equal numbers of CD34+ and CD34− LKS cells (Figure 12c, right). Sixteen weeks after tertiary transplant, total peripheral blood leukocytes decreased five fold with an even more significant reduction in the frequency of B cells (Figure 12d). Additionally, there was a striking reduction and, in some KO FL recipients, a complete absence, of CD45.2+ LKS cells, CD34− LKS cells, and LKS CD34− SLAM long term HSC at this time point (Figure 12e). These serial transplantation experiments suggest that the absence of Tle4 leads to reduced self-renewal of HSC.
Figure 12. Tle4 null fetal liver HSCs have impaired B cell development and exhaust with serial transplantation.

(a) Schematic of serial FL transplantation experimental design. (b) Peripheral blood and LKS analysis of FL transplant recipients 16 weeks after transplant revealed peripheral leukopenia (WBC) and specifically B cell (B220+) lymphopenia in the absence of Tle4 with no difference in LKS, LKS, CD34+, LKS CD34− populations (n = 10 per genotype for blood analysis, n = 6–7 per genotype for LKS analysis, ***: P<.0001). (c) Peripheral blood and LKS analysis 16 weeks after secondary transplantation also indicates leukopenia and lymphopenia in Tle4 null cells, but at this time also a significant decrease in LKS and LKS, CD34− populations (n = 5 KO recipients, n = 10 WT recipients for blood analysis, n = 5 mice per group for LKS analysis, *: P<.05 **: P<.001). (d) Peripheral blood analysis and LKS analysis 16 weeks after tertiary transplantation again showed leukopenia, B-cell lymphopenia, and a profound decrease in all HSC containing populations (n = 4 per genotype for blood and LKS analysis, *: P<.05, **: P<.01, ***: P<.001). (e) Representative flow cytometry plots showing progressive loss of Tle4 null HSCs over successive transplantation.
Discussion
In Drosophila, Groucho is a master transcriptional regulator expressed ubiquitously throughout development and influences multiple key developmental processes [7]. The precise roles of the various TLE homologues in vertebrates are not very well understood. To characterize those functions specific to Tle4, we developed a novel Tle4 null mouse model. Our studies have identified the critical importance of Tle4 in bone calcification, bone marrow niche formation, BM cellularity, B cell development, HSPC self-renewal capacity, and thymic and splenic architecture.
One of the striking abnormalities in these KO mice is a decreased calcification of the skeleton. This impaired ossification is apparent in both membranous bone (flat skull bones) as well as endochondral bones (long bones) by birth. The phenotype of impaired mineralization and abnormal growth plates in Tle4 null mice is similar to that reported in mice lacking Grg/Aes, a truncated member of the Groucho/TLE gene family [20]. However, the defects in Tle4 null mice are more profound and worsen with age instead of phenotypic recovery seen in Grg5 null mice. Given the reported function of Grg5 as a dominant negative inhibitor of Tle proteins, it might be surprising to see a similar phenotype between loss of Grg5 and Tle4, however, this dominant negative effect is only demonstrated in some over-expression or mis-expression studies and evidence points to other roles for Grg5 independent of its ability to inhibit the Tles, and in some contexts has similar repressive functions as the Tles [21]. The Groucho/TLE proteins are capable of interacting with multiple signaling pathways and may alter the function of key proteins important for bone formation, including Runx2/Cbfa1, a critical regulator of bone development and maturation [14], [22]. The apparent resorption of trabeculae seen in 4 week old mice under the growth plate suggests additional abnormalities in addition to bone formation. Further investigations are underway to better characterize the nature of this defect in bone development and maintenance.
Several reports have described the role of the BM microenvironment on hematopoiesis [23], [24]. Osteoblasts have a well-defined role in supporting B lymphopoiesis via expression of the heterotrimeric G protein alpha subunit G(s)alpha [25], [26]. While we have not yet determined whether osteoblasts are specifically affected in our model per se, it is reasonable to assume that the compromise of trabecular bone is at least contributory to the observed B cell and HSPC defects. The inability of Tle4 null stromal cells cultured in vitro to maintain WT HSC suggests that the hematopoietic phenotype seen in KO mice may derive in part from niche-induced deregulation. Furthermore, as evidenced by TUNEL staining of bones harvested from WT and KO mice, it is clear that the absence of Tle4 has an effect on the viability and integrity of the BM niche and stroma, Further experiments are needed to better characterize the nature of this defective stromal support of HSPC.
Loss of Tle4 appears to significantly impair LSK differentiation into granulocyte, monocyte, macrophage progenitors and LSK self-renewal, at least in part due to increased cellular apoptosis. The finding of preserved numbers of long term stem cells as marked by CD34+ LSK CD48− CD150+ in two week old Tle4 knockout mice, despite decreases in more mature lineages, further studies are need to understand the mechanisms of this preservation.
Concurrently, BM and fetal liver serial transplantation experiments demonstrate a robust HSPC-intrinsic effect of Tle4 deletion. In both transplant models, mice receiving KO HSPC develop peripheral leukopenia. Moreover, this finding in FL serial transplantation illustrates the potential HSPC-intrinsic defects of Tle4 loss leading to decreased capacity of HSPC self-renewal. Additionally, our study provides the first direct in vivo evidence of a role of Tle4 on B-cell development, an effect previously inferred based on interactions of Tle4 and Pax5 [28],[29]. The somewhat distinct block in B-cell differentiation seen with Tle4 loss compared to that reported with Pax5 loss suggests Tle4 may exert some B-cell effects independent from Pax5, although we can’t exclude potential animal models differences as accounting for this effect. Taken together, our data demonstrates the critical importance of Tle4 in regulating various developmental processes central to bone maturation, medullary hematopoiesis, and HSPC maintenance. These findings may have significant implications for understanding hematopoiesis in both normal and disease states. Moreover, our observations provide further insight and affirmation to previous findings that implicate Tle4 as a critical regulator of leukemia and other states of hematological dysregulation.
Conclusion
In summary, by the development of the first model for Tle4 deletion in mammals, our data provide evidence for an essential role for Tle4 in mammalian bone and blood development. Tle4 deficient B cells exhibit intrinsic developmental defects and HSC exhibit stem cell depletion after serial transplantation. Tle4 null bone marrow stromal cells fail to support hematopoiesis in vitro, suggesting a potentially novel extrinsic role of Tle4 in the regulation of hematopoiesis. Tle4 null mice have profound defects in bone mineralization and growth plate organization, which apparently affects skeletal growth.
Our work has shed light on a novel regulatory function for this corepressor in normal hematopoiesis and bone development. As such, elucidating the regulatory mechanisms controlled by Tle4 offers a significant challenge and opportunity for expanding our understanding of bone development and the multicellular orchestration of hematopoiesis. This work further potentially offers novel insight into the transcriptional processes underlying malignant transformation.
Acknowledgments
The authors thank Laura Prickett-Rice, Katherine Folz-Donohue, Meredith Weglarz, and David Dombkowski for assistance with flow cytometry and sorting. We thank Dr. Yiyun Zhang and Dr. Joanna Yeh for their helpful comments and discourse.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by National Institutes of Health grants K08 CA138916-02 (DSK), R01 CA115772 (DAS, JCW, XC, JW), and T32 CA071345 (JW) as well as funding by Hyundai Hope on Wheels (DAS; http://www.hyundaihopeonwheels.org) and the Mattina Proctor Foundation (DAS; 75 Federal St, Boston, Massachusetts 02110). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Nwajei F, Konopleva M (2013) The bone marrow microenvironment as niche retreats for hematopoietic and leukemic stem cells. Adv Hematol 2013: 953982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Grimwade D, Walker H, Oliver F, Wheatley K, Harrison C, et al. (1998) The importance of diagnostic cytogenetics on outcome in AML: analysis of 1,612 patients entered into the MRC AML 10 trial. The Medical Research Council Adult and Children’s Leukaemia Working Parties. Blood 92: 2322–2333. [PubMed] [Google Scholar]
- 3. Dayyani F, Wang J, Yeh JR, Ahn EY, Tobey E, et al. (2008) Loss of TLE1 and TLE4 from the del(9q) commonly deleted region in AML cooperates with AML1-ETO to affect myeloid cell proliferation and survival. Blood 111: 4338–4347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fraga MF, Berdasco M, Ballestar E, Ropero S, Lopez-Nieva P, et al. (2008) Epigenetic inactivation of the Groucho homologue gene TLE1 in hematologic malignancies. Cancer Res 68: 4116–4122. [DOI] [PubMed] [Google Scholar]
- 5. Paroush Z, Finley RL Jr, Kidd T, Wainwright SM, Ingham PW, et al. (1994) Groucho is required for Drosophila neurogenesis, segmentation, and sex determination and interacts directly with hairy-related bHLH proteins. Cell 79: 805–815. [DOI] [PubMed] [Google Scholar]
- 6. Turki-Judeh W, Courey AJ (2012) Groucho: a corepressor with instructive roles in development. Curr Top Dev Biol 98: 65–96. [DOI] [PubMed] [Google Scholar]
- 7. Jennings BH, Ish-Horowicz D (2008) The Groucho/TLE/Grg family of transcriptional co-repressors. Genome Biol 9: 205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brunquell C, Biliran H, Jennings S, Ireland SK, Chen R, et al. (2012) TLE1 is an anoikis regulator and is downregulated by Bit1 in breast cancer cells. Mol Cancer Res 10: 1482–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Seo SW, Lee H, Lee HI, Kim HS (2011) The role of TLE1 in synovial sarcoma. J Orthop Res 29: 1131–1136. [DOI] [PubMed] [Google Scholar]
- 10. Krause DS, Lazarides K, von Andrian UH, Van Etten RA (2006) Requirement for CD44 in homing and engraftment of BCR-ABL-expressing leukemic stem cells. Nat Med 12: 1175–1180. [DOI] [PubMed] [Google Scholar]
- 11. Mukherjee S, Raje N, Schoonmaker JA, Liu JC, Hideshima T, et al. (2008) Pharmacologic targeting of a stem/progenitor population in vivo is associated with enhanced bone regeneration in mice. J Clin Invest 118: 491–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krause DS, Fulzele K, Catic A, Sun CC, Dombkowski D, et al.. (2013) Differential regulation of myeloid leukemias by the bone marrow microenvironment. Nat Med in press. [DOI] [PMC free article] [PubMed]
- 13. Chen G, Nguyen PH, Courey AJ (1998) A role for Groucho tetramerization in transcriptional repression. Mol Cell Biol 18: 7259–7268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. McLarren KW, Lo R, Grbavec D, Thirunavukkarasu K, Karsenty G, et al. (2000) The mammalian basic helix loop helix protein HES-1 binds to and modulates the transactivating function of the runt-related factor Cbfa1. J Biol Chem 275: 530–538. [DOI] [PubMed] [Google Scholar]
- 15. Hardy RR, Hayakawa K (2001) B cell development pathways. Annu Rev Immunol 19: 595–621. [DOI] [PubMed] [Google Scholar]
- 16. Nutt SL, Urbanek P, Rolink A, Busslinger M (1997) Essential functions of Pax5 (BSAP) in pro-B cell development: difference between fetal and adult B lymphopoiesis and reduced V-to-DJ recombination at the IgH locus. Genes Dev 11: 476–491. [DOI] [PubMed] [Google Scholar]
- 17. Walkley CR, Olsen GH, Dworkin S, Fabb SA, Swann J, et al. (2007) A microenvironment-induced myeloproliferative syndrome caused by retinoic acid receptor gamma deficiency. Cell 129: 1097–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fulzele K, Krause DS, Panaroni C, Saini V, Barry KJ, et al. (2013) Myelopoiesis is regulated by osteocytes through Gsalpha-dependent signaling. Blood 121: 930–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Milili M, Gauthier L, Veran J, Mattei MG, Schiff C (2002) A new Groucho TLE4 protein may regulate the repressive activity of Pax5 in human B lymphocytes. Immunology 106: 447–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang WF, Wang YG, Reginato AM, Plotkina S, Gridley T, et al. (2002) Growth defect in Grg5 null mice is associated with reduced Ihh signaling in growth plates. Dev Dyn 224: 79–89. [DOI] [PubMed] [Google Scholar]
- 21. Beagle B, Johnson GV (2010) AES/GRG5: more than just a dominant-negative TLE/GRG family member. Dev Dyn 239: 2795–2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Choi JY, Pratap J, Javed A, Zaidi SK, Xing L, et al. (2001) Subnuclear targeting of Runx/Cbfa/AML factors is essential for tissue-specific differentiation during embryonic development. Proc Natl Acad Sci U S A 98: 8650–8655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Calvi LM, Adams GB, Weibrecht KW, Weber JM, Olson DP, et al. (2003) Osteoblastic cells regulate the haematopoietic stem cell niche. Nature 425: 841–846. [DOI] [PubMed] [Google Scholar]
- 24. Asada N, Katayama Y, Sato M, Minagawa K, Wakahashi K, et al. (2013) Matrix-embedded osteocytes regulate mobilization of hematopoietic stem/progenitor cells. Cell Stem Cell 12: 737–747. [DOI] [PubMed] [Google Scholar]
- 25. Zhu J, Garrett R, Jung Y, Zhang Y, Kim N, et al. (2007) Osteoblasts support B-lymphocyte commitment and differentiation from hematopoietic stem cells. Blood 109: 3706–3712. [DOI] [PubMed] [Google Scholar]
- 26. Wu JY, Purton LE, Rodda SJ, Chen M, Weinstein LS, et al. (2008) Osteoblastic regulation of B lymphopoiesis is mediated by Gs{alpha}-dependent signaling pathways. Proc Natl Acad Sci U S A 105: 16976–16981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ding L, Saunders TL, Enikolopov G, Morrison SJ (2012) Endothelial and perivascular cells maintain haematopoietic stem cells. Nature 481: 457–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Eberhard D, Jimenez G, Heavey B, Busslinger M (2000) Transcriptional repression by Pax5 (BSAP) through interaction with corepressors of the Groucho family. EMBO J 19: 2292–2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Linderson Y, Eberhard D, Malin S, Johansson A, Busslinger M, et al. (2004) Corecruitment of the Grg4 repressor by PU.1 is critical for Pax5-mediated repression of B-cell-specific genes. EMBO Rep 5: 291–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.