

Abstract
Objective
Inhibitors of the 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase enzyme (statins) are cholesterol-lowering drugs that have shown promise as therapeutic agents in various animal models of autoimmune disease. The results of initial clinical trials with statins in multiple sclerosis and rheumatoid arthritis have also been encouraging. In this study, we attempted to treat a widely studied murine model of spontaneous systemic lupus erythematosus (SLE) with atorvastatin.
Methods
(NZB × NZW)F1 (NZB/NZW) mice received daily oral doses of atorvastatin for 20 weeks. The mice were monitored weekly for survival and proteinuria. Anti–double-stranded DNA (anti-dsDNA) antibody levels in sera were determined by enzyme-linked immunosorbent assay (ELISA). T lymphocyte cytokine production in vitro, as well as cytokine levels in vivo, were measured by ELISA. T cell proliferation was assessed by thymidine incorporation assay. Serum cholesterol levels were determined using a standard fluorometric assay. Kidney tissue was harvested and evaluated for pathologic changes.
Results
In NZB/NZW mice, oral atorvastatin had significant effects on T cell proliferation and cytokine production in vitro. Atorvastatin also induced significant increases in serum levels of interleukin-4. However, atorvastatin treatment in NZB/NZW mice had no significant impact on proteinuria, survival, serum anti-dsDNA antibody and cholesterol levels, or extent of renal disease.
Conclusion
Monotherapy with oral atorvastatin has no protective effects in a murine model of spontaneous SLE. The efficacy of atorvastatin in human SLE remains to be determined.
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease that affects more than 1 million people in the US. The clinically heterogeneous nature of SLE renders therapeutic intervention particularly challenging. Conventional approaches include the use of corticosteroids, cytotoxic agents such as cyclophosphamide and azathioprine, and, more recently, investigational agents such as mycophenolate mofetil and rituximab. These agents have been used with varying success and are associated with a considerable number of side effects (1–3).
In addition to the myriad of clinical and laboratory manifestations that comprise the diagnostic criteria for SLE and related connective tissue diseases, it has become increasingly clear that patients with SLE have increased morbidity and mortality resulting from accelerated atherosclerosis (4). This realization has led to efforts by clinicians to reduce corticosteroid use, as well as to recommend patient lifestyle modifications.
Statins, or 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitors, are cholesterol-lowering drugs that have displayed immunomodulatory effects in numerous models of inflammatory and autoimmune disease, including multiple sclerosis (MS) and rheumatoid arthritis. Although side effects have been reported, statins are generally well tolerated in humans, making these agents ideal for potential use as therapeutic agents in immune-mediated diseases. In this study, our goal was to determine whether monotherapy with atorvastatin could suppress clinical disease in the (NZB × NZW)F1 (NZB/NZW) mouse model of spontaneous SLE.
Materials and Methods
Mice
Female NZB/NZW mice were purchased from The Jackson Laboratory (Bar Harbor, ME) and maintained under standard conditions in the Research Animal Facility at Stanford University. Experiments were conducted in accordance with approved Institutional Animal Care and Use Committee and National Institutes of Health (NIH) guidelines.
Reagents
Atorvastatin (prescription formulation; Pfizer, New York, NY) was brought into suspension in phosphate buffered saline (PBS) at 0.4 mg/ml, and a 0.5-ml volume (equivalent to 10 mg/kg) was administered via oral gavage, once daily, using 20-mm feeding needles (Popper & Sons, New Hyde Park, NY). PBS was administered as a vehicle control. Treatment of the mice (n = 15) began at 20 weeks of age, and the surviving mice were killed at 40 weeks of age.
Renal histopathologic analysis
Proteinuria was assessed once weekly using Albustix reagent strips for urinalysis (Bayer, Elkhart, IN). At the end of the experiment, kidneys were harvested from surviving mice and fixed in 10% buffered formalin. Periodic acid-Schiff (PAS) staining was performed on paraffin-embedded sections, and kidney damage was scored according to standard NIH activity and chronicity indices (5) in a blinded manner by one of the authors (JPH).
T cell proliferation assays
Splenocytes were negatively selected for T cells via an enrichment column (R&D Systems, Minneapolis, MN). T cells (>95% purity, as determined by flow cytometric analysis [data not shown]) were cultured for 48 hours at 105 cells per well in 96-well Nunc plates (Nalgene, Rochester, NY) coated with anti-CD3 monoclonal antibody (2C11; BD PharMingen, San Diego, CA) and anti-CD28 monoclonal antibody (37.51; BD PharMingen), each at a concentration of 5 μg/ml. Culture medium consisted of RPMI 1640 (Gibco, Carlsbad, CA) supplemented with L-glutamine (2 mM), sodium pyruvate (1 mM), nonessential amino acids (0.1 mM), penicillin (100 units/ml), streptomycin (0.1 mg/ml), 2-mercaptoethanol (5 × 10−5M), and 10% fetal calf serum. Wells were pulsed with 1 μCi 3H-thymidine (MP Biomedicals, Solon, OH) for the final 16 hours of culture, and incorporated radioactivity was measured using a Betaplate scintillation counter (Wallac, Gaithersburg, MD).
Enzyme-linked immunosorbent assay (ELISA)
Cytokine levels in mouse serum (diluted 1:10) were determined by ELISA kits (BD PharMingen). Cytokine levels in T cell culture supernatants were determined after anti-CD3/CD28 stimulation for 48 hours or 72 hours. Anti-double-stranded DNA (anti-dsDNA) antibody levels were determined using an ELISA kit (Alpha Diagnostic, San Antonio, TX). Mouse sera were diluted at 1:100, followed by the addition of horseradish peroxidase-conjugated goat anti-mouse antibodies specific for IgG (Southern Biotechnology, Birmingham, AL) at a dilution of 1:5,000.
Serum cholesterol assays
Free cholesterol and cholesterol ester levels in serum were determined using the Amplex Red Cholesterol Assay Kit (Invitrogen, Carlsbad, CA).
Statistical analysis
Statistical analysis was performed using GraphPad Prism software (San Diego, CA). One-way analysis of variance (ANOVA) was used for comparison of proteinuria data, and Kaplan-Meier survival curve data were analyzed using the log-rank test. All other comparisons were performed using Student's unpaired t-test.
Results
Proteinuria
NZB/NZW mice spontaneously develop a syndrome that closely resembles human SLE, marked by the production of antinuclear antibodies and renal damage. In order to examine the therapeutic efficacy of an HMG-CoA reductase inhibitor in murine lupus, we administered atorvastatin to lupus-prone mice via oral gavage, on a daily basis. Efficacy in suppressing clinical experimental autoimmune encephalomyelitis (EAE), an animal model of human MS (6), has been demonstrated with daily oral doses of 1 mg/kg and 10 mg/kg. For studies in the NZB/NZW mouse model of SLE, we chose a daily oral dose of 10 mg/kg, a dose that is significantly higher than the recommended maximum equivalent dose (80 mg) for humans with hypercholesterolemia (7). Treatment of the mice (n = 15) was initiated at 20 weeks of age, and the mice were regularly monitored for proteinuria, an indicator of kidney damage. As shown in Figure 1A, daily atorvastatin treatment for 20 weeks had no significant impact on either the age at onset or the frequency of proteinuria in NZB/NZW mice. We also did not observe any adverse physical signs in atorvastatin-treated mice.
Figure 1.
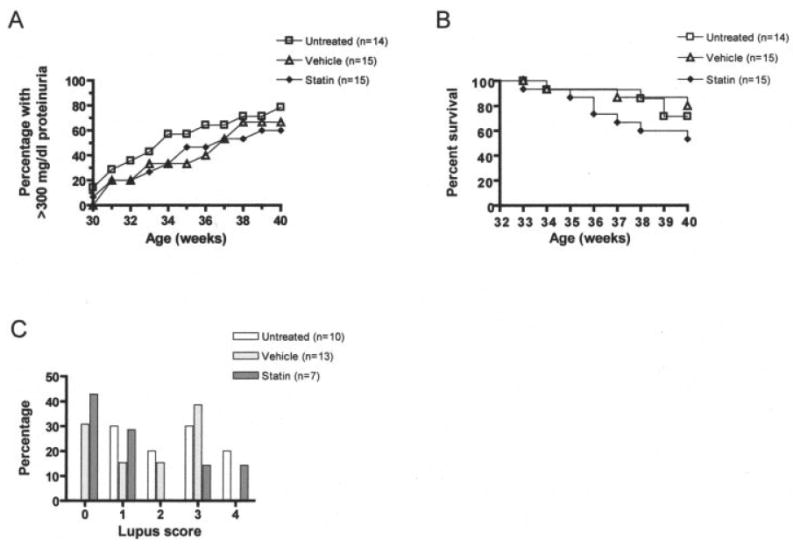
Proteinuria, survival, and lupus scores in female (NZB × NZW)F1 (NZB/NZW) mice treated with atorvastatin, untreated mice, and vehicle-treated mice. A, Proteinuria data, expressed as the percentage of mice with severe proteinuria (≥300 mg/dl) as a function of age. B, Survival data, displayed in Kaplan-Meier curve form. C, Pathologic changes in renal tissue. Periodic acid–Schiff-stained tissue sections were scored on a semiquantitative scale from 0 to 4, where 0 = no glomeruli involved, 1 = up to 25% of the glomeruli involved, 2 = 25–50% of the glomeruli involved, 3 = 51–75% of the glomeruli involved, and 4 = >75% of the glomeruli involved.
Survival
We also monitored SLE mice for survival. As shown in Figure 1B, we observed slightly increased mortality in atorvastatin-treated NZB/NZW mice, with 53.3% survival at 40 weeks, compared with >70% survival in untreated NZB/NZW mice and 80% survival in vehicle-treated control mice. However, these differences were not statistically significant.
Renal pathology
PAS-stained kidney tissue specimens from surviving SLE mice were evaluated for pathologic changes, by a single observer (JPH) who had no knowledge of the treatment a given mouse had received. There appeared to be a slight trend toward reduced renal disease in atorvastatin-treated NZB/NZW mice (compared with untreated controls). More than 40% of atorvastatin-treated NZB/NZW mice analyzed displayed no glomerular involvement, whereas all untreated controls displayed at least some degree of renal pathology (Figure 1C). However, these results were not statistically significant (as determined by ANOVA). Furthermore, any apparent ameliorating effects of atorvastatin treatment on kidney disease should be weighed against the increased mortality observed in atorvastatin-treated NZB/NZW mice. NZB/NZW mice were also evaluated using parameters of renal disease that are representative of the most significant pathologic abnormalities in NZB/NZW mice with advanced lupus nephritis. These indices included grading of glomerulosclerosis, tubular dilatation, tubular atrophy, interstitial fibrosis, and perivascular infiltration, none of which were significantly impacted by atorvastatin treatment (data not shown).
Anti-dsDNA antibody levels
We performed ELISAs to determine whether oral atorvastatin had any effect on serum levels of anti-dsDNA antibodies in murine lupus. As shown in Figure 2A, anti-dsDNA antibody levels in sera derived from 40-week-old atorvastatin-treated NZB/NZW mice did not differ significantly from those in sera from untreated or vehicle-treated controls. These results demonstrate that oral atorvastatin does not inhibit anti-dsDNA antibody production in murine SLE.
Figure 2.
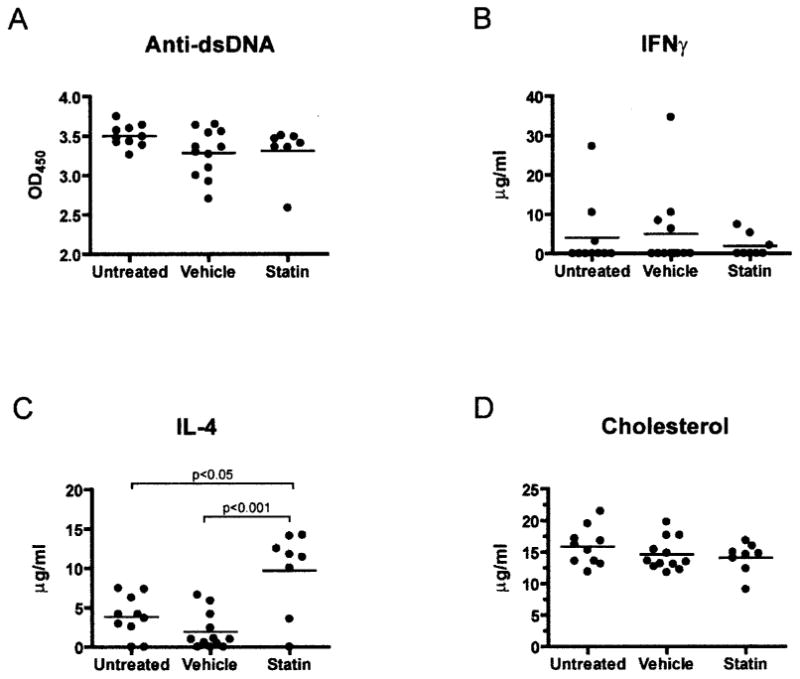
A, IgG anti–double-stranded DNA (anti-dsDNA) levels in serum collected from (NZB × NZW)F1 (NZB/NZW) mice at 40 weeks of age, as measured by enzyme-linked immunosorbent assay (ELISA). B and C, Serum levels of interferon-γ (IFNγ) (B) and interleukin-4 (IL-4) (C), as determined by ELISA. D, Cholesterol levels in serum collected from NZB/NZW mice at 40 weeks of age, assessed as described in Materials and Methods. Bars show the mean. OD450 = optical density at 450 nm.
Serum cytokine levels determined by ELISA
Treatment with oral atorvastatin is associated with induction of Th2 responses in the EAE model of MS (6). To address whether atorvastatin could cause immune deviation in murine lupus, we measured cytokine levels in sera derived from NZB/NZW mice at 40 weeks of age. Serum levels of interferon-γ)/(IFNγ) were below the limit of detection for most NZB/NZW samples (Figure 2B), and serum levels of tumor necrosis factor (TNF) were not significantly affected by atorvastatin treatment (data not shown). However, oral atorvastatin induced a significant increase in serum levels of interleukin-4 (IL-4) (Figure 2C). These results suggest that oral atorvastatin promotes induction of Th2 responses in vivo in NZB/NZW mice. However, it is important to note that these results are confined to surviving mice; it is possible that atorvastatin induced a different cytokine profile in mice that died before the end of the experiment.
Serum cholesterol levels
As inhibitors of HMG-CoA reductase, statins have been shown to modulate serum cholesterol levels in numerous human and animal models. To determine whether oral atorvastatin treatment modulated serum lipid levels in SLE mice, sera from NZB/NZW mice were assayed for levels of cholesterol at 40 weeks of age. Notably, compared with the effect of atorvastatin in the untreated and vehicle-treated mice, atorvastatin treatment had no measurable impact on serum cholesterol levels in NZB/NZW mice (Figure 2D). These results are consistent with previous observations that statins are capable of influencing immune responses in the absence of cholesterol-lowering activity (8).
Finding of in vitro T cell assays
Oral atorvastatin treatment had been reported to suppress lymphocyte antigen-specific proliferative and cytokine recall responses in EAE (6). One complicating feature of the NZB/NZW model is that numerous T cell epitopes have been implicated in the initiation and progression of disease. Thus, we analyzed murine SLE T cell responses to mitogenic stimulation. Upon termination of the experiment, we harvested splenocytes from surviving NZB/NZW mice. We enriched for T cells via negative selection, followed by incubation in the presence of plate-bound anti-CD3 and anti-CD28 antibodies. As shown in Figure 3, anti-CD3/CD28–stimulated T cells from atorvastatin-treated NZB/NZW mice proliferated more than those from their untreated counterparts. We also assayed supernatants from cultures of anti-CD3/CD28– stimulated T cells for cytokine levels. Consistent with observations made in the EAE model, T cells from atorvastatin-treated NZB/NZW mice produced higher levels of IL-4 and IL-10 compared with untreated mice, suggesting induction of Th2 and/or regulatory responses. Culture supernatants from atorvastatin-treated animals also contained elevated levels of IL-6 and TNF, both of which are commonly associated with proinflammatory responses. Interestingly, atorvastatin treatment did not impact in vitro production of IFNγ by T cells in NZB/NZW mice. Taken together, the data demonstrate that oral administration of atorvastatin influences in vitro T cell responses in the NZB/NZW mouse model of spontaneous SLE.
Figure 3.
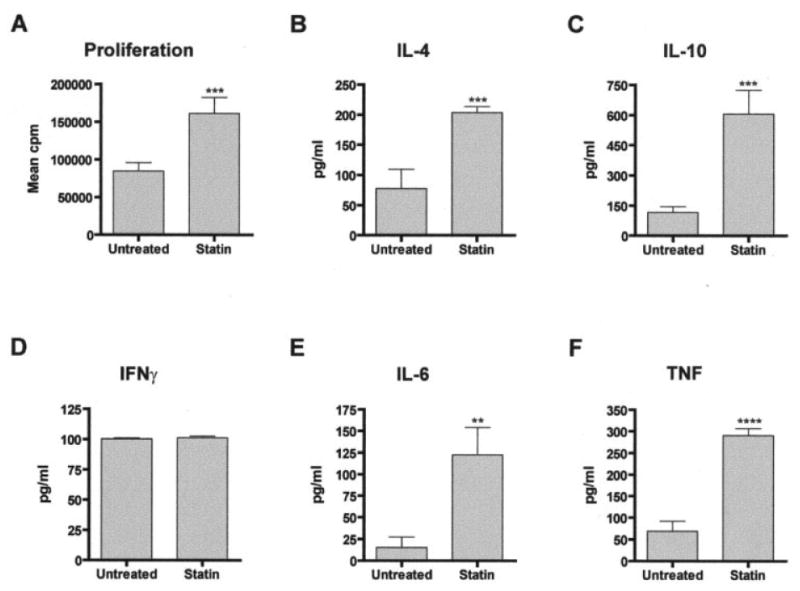
In vitro T cell assays. Splenic T cells from untreated and atorvastatin-treated NZB/NZW mice were stimulated with anti-CD3 and anti-CD28 antibodies, as described in Materials and Methods. A, Proliferation, as assessed by standard thymidine incorporation assay. B–F, Cytokine levels. Levels of IL-4 (B), IL-10 (C), IFNγ (D), IL-6 (E), and tumor necrosis factor (TNF) (F) were measured in cell culture supernatants by ELISA. Values are the mean and SD. ** = P < 0.01; *** = P < 0.005; **** = P < 0.001, by Student's t-test. See Figure 2 for other definitions.
Discussion
Statins have gained considerable attention as potential therapeutic agents in numerous diseases that are considered to be mediated by the immune system. In this study, we sought to determine whether atorvastatin had any beneficial effects on lupus nephritis in the NZB/NZW mouse model of SLE. In NZB/NZW mice, we observed that daily oral administration of high doses of atorvastatin had no significant impact on 4 major parameters of SLE disease: proteinuria, survival, anti-dsDNA antibodies, and renal disease.
In contrast to our findings, Lawman et al (9) noted that atorvastatin inhibited development of disease in NZB/NZW mice when it was administered via intra-peritoneal injection at a dose of 30 mg/kg. For our SLE studies, we administered atorvastatin orally rather than intraperitoneally. It has been reported that oral atorvastatin effectively suppresses clinical EAE at doses as low as 0.1 mg/kg, with more dramatic suppression occurring at a dose of 10 mg/kg (6). Thus, although the biologic effects of statins appear to be dose dependent in the EAE model, we used a dose (10 mg/kg) that is considerably higher than the recommended maximum equivalent dose for humans with hypercholesterolemia (7). Despite this high dose, we observed no significant impact on disease outcome in NZB/NZW mice. Furthermore, serum cholesterol levels were unaltered by oral atorvastatin treatment. Conversely, Lawman and colleagues demonstrated that intraperitoneal administration of atorvastatin reduced serum cholesterol levels in NZB/NZW mice. The route of atorvastatin administration may influence its absorption and distribution. Although detailed pharmacokinetics analysis of intraperitoneal versus oral atorvastatin administration may elucidate potential reasons for these disparate results, daily oral administration, as used in our study, more closely models how atorvastatin is administered in humans.
Although we observed no impact of atorvastatin treatment on clinical disease, we performed several assays in an effort to determine whether atorvastatin possessed immunomodulatory activity in murine SLE. Notably, sera derived from atorvastatin-treated NZB/NZW mice contained elevated levels of IL-4, consistent with systemic induction of Th2 responses. We also analyzed T cell proliferation and cytokine production in vitro. Compared with the levels in untreated controls, levels of IL-4, IL-6, IL-10, and TNF were higher in culture supernatants derived from atorvastatin-treated NZB/NZW T cells stimulated with anti-CD3/CD28 antibodies. Taken together, these results suggest that even though atorvastatin was biologically active in vivo in this murine model of SLE, atorvastatin still failed to treat the disease.
Several clinical trials are under way that are investigating the efficacy of statins in treating autoimmune diseases. Vollmer and colleagues (10) have reported on the efficacy of oral simvastatin in the treatment of 30 patients in whom relapsing–remitting MS was diagnosed, and a larger phase II study of atorvastatin therapy in patients with clinically isolated syndrome and high risk of conversion to MS is currently ongoing. Statin therapy has also shown beneficial effects in patients with inflammatory rheumatic diseases. A 1-year trial of atorvastatin demonstrated reductions in proteinuria, cholesterol levels, and the rate of progression of chronic kidney disease in SLE patients with idiopathic chronic glomerulonephritis and proteinuria, abnormal creatinine clearance, and marked hypercholesterolemia (11). No randomized studies have yet been reported in patients with SLE, although a very small trial of simvastatin demonstrated rapid reduction of proteinuria levels in 3 patients with SLE (12).
It is important to note that treatment with statins has not produced universally positive results. Atorvastatin, administered orally or intraperitoneally, had no efficacy in 2 distinct mouse models of diabetes (13). Simvistatin administered intraperitoneally had no effects on lymphadenopathy, renal disease, or proinflammatory cytokine production in the gld mouse model of autoimmunity (14). We also observed that atorvastatin treatment had no significant impact on proteinuria or survival in the pristane-induced BALB/c mouse model of SLE and in the MRL/lpr mouse model of spontaneous SLE (data not shown). It is noteworthy that reported failures to modulate autoimmune disease with statins in animal models have generally occurred in settings in which disease is spontaneous, such as diabetes and SLE. Success in animal models treated with statins occurs in diseases that are actively induced with Freund's complete adjuvant (CFA), such as EAE and collagen-induced arthritis. The question of whether the capacity of CFA to activate innate immunity is countered by a suppressive effect of statins on the innate immune response remains unanswered. The relative effect of pristane on the innate and adaptive immune response is also not completely understood.
Although statin therapy is associated with relatively few side effects in humans, dose-related complications can arise. These can include SLE-like syndromes, vasculitis, hepatitis, rhabdomyolysis, and myositis. We observed slightly increased mortality in atorvastatin-treated NZB/NZW mice, although this finding was not statistically significant. Nevertheless, it is possible that dose-related toxicity can occur in rodent models. A recent study showed that increased myoglobinuria developed in rats treated with atorvastatin at a dose of 10 mg/kg once daily for 2 months (15). However, it has not been established whether atorvastatin has any deleterious effects on renal function in NZB/NZW mice.
In summary, our results demonstrate that oral atorvastatin does not ameliorate murine SLE. Our data and the published literature to date suggest that the use of atorvastatin or other statins as a therapy for autoimmunity or accelerated atherosclerosis in human SLE should be approached with caution until the results of large, prospective, randomized trials in humans with SLE are reported.
Acknowledgments
We are grateful to Liana Gefter for critical review of the manuscript.
Dr. Graham's work was supported by the NIH (National Research Service Award fellowship AI10663-02). Dr. Steinman's work was supported by grants from the NIH (R01-AI-59709) and the National Multiple Sclerosis Society. Dr. Utz's work was supported by the Dana Foundation, the Floren Family Trust, the Northern California Chapter of the Arthritis Foundation, the National Multiple Sclerosis Society, and the NIH. Dr. Ho is recipient of an Arthritis Foundation Northern California Chapter Grant.
Biography
Dr. Utz is a member of the Scientific Advisory Board of Monogram Biosciences and XDx, and is a cofounder and consultant at Bayhill Therapeutics. Dr. Utz has received consulting fees, speaking fees, and/or honoraria from UCB, Johnson & Johnson, Avanir, Genentech, Biogen Idec, AstraZeneca Pharmaceuticals, Monogram Biosciences, and XDx (less than $10,000 each), and owns stock and/or stock options in Bayhill Therapeutics.
Dr. Steinman is a cofounder and consultant at Bayhill Therapeutics and owns stock and/or stock options in Bayhill Therapeutics.
Footnotes
Author Contributions: Dr. Ho had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study design. Graham, Steinman, Utz, Ho.
Acquisition of data. Graham, Lee, Utz, Ho.
Analysis and interpretation of data. Graham, Lee, Higgins, Steinman, Utz, Ho.
Manuscript preparation. Graham, Lee, Utz, Ho.
Statistical analysis. Graham, Steinman, Utz, Ho.
References
- 1.Petri M. Cyclophosphamide: new approaches for systemic lupus erythematosus. Lupus. 2004;13:366–71. doi: 10.1191/0961203303lu1028oa. review. [DOI] [PubMed] [Google Scholar]
- 2.Byrd J, Waselenko J, Maneatis T, Murphy T, Ward F, Monahan B, et al. Rituximab therapy in hematologic malignancy patients with circulating blood tumor cells: association with increased infusion-related side effects and rapid blood tumor clearance. J Clin Oncol. 1999;17:791–5. doi: 10.1200/JCO.1999.17.3.791. [DOI] [PubMed] [Google Scholar]
- 3.Oellerich M, Shipkova M, Schutz E, Wieland E, Weber L, Tonshoff B, et al. German Study Group on Mycophenolate Mofetil Therapy in Pediatric Renal Transplant Recipients Pharmacokinetic and metabolic investigations of mycophenolic acid in pediatric patients after renal transplantation: implications for therapeutic drug monitoring. Ther Drug Monit. 2000;22:20–6. doi: 10.1097/00007691-200002000-00004. review. [DOI] [PubMed] [Google Scholar]
- 4.Bruce I. Atherogenesis and autoimmune disease: the model of lupus. Lupus. 2005;14:687–90. doi: 10.1191/0961203305lu2201oa. review. [DOI] [PubMed] [Google Scholar]
- 5.Austin HA, Muenz LR, Joyce KM, Antonovych TT, Balow JE. Diffuse proliferative lupus nephritis: identification of specific pathologic features affecting renal outcome. Kidney Int. 1984;25:689–95. doi: 10.1038/ki.1984.75. [DOI] [PubMed] [Google Scholar]
- 6.Youssef S, Stuve O, Patarroyo JC, Ruiz PJ, Radosevich JL, Hur EM, et al. The HMG-CoA reductase inhibitor, atorvastatin, promotes a Th2 bias and reverses paralysis in central nervous system autoimmune disease. Nature. 2002;420:78–84. doi: 10.1038/nature01158. [DOI] [PubMed] [Google Scholar]
- 7.Bernini F, Poli A, Paoletti R. Safety of HMG-CoA reductase inhibitors: focus on atorvastatin. Cardiovasc Drugs Ther. 2001;15:211–8. doi: 10.1023/a:1011908004965. review. [DOI] [PubMed] [Google Scholar]
- 8.Dunn SE, Youssef S, Goldstein MJ, Prod'homme T, Weber MS, Zamvil SS, et al. Isoprenoids determine Th1/Th2 fate in pathogenic T cells, providing a mechanism of modulation of autoimmunity by atorvastatin. J Exp Med. 2006;203:401–12. doi: 10.1084/jem.20051129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lawman S, Mauri C, Jury EC, Cook HT, Ehrenstein MR. Atorvastatin inhibits autoreactive B cell activation and delays lupus development in New Zealand black/white F1 mice. J Immunol. 2004;173:7641–6. doi: 10.4049/jimmunol.173.12.7641. [DOI] [PubMed] [Google Scholar]
- 10.Vollmer T, Key L, Durkalski V, Tyor W, Corboy J, Markovic-Plese S, et al. Oral simvastatin treatment in relapsing-remitting multiple sclerosis. Lancet. 2004;363:1607–8. doi: 10.1016/S0140-6736(04)16205-3. [DOI] [PubMed] [Google Scholar]
- 11.Bianchi S, Bigazzi R, Caiazza A, Campese VM. A controlled, prospective study of the effects of atorvastatin on proteinuria and progression of kidney disease. Am J Kidney Dis. 2003;41:565–70. doi: 10.1053/ajkd.2003.50140. [DOI] [PubMed] [Google Scholar]
- 12.Abud-Mendoza C, de la Fuente H, Cuevas-Orta E, Baranda L, Cruz-Rizo J, Gonzalez-Amaro R. Therapy with statins in patients with refractory rheumatic diseases: a preliminary study. Lupus. 2003;12:607–11. doi: 10.1191/0961203303lu429oa. [DOI] [PubMed] [Google Scholar]
- 13.Palomer X, Calpe-Berdiel L, Verdaguer J, Carrillo J, Pastor X, Mauricio D, et al. Atorvastatin does not decrease or delay diabetes onset in two different mouse models of type 1 diabetes. Diabetologia. 2005;48:1671–3. doi: 10.1007/s00125-005-1834-z. [DOI] [PubMed] [Google Scholar]
- 14.Aprahamian T, Bonegio R, Rizzo J, Perlman H, Lefer DJ, Rifkin IR, et al. Simvastatin treatment ameliorates autoimmune disease associated with accelerated atherosclerosis in a murine lupus model. J Immunol. 2006;177:3028–34. doi: 10.4049/jimmunol.177.5.3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pierno S, Didonna M, Cippone V, De Luca A, Pisoni M, Frigeri A, et al. Effects of chronic treatment with statins and fenofibrate on rat skeletal muscle: a biochemical, histological and electrophysiological study. Br J Pharmacol. 2006;149:909–19. doi: 10.1038/sj.bjp.0706917. [DOI] [PMC free article] [PubMed] [Google Scholar]