

Abstract
We used the 4C-Seq technique to characterize the genome-wide patterns of spatial contacts of several CpG islands located on chromosome 14 in cultured chicken lymphoid and erythroid cells. We observed a clear tendency for the spatial clustering of CpG islands present on the same and different chromosomes, regardless of the presence or absence of promoters within these CpG islands. Accordingly, we observed preferential spatial contacts between Sp1 binding motifs and other GC-rich genomic elements, including the DNA sequence motifs capable of forming G-quadruplexes. However, an anchor placed in a gene/CpG island-poor area formed spatial contacts with other gene/CpG island-poor areas on chromosome 14 and other chromosomes. These results corroborate the two-compartment model of the spatial organization of interphase chromosomes and suggest that the clustering of CpG islands constitutes an important determinant of the 3D organization of the eukaryotic genome in the cell nucleus. Using the ChIP-Seq technique, we mapped the genome-wide CTCF deposition sites in the chicken lymphoid and erythroid cells that were used for the 4C analysis. We observed a good correlation between the density of CTCF deposition sites and the level of 4C signals for the anchors located in CpG islands but not for an anchor located in a gene desert. It is thus possible that CTCF contributes to the clustering of CpG islands observed in our experiments.
Keywords: CpG island, housekeeping gene, CTCF, genome spatial organization, 4C
Introduction
Recent studies of the 3D organization of the eukaryotic genome demonstrated that the spatial interactions between distant regulatory elements are important for the control of gene expression.1-3 In particular, it has been shown that the juxtaposition of gene promoters and distant regulatory elements plays a pivotal role in the mediation of cell lineage- and development stage-specific gene expression.4-6 The most convincing observations demonstrating the importance of the spatial interactions of distal regulatory elements with the promoters of target genes were made in experiments aimed at elucidating the patterns of regulation of globin gene expression in vertebrates.4,5,7-11 However, the mechanisms supporting the functionally dependent spatial organization of interphase chromosomes are poorly understood. It has been proposed that remote regulatory elements (enhancers) and promoters of target genes are assembled into a common activating complex (active chromatin hub, ACH) that is stabilized by DNA-protein and protein-protein interactions.4,5 However, it seems equally possible that the juxtaposition of remote enhances and promoters is mediated by the shaping of a chromatin domain, i.e., by the specific folding of a chromatin fiber, which can be influenced by a number of factors that govern the path of a chromatin fiber within the folded interphase chromosome.12,13 Recent studies have demonstrated that the 3D organization of the eukaryotic genome is closely linked to the spatial compartmentalization of the eukaryotic cell nucleus.13-15 In this respect, transcription factories16-18 are a good example because they may be considered as assemblies of genome regulatory elements (i.e., chromatin hubs) and as nuclear compartments where RNA polymerase II molecules and accessory proteins (transcription factors, chromatin remodeling complexes, etc.) are concentrated.19,20 It was proposed that the organization of the eukaryotic genome in transcription factories constitutes a major determinant of the large-scale spatial organization of interphase chromosomes.21 The principles of gene organization in transcription factories have been extensively studied but remain unclear. It was reported that functionally related genes tend to be attracted to common transcription factories,22,23 consistent with the model of specialized transcription factories.24 However, the data supporting this model are rather controversial. For example, it was demonstrated that the probability of finding more than two genes possessing the same tissue specificity in the same transcription factory was less than expected based on the random partitioning of the expressed genes among all transcription factories.22,23 Furthermore, there are well-documented examples of transcription factories that contain both tissue-specific and housekeeping genes.8,25,26
Notably, even in differentiated cells, the average number of transcribed housekeeping genes significantly surpasses the number of transcribed tissue-specific genes. Thus, if the assembly of transcribed genes into transcription factories indeed directs the folding of the interphase genome, it is reasonable to assume that this folding is first directed by the assembly of the housekeeping genes into such factories. To identify and understand the principles underlying the assembly of the housekeeping and tissue-specific genes into transcription factories, we analyzed the pattern of long-range spatial interactions of the chicken housekeeping gene NPRL3,27 which is located on chromosome 14 upstream of a cluster of α-globin genes. The NPRL3 gene resides within an extended region of genomic synteny and is highly conserved in vertebrates.28,29 The promoter of NPRL3 is associated with a CpG island (CGI),30 which also harbors a replication origin.31 In chicken erythroid cells, this CGI is recruited to the active chromatin hub that controls the expression of the α-globin genes.7 Using the 4C technique32 followed by deep sequencing analysis, we mapped the full spectrum of contacts of the DNA fragment harboring the NPRL3 promoter with other regions on chromosome 14 and with other chromosomes in lymphoid (DT40) and erythroid (HD3) chicken cells. The results suggest that the interaction of CGIs constitutes an important determinant of the 3D organization of interphase chromosomes. Correspondingly, the association of housekeeping genes is likely to constitute the basis for the formation of the majority of the transcription factories. This conclusion was further corroborated by analysis of the interaction profiles of several other CGIs scattered along the chicken chromosome 14.
Results
Experimental design
We applied the 4C-Seq technique to map the long-range interactions of a CGI harboring the promoter of the housekeeping gene NPRL3 on a genome-wide scale. The experiments were performed in parallel on cultured lymphoid (DT40) and erythroid (HD3) chicken cells. The transcription level of the NPRL3 gene is nearly the same in these two cell lines.26 The HindIII restriction enzyme was used to prepare the initial 3C material, and DpnII was used to make the 4C libraries. Two independent 4C experiments were performed with each cell line. Comparison of the results demonstrated the good reproducibility of the 4C data (see below). The 4C libraries were analyzed using massive parallel sequencing (45–64 × 106 paired end reads, i.e., 22–32 million read pairs in each experiment). The sequencing data were mapped to the chicken genome (assembly galGal4). Only the reads containing HindIII-ligation junctions were taken into consideration. During the preparation of the initial 3C library, both ends of each HindIII restriction fragment may be ligated to the bait. We summarized the number of reads that independently mapped to the ends of each HindIII fragment and used the sum of these numbers as a parameter that reflected the overall probability of ligation of the corresponding HindIII fragment to the bait. Further analysis was performed as described.33 The clusters of interacting fragments were identified using a sliding window approach33 (window size = 3–200 HindIII restriction fragments) and by binning the genome into 100 Kb fragments (see Materials and Methods). Figure 1A and B show the distribution of the raw 4C signals on chromosome 14 (harboring the bait) and the P values calculated as described in Materials and Methods. The distribution of the P values is also presented as domainograms (Fig. 1C). It is evident that the distributions of the 4C signals are quite similar in the two biological replicates and differ in the two cell lines studied (cluster dendrogram to the right of the graphs showing the distribution of the raw 4C signals in Fig. 1A). Taking into account the similarity of the two biological replicates (the Pearson correlation coefficients of 0.878 for DT40 and 0.978 for HD3 cells), the reads obtained in the two parallel experiments were combined during further analysis.
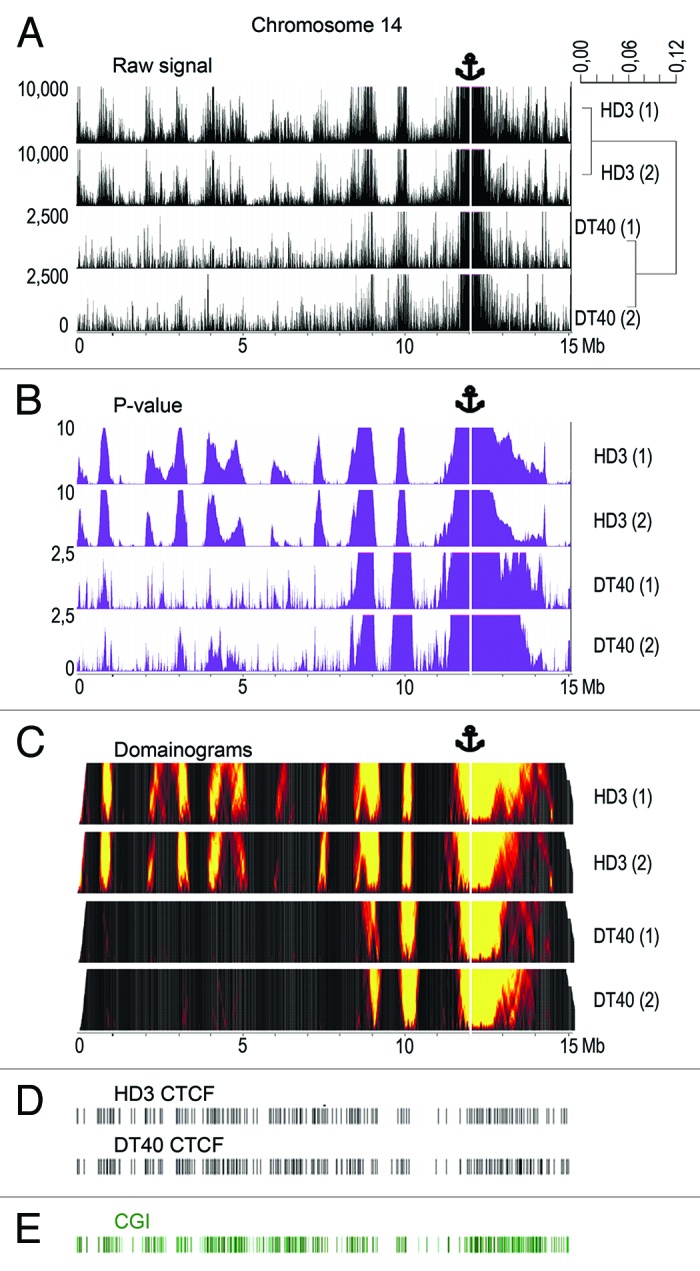
Figure 1. The distribution of NPRL3–4C signals, CGIs, and CTCF binding sites along chicken chromosome 14. (A) The distribution of the raw 4C signal in chromosome 14 obtained using the NPRL3 anchor in chicken HD3 and DT40 cells. The sequenced reads were aligned to the genome near HindIII sites. The distances along chromosome 14 are presented in Mb, according to the galGal4 assembly (UCSC). The values on y-axis represent the number of 4C reads calculated as described in Material and Methods section. The position of the viewpoint is indicated by the anchor sign above the graphs and by the vertical white line. The dendrogram to the right of the graphs shows the similarity between the biological replicates and the differences between the HD3 and DT40 cells. (B) Same as A, but P values are shown. The window with the most significant excess of signal over the background was chosen to determine the P value for each genomic position on chromosome 14 (see Materials and Methods). The y-axis shows the –log10 of the most significant P value for the chosen window. (C) Domainogram plots70 demonstrating the distribution of the NPRL3–4C signals along chromosome 14 in HD3 and DT40 cells. The intensity of the 4C signal (the fraction of restriction fragments with non-zero coverage) in small windows of variable size (3–200 HindIII restriction fragments) was compared with a large background window (300 HindIII restriction fragments). The color range represents the significances of the differences; the x-axis corresponds to the genomic coordinate and the y-axis shows the size of the small window (the higher the coordinate, the larger the window). (D) The distribution of the CTCF deposition sites along chicken chromosome 14. (E) The distribution of the annotated CGIs along chicken chromosome 14 (according to the galGal4 assembly).
Interaction of the bait with distant regions located on the same and other chromosomes
We sequenced approximately 15 × 106 HindIII-ligation junctions for each sample. Figure 2A shows the total numbers and percentages of reads that mapped to chromosome 14 (harboring the bait) and to other chromosomes. The results obtained in the 2 biological replicates were quite similar and are summarized in Figure 2A. Of note are the striking differences in the distribution of reads between chromosome 14 and the other chromosomes in the DT40 and HD3 cells. In the DT40 cells, only approximately 14% of all reads mapped to chromosome 14, whereas 86% of the reads were distributed without much preference over the other chromosomes. The average numbers of reads per Kb were similar for long and short chromosomes (Fig. 2B). In contrast, in the HD3 cells, ~63% of all reads mapped to chromosome 14. Furthermore, the distribution of the other reads (~37% of all reads) across other chromosomes was strikingly non-random, with a clear preference for small chromosomes (Fig. 2B). This result likely reflects the more compact organization of these chromosomes in the erythroid cells and the more pronounced segregation of the long and short chromosomes in the nuclear space.

Figure 2. Chromosome coverage by the NPRL3-4C reads. (A) The fraction of reads that mapped to the bait-containing chromosome 14. The numbers show the combined numbers of reads that mapped in two independent biological experiments. (B) Chromosome sizes (blue columns) and the mean coverage of chromosomes by the 4C reads (reads per kilobase). The red and green columns represent the data from the HD3 and DT40 cells, respectively.
Interaction between CGIs
We analyzed the possible correlation between the positions of interacting DNA fragments with different genomic features. Among the genomic features considered (GC content, CGIs, gene density, transcription factor binding sites), the most obvious correlation was observed between the 4C signal and the presence of CGIs (Fig. 3). This correlation was especially evident when the interacting sites in all chromosomes or all chromosomes except the bait-containing chromosome 14 were analyzed (Fig. 3A and B). Although the correlation was also evident in the case of chromosome 14, the standard deviation of the correlation coefficient was more pronounced, possibly because of the relatively small number of bins (Fig. 3C). Notably, the positive correlation between the 4C signal and the density of CGIs was observed in both DT40 and HD3 cells (Fig. 3). Considering that the viewpoint contained a CGI, we concluded that there is a tendency for the clustering of CGIs present on the same and different chromosomes. Consistently, we observed a clear positive correlation between the intensity of the 4C signal and the presence of the GC-rich binding motifs for Sp1 that are frequently found in CGIs (Fig. 4A). The same tendency was observed in both DT40 and HD3 cells (Fig. 4A). Taking into account the fact that CTCF participates in establishing contacts between remote genomic elements,34,35 we have studied possible correlation between the intensity of the 4C signal and the presence of binding motifs for CTCF. Such a correlation was indeed observed (Fig. 4B). It should be noted, however, that reported CTCF binding motifs differ slightly in cells of different origins.36-38 Thus, we mapped the actual CTCF binding sites in both HD3 and DT40 cells. A low-resolution map of the CTCF binding sites in chromosome 14 is presented in Figure 1D. The results of the analysis (Fig. 4C) demonstrated a good correlation between the positions of the experimentally determined CTCF binding sites and the 4C signal.

Figure 3. Scatterplots and box-plots showing the relationship between the NPRL3–4C signal (y-axis) in HD3 and DT40 cells and the density of CGIs (x-axis). The points represent non-overlapping 100 Kb genomic bins (see Materials and Methods). The region surrounding the anchor (NPRL3 anchor - chr14: 11500000–12800000) was not taken into account. The linear regression lines and the confidence intervals for the prediction are shown. The points at which the 4C signal was higher than 2000 reads per bin for all chromosomes and higher than 10 000 reads per bin for chromosome 14 were discarded. The box-plots contain the points split into 5 groups of equal size according to their x-value.

Figure 4. Scatterplots and box-plots showing the relationship between the NPRL3-4C signal (y-axis) in HD3 and DT40 cells and the density of various genomic features (x-axis). (A–F) The correlation of the NPRL3–4C signal with the Sp1 motifs, CTCF motifs, CTCF deposition sites, as determined by ChIP-Seq, NF-E2 binding motifs, GATA1 binding motifs, and G-quadruplex motifs is shown. Other designations are as in Figure 3.
Surprisingly, in erythroid cells (HD3), there was a negative correlation of the 4C signal with the predicted binding sites for the erythroid-specific transcription factors (NF-E2 and GATA1). In this respect, erythroid cells did not differ from lymphoid cells (Fig. 4D and E). We reasoned that the observed positive and negative correlations between the 4C signals and the densities of binding motifs for different transcription factors might simply reflect the differences in the GC contents of the transcription factor binding motifs. To test this hypothesis, we performed a permutation analysis of the transcription factor binding motifs. This analysis was performed only for HD3 cells because all the identified correlations were more pronounced in these cells (Figs. 3 and 4). The results of the analysis were consistent with our hypothesis (Fig. 5, anchor NPRL3). Indeed, the intensity of the 4C signal correlated well just with the GC content. However, the correlation of the 4C signal with the content of CGI appeared to be more pronounced. Besides, the observed anticorrelation of the 4C signal with the density of the NF-E2 and GATA1 motifs was diminished upon motif shuffling. Of potential interest is the positive correlation of the 4C signal with the G-quadruplex motif density (Figs. 4F and 5). G-quadruplexes were reported to be important elements of eukaryotic DNA replication origins.39,40 The spatial clustering of CGIs may thus be necessary for both transcription and DNA replication.
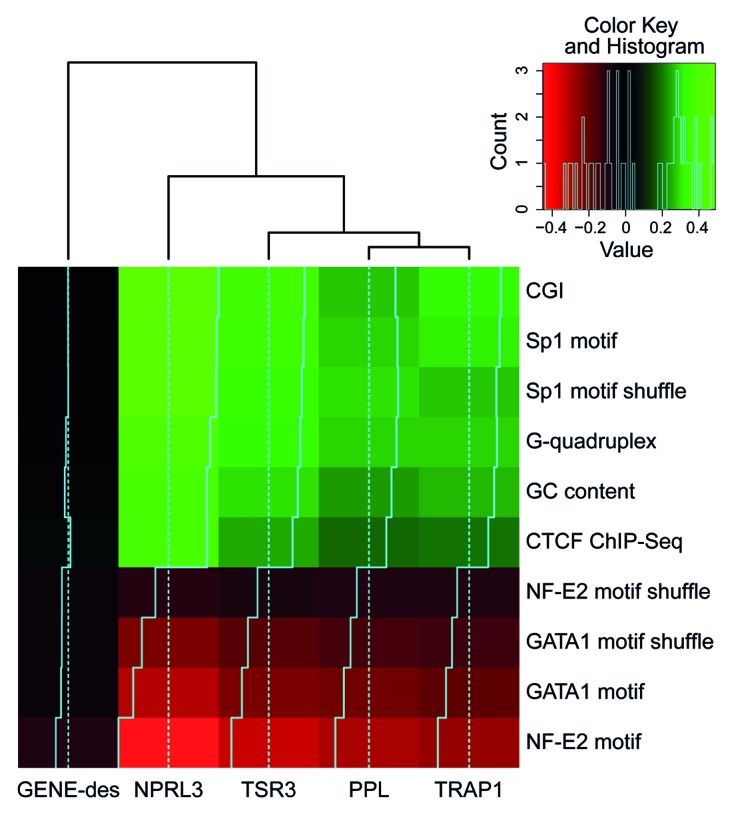
Figure 5. Heat map representing the correlations between the density of various genomic features (rows) and the 4C signal for different anchors (columns). As a control for the motif nucleotide content, motifs with shuffled positions were analyzed. The color represents the correlation value; black corresponds to no correlation, and light green or red correspond to positive or negative correlation, respectively. The histogram in the top-right corner shows the distribution of the correlation values presented in the heat map.
Spatial segregation of the active and inactive genomic compartments
To verify the importance of the observations made using the bait fixed on the NPRL3 promoter, an additional 4C experiment with several baits was performed using HD3 cells. In this experiment, the anchors were placed on different CGIs on chromosome 14 that demonstrated a strong interaction with the NPRL3 anchor (Fig. 6, anchors TSR3, TRAP1, PPL). The fourth anchor (anchor GENE-Des) was placed in a gene-poor area that did not interact with the NPRL3 promoter (Fig. 6). The fifth anchor was again placed on the NPRL3 promoter to relate the new set of data with the one described above (Fig. 6, anchor NPRL3). Two independent biological replicates were analyzed, and for each replicate ~10 million interacting fragments were sequenced and uniquely assigned to one of the anchors. The similarity between the biological replicates was very high (Pearson’s correlation coefficients of 0.97 or more for the anchors TSR3, GENE-Des, NPRL3, and PPL and 0.64 for the anchor TRAP1). Nevertheless, taking into account the latter value, we analyzed the two sets of data separately. Unless specifically indicated, the data from experiment 1 (deposited in GEO database under accession number GSM1255519) were analyzed. The profiles of the 4C signals in chromosome 14 for each of the four selected anchors are presented in Figure 6. The anchors TSR3, TRAP1, and PPL yielded 4C patterns relatively similar to these observed with the anchor NPRL3. The anchor located in a gene desert (GENE-Des) also showed some long-distance interactions along chromosome 14. However, the profile of these interactions differed significantly from those observed with the other anchors (Fig. 6). The Pearson’s correlation coefficients for the profiles of the 4C signal observed on chromosome 14 using the NPRL3 anchor (in two biological replicates) and the TSR3, TRAP1 and PPL anchors (in two biological replicates) were ≥0.288. In contrast, the profiles of the 4C signals observed with the GENE-Des anchor showed no similarity with the profiles observed with the NPRL3 anchor (the Pearson correlation coefficients < 0) (Table 1).
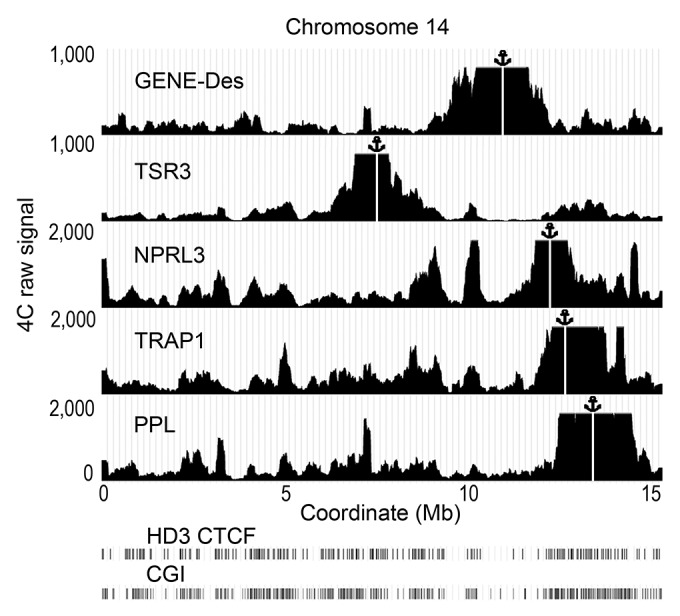
Figure 6. Distribution of the raw 4C signals on chromosome 14 obtained using the anchors situated in CGIs (NPRL3, TSR3, TRAP, PPL) and an anchor situated in a CGI-poor area (GENE-Des) in HD3 cells. The sequenced reads were aligned to the genome near HindIII sites. The distances along chromosome 14 are presented in Mb according to the galGal4 assembly (UCSC). The position of the viewpoint is indicated by the anchor sign above the graphs and by the vertical white line. The positions of CGIs and the CTCF deposition sites mapped by ChIP-Seq in HD3 cells are shown below the graphs demonstrating the distribution of the 4C signals (see the Fig. 1 legend for details). Note that in this illustration, the raw 4С signal was smoothed with moving average approach (200 Kb sliding window width).
Table 1. Comparison of the profiles of 4C signals obtained using the anchors GENE-Des, TSR3, TRAP, and PPL with the profiles obtained using the anchor NPRL3.
| NPRL3 (1) | NPRL3 (2) | |
|---|---|---|
| Gene-Des (1) | -0.063 | -0.016 |
| Gene-Des (2) | -0.141 | -0.094 |
| TSR3 (1) | 0.335 | 0.288 |
| TSR3 (2) | 0.328 | 0.289 |
| NPRL3 (1) | 1 | 0.976 |
| NPRL3 (2) | 0.976 | 1 |
| TRAP1 (1) | 0.631 | 0.570 |
| TRAP1 (2) | 0.568 | 0.490 |
| PPL (1) | 0.440 | 0.384 |
| PPL (2) | 0.481 | 0.399 |
Pearson correlation coefficients are shown. The results of the 2 independent biological experiments were treated independently.
Similar to the NPRL3 anchor, the TSR3, TRAP1 and PPL anchors demonstrated preferential long-distance interactions with CGIs, Sp1 binding motifs and CTCF binding sites but not with NF-E2 and GATA1 binding motifs (Figs. S1, S3, and S4). Notably, no such correlations were observed with the GENE-Des anchor (Fig. 5; Fig. S2).
Distribution of the 4C signal in the vicinity of promoters and CGIs
CGIs frequently harbor active promoters. It might happen that the clustering of CGIs observed in the above analysis was entirely due to the presence of these active promoters. Alternatively, the clustering of CGIs can occur regardless of the presence of active promoters. In an attempt to clarify the situation, we analyzed the distribution of the 4C signal around CGIs and promoters, using the data obtained with NPRL3 anchor fragment in HD3 cells. We first plotted 4C profiles around each CGI found in the chicken genome, superposed and averaged them. The obtained 4C curve had a clear peak on a CGI (Fig. 7A), in agreement with the correlation analysis. On the contrary, the regions between CGIs demonstrated a decline in the 4C curve (Fig. 7B). We next performed similar analysis on the transcription start sites (TSSs) and observed a peak slightly upstream of a TSS (i.e., on presumed promoters), although in this case the absolute level of the 4C signal was lower (Fig. 7C). To interpret these data it is necessary to keep in mind that many promoters co-localize with CGIs. It is true for the NPRL3 gene promoter used as an anchor in this analysis, and we found that of 16950 TSSs identified in the chicken genome, 5650 were located within CGIs (Fig. 7E), and 4911 of 19309 CGIs present in chicken genome, harbored a TSS (Fig. 7F). In order to find out whether a CGI by itself or a promoter nested within a CGI contributes more to the observed clustering of CGIs, we separately analyzed promoters associated with CGIs and promoters lacking such association. All TSSs were subdivided into 5 groups according to the distance to the nearest CGI that could be found upstream or downstream of the TSS. Then averaged 4C profiles were plotted separately for each of these groups. As seen in Figure 7G, TSSs co-localizing or neighboring CGIs demonstrated higher frequency of interaction with the anchor restriction fragment. Furthermore, the 4C signal curves showed a peak at presumed promoters (up to 5 Kb upstream of a TSS) (Fig. 7G, red, yellow, and green curves). TSSs located away from CGIs showed lower interaction frequency, with no peak on presumed promoters (Fig. 7G, blue and violet curves). We analyzed in a similar way the CGIs that were grouped according to the distance to the nearest promoter. In this case, the dependence of 4C signal on CGI-promoter distance was not pronounced. CGIs harboring promoters and CGIs located at a distance of 1.5–10 Kb from promoters demonstrated comparable levels of the 4C signal with a peak on a CGI (Fig. 7H, red, yellow and green curves). Remarkably, even remote CGIs (˃25 Kb from the nearest promoter) demonstrated a peak on the 4C curve (Fig. 7H, violet curve). Taken together, these data suggest that the clustering of CGIs in the nuclear space is not directly related to the presence of active promoters within CGIs. Promoterless CGIs participate in the clustering as well as CGIs harboring promoters.

Figure 7. Distribution of the 4C signal in the vicinity of promoters and CGIs observed in experiments with NPRL3 anchor. (A, C, and D) Averaged 4C signal profiles for 40 consecutive 1 Kb bins surrounding various genomic features: CGIs (A), TSSs (С) or CTCF sites (D). Х-axis represent genomic coordinate relative to a genomic feature, y-axis represents averaged 4C signal value, gray lines represent the profiles for bootstrapped sets of genomic features (see Material and Methods section for details). (B) Averaged 4C signal profiles between consecutive CGIs. Х-axis represents relative location in a region between CGIs. (E) Fraction of TSSs overlapped by a CGI and (F) fraction of CGIs overlapping a TSS. (G, H) Plots similar to (A–D), showing averaged 4C signals in the vicinity of TSSs grouped by the distance of a TSS to the nearest CGI (G) and in the vicinity of CGIs grouped by the distance of a CGI to the nearest TSS (H). See the legend below the plots for the distances thresholds.
As was mentioned above, the clustering of CGIs may be at least in part mediated by interaction of bound CTCF molecules. Indeed, analysis of the distribution of the 4C signals revealed some increase in the vicinity of CTCF deposition sites (Fig. 7D).
The observations made with NPRL3 anchor in HD3 cells were reproduced upon similar analysis of the 4C data obtained with TSR3, TRAP1 and PPL anchors in HD3 cells (data not shown). However, the opposite trends were identified in the analysis of the 4C data obtained with the GENE-Des anchor (Fig. 8). In this case, CGIs and promoters demonstrated a decline in the 4C curve (Fig. 8A and C), while the regions between CGIs were characterized by an elevated 4C signal (Fig. 8B).

Figure 8. Distribution of the 4C signal in the vicinity of promoters and CGIs observed in experiments with GENE-Des anchor. Plots similar to shown in Figure 7A–C, obtained in the experiment with the GENE-Des anchor.
Discussion
The spatial organization of the eukaryotic genome appears to play an important role in the regulation of gene expression.1,5 Based on the analysis of Hi-C data, Dekker and collaborators proposed that the open and closed chromatin domains are spatially segregated to form 2 different compartments.41,42 These authors also noted that the gene-rich small chromosomes tend to be located closer to each other in the nuclear space. The segregation of gene-poor long and gene-rich short chromosomes was observed previously in chicken cells where all the gene-rich micro-chromosomes are concentrated close to the center of the nucleus.43 Therefore, it is not surprising that our analysis detected the preferential interaction of chicken chromosome 14 with other short chromosomes, at least in erythroid cells. Our results support the hypothesis that active genomic regions form a distinct spatial compartment. Indeed, the anchors placed on CGIs were found to be involved in spatial interactions with CGI-rich regions in the same and other chromosomes. Conversely, the anchor placed in a gene/CGI-poor area did not show a preferential spatial interaction with CGI-rich areas. A model for chromosome folding that allows the preferential interaction of all gene-rich areas is not immediately apparent. However, it is noteworthy that the 4C and other C-methods only allow for the determination of the average pattern of interactions in a cell population. This pattern is likely to represent the superimposition of a number of alternative configurations existing in individual cells.44,45
Our data strongly suggest that the spatial association (clustering) of CGIs may constitute a driving force for the spatial segregation of active genomic regions. The biological significance of the CGIs spatial clustering and the mechanisms supporting this clustering are far from evident. In recent years, the association of transcribed genes into transcription factories has gained much attention.19-21,46 Notably, the housekeeping genes constitute the majority of expressed genes, even in specialized cells, and the promoters of most housekeeping genes are located in CGIs.47 Assuming that there is not much specificity in the assembly of genes into transcription factories, each transcription factory might be expected to contain one or several housekeeping genes. Still, detailed analysis of the distribution of the 4C signal around promoters associated and non-associated with CGIs and CGIs that do not harbor promoters suggests that CGIs participate in the clustering regardless of the presence of a promoter (Fig. 7).
In erythroid cells, the erythroid-specific genes demonstrate a tendency to share transcription factories.22,23 However, the probability of the association of more than 2 erythroid-specific genes in the same transcription factory was lesser than the expected probability based on the random distribution of genes among all transcription factories. Considering that an average transcription factory contains 8 elongating RNA polymerase II molecules48 (up to 30 molecules, according to other estimates49,50), it is likely that in erythroid cells, the transcribed housekeeping genes constitute the cores of all transcription factories. Our experimental system can be used to estimate the relative contribution of the association between the housekeeping and tissue-specific genes to the 3D genome architecture. The viewpoint selected for the initial 4C analysis was fixed in the CGI located just upstream (~5 Kb) of the cluster of α-globin genes that are not active in lymphoid cells. Correspondingly, the associations between the erythroid-specific genes are not likely to contribute to the spectrum of 4C signals observed in these cells. Conversely, such a contribution can be expected in the erythroid (HD3) cells. Nevertheless, in both the lymphoid and erythroid cells, we were only able to detect the correlation of the 4C signals with CGIs and the binding motifs for the ubiquitous transcription factor Sp1 but not with the predicted binding sites for erythroid-specific transcription factors. Therefore, it is likely that in erythroid cells, the contribution of the associations between CGIs to the 3D genome architecture is more important than the contribution of the associations between erythroid-specific genes. This conclusion is corroborated by the observation that in mouse and chicken erythroid cells, the α-globin genes were recruited to transcription factories mediating the transcription of housekeeping genes.8,51
When considering the possible biological significance of the CGIs clustering, it is noteworthy that CGIs frequently contain origins of DNA replication.52 Furthermore, it appears that the most effective replication origins are located within CGIs that harbor the promoters of active genes.53 Replication origins and their bound protein complexes are assembled in nuclear foci,54 which are likely converted into replication factories55,56 upon origin firing. This suggests that the clustering of CGIs may be essential for the spatial organization of replication units; this hypothesis deserves further study. Nevertheless, our results provide indirect evidence for a possible link between the clustering of CGIs and the spatial organization of replication units. We found that the NPRL3 anchor located within a CGI harboring a replication origin31 established long-distance interactions with regions containing G-quadruplex motifs, which are frequently present within CGIs and appear to be important elements of DNA replication origins in higher eukaryotes.39 The other anchors located in CGIs also demonstrated preferential interactions with G-quadruplex motifs. Therefore, it is possible that the CGIs clustering reflects the organization of replication origins into replication factories.
The hypotheses that the CGIs clustering is essential for the spatial organization of replication origins and that this clustering reflects the association of housekeeping genes into transcription factories are not mutually exclusive. In both cases, the clustering of CGIs may be supported by depletion-attraction, as discussed previously.57-59 It is also possible that other additional interactions stabilize the associations of CGIs. Sp1 was reported to participate in the mediation of long-range interactions of remote genomic elements,60 and we observed a good correlation between the 4C signals and the presence of Sp1 binding sites. The CGIs frequently contain CTCF binding sites, and the role of CTCF in supporting the 3D genome architecture is well established.35,61-66 Consistently, we observed a good genome-wide correlation between the 4C signals and the CTCF binding sites that were mapped using the ChIP-Seq procedure. This correlation was most evident at low scale resolution suggesting that the presence of CTCF binding sites within the transcriptionally active part of the genome may contribute to the spatial separation of this part of the genome. In mammals, the transcriptionally active part of the genome is enriched in SINE-type repeats. Some of these repeats have been reported to contain CTCF binding sites.67 Unfortunately, it is not clear whether repetitive elements present in the chicken genome are rich in CTCF binding sites.
Materials and Methods
The 4C-Seq and ChIP-Seq data were deposited in GEO (GSE51939).
Accession numbers for 4C-Seq data: “bait” NPRL3 in HD3 cells (replicate 1 GSM1255515; replicate 2 GSM1255516); “bait” NPRL3 in DT40 cells (replicate 1 GSM1255517; replicate 2 GSM1255518); “bait” TSR3, GENE-Des, TRAP1, PPL in HD3 cells (replicate 1 GSM1255519; replicate 2 GSM1255520).
Accession numbers for CTCF ChIP-Seq data: HD3 input control GSM1253764; CTCF HD3 non-induced cells GSM1253765; CTCF HD3 induced cells GSM1253766; CTCF DT40 cells GSM1253767.
Cell culture
The avian erythroblastosis virus-transformed chicken erythroblast cell line HD3 (clone A6 of the line LSCC68,69) and the DT40 lymphoid cell line (CRL-2111, ATCC) were grown in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 2% chicken serum and 8% fetal bovine serum at 37 °C with 5% CO2. For DT40 cells, the medium also contained 50 μM β-mercaptoethanol.
4C-Seq analysis
The 4C procedure was performed as previously described70 with minor modifications. Briefly, 107 cells were fixed with 2% formaldehyde in DMEM medium supplemented with 10% fetal bovine serum (FBS) for 10 min at room temperature, and the reaction was stopped by the addition of glycine to a final concentration of 0.125 M. After washing with PBS/10% FBS, the fixed cells were incubated for 10 min in an ice-cold lysis solution (10 mM Tris pH 8.0, 10 mM NaCl, 0.2% Nonidet P40, and a protease inhibitor cocktail [Thermo Scientific]) at a concentration of 2 × 107 cells/ml to release the nuclei. The nuclei were harvested and suspended in 0.5 ml of 1.2× restriction buffer 2 (New England Biolabs). SDS was added to a final concentration of 0.3%, and the solution was incubated on a shaker for 1 h at 37 °C. Triton X-100 was added to a final concentration of 1.8%, and the solution was incubated for 1 h at 37 °C to sequester the SDS. The DNA was digested by overnight incubation with 800 units of HindIII (New England Biolabs) at 37 °C on a shaker. The restriction endonuclease was inactivated by the addition of SDS to a final concentration of 1.6% and incubation for 20 min at 65 °C. The solution was diluted by adding 7 ml of 1 × ligation buffer (Thermo Scientific). Triton X-100 was added to a final concentration of 1%, and the solution was incubated at 37 °C for 1 h on a shaker. Next, 100 units of T4 DNA Ligase (Thermo Scientific) was added, and the DNA was ligated for 4.5 h at 16 °C and subsequently for 30 min at room temperature with slow agitation. The cross-links were reversed by incubation at 65 °C for 16 h in the presence of Proteinase K (40 μg/ml). After cross-link reversion, RNase A was added to a final concentration of 40 μg/ml, and the RNA was digested for 45 min at 37 °C. The DNA was purified by extraction with phenol followed by precipitation with ethanol. To further purify the DNA, the samples were processed with the QIAEX II Gel Extraction Kit (Qiagen). The concentration of DNA was determined using a fluorometric assay (Qubit, Invitrogen).
Fifty micrograms (100 ng/µl) of a ligated 3C DNA template was digested overnight with 200 units of DpnII (New England Biolabs). The restriction endonuclease was inactivated by incubation for 20 min at 65 °C, and the DNA was purified by phenol-chloroform extraction and ethanol precipitation. Next, the DNA was ligated using 100 units of T4 DNA Ligase (Thermo Scientific) in 14 ml of 1× ligation buffer (Thermo Scientific) for 4 h at 16 °C. The ligation products were precipitated with ethanol and further purified using the QIAquick PCR purification kit (Qiagen). To linearize the circular molecules of interest, the DNA was treated with a specific tertiary restriction enzyme: EcoNI for the TSR3 bait, NcoI for the GENE-Des, TRAP1 and PPL baits, and EcoRV for the NPRL3 bait. The digested products were purified using the QIAquick PCR purification kit (Qiagen).
The PCR reactions were performed using the Expand Long Template PCR system (Roche). Each 50 µl PCR reaction contained 120 ng of the DNA template, 1 × PCR buffer 1, 0.3 µM of each of the primers, 0.35 mM of each dNTP, and 3.75 units of the Expand Long enzyme mix (Roche). The sequences of the HindIII / DpnII 4C primers are as follows (5′-3′): TSR3 bait CACTCATCTC CCCGTACTTT G / AAGTTTCTTT TAATTTGGAG ACTTTC, GENE-Des bait AATTTGTGAA GCAGTTGTAT GTAGTC / TCTTCTCCAC ATAATCCCAC ACT, NPRL3 bait GCCAGGATAT AGATTCTGCT TT / CCTCTGACAT AATTGCCGAT AG, TRAP1 bait CCAGAGATTC TCAAATCACA GCA / CTATGGGGAC AAGTGAGGAA CAG, and PPL bait AAAGCATCTC CTCTCCCTGA AG / GTCTCCCACA GTCACTCCTC CT. The PCR amplification was performed in the linear range as follows: an initial denaturation for 2 min at 94 °C, 30 cycles of 15 s at 94 °C, 1 min at 57 °C, and 3 min at 68 °C, and a final elongation for 4 min at 68 °C. The PCR products were purified and concentrated using the QIAquick PCR purification kit (Qiagen). In a multibait experiment, 4C libraries obtained with different pairs of primers were mixed in equal weights and further processed as one sample. After separation of the fragments by size on 1.5% agarose gels, two zones were excised: 70–400 bp (S) and 400–1500 bp (L). Following extraction with the QIAquick gel extraction kit (Qiagen), the fragments of the L fraction were sonicated to reduce the fragment size to 100–500 nucleotides using a Covaris S220 instrument with the following parameters: time 300 s, duty cycle 10%, and peak power 23 W. Next, the S and L fractions were processed using the TruSeq DNA sample prep kit v.2 (Illumina), and post-PCR size selection was performed using agarose gels (fragments with length 200–600 nucleotides were selected). After purification, the library concentrations were measured using the Qubit fluorometer (Invitrogen), and real-time PCR was performed using primers complementary to the distal regions of the Illumina adapters (I-qPCR-1.1: AATGATACGG CGACCACCGA GAT and I-qPCR-2.1: CAAGCAGAAG ACGGCATACG A). Next, the libraries from the S and L fractions were combined in equal amounts, diluted to 2 nM, denatured using 0.1 M NaOH and subsequently diluted to a final concentration of 10 pM using HT1 buffer (Illumina). The cluster generation was performed using the cBot instrument and the TruSeq PE Cluster Kit v3 reagents. The sequencing was performed on a HiSeq2000 instrument using the TruSeq SBS Kit v3 reagents (Illumina), with read lengths of 101 nucleotides from each end.
Analysis of the 4C data
The paired-end Illumina sequencing reads were preprocessed by trimming the subsequence originating from the bait region. From each pair of forward and reverse reads, only the read containing the HindIII ligation site was chosen, while its pair was discarded. The bait-originating subsequence of the read located before the HindIII site was used to classify the reads by the 4C anchor. A list of the reference bait-originated subsequences is presented in Table S1. Those bait-originating fragments were trimmed to take into analysis the sequences located after the HindIII site because they presumably originated from the DNA regions interacting with the anchor. The preprocessed reads were mapped to the reference genome galGal4 using the bowtie aligner.71 The reference genome (galGal4) was digested in silico with HindIII, and the regions between the restriction sites were chosen for further analysis.
Proper 4C reads should map either upstream or downstream of the HindIII sites, and one end of each read should precisely coincide with the restriction site. For every restriction fragment, the number of reads that mapped to its left or right end was added; the resulting value was considered the raw 4C signal for that restriction fragment. It should be noted that because the ligation events could occur only at the ends of restriction fragments, no linear dependence was assumed between the number of reads per fragment and the fragment length. To smooth the signal, the genome was binned into 100 Kb fragments. The average coverage by the 4C signal was calculated for every fragment.
To identify the bait-interacting domains on different scales, we used the domainogram approach.70 The fraction of the restriction fragments with non-zero coverage was assumed to represent the overall 4C signal intensity in a genomic window. The fraction of the restriction fragments with non-zero coverage was compared between small windows of variable size (3–200 HindIII restriction fragments) and a large background window (300 HindIII restriction fragments). These comparisons yielded Fisher test P values. The domainograms (Fig. 1C) were plotted to represent those P values: the pixel color represents the P value, the pixel x-coordinate represents the genomic coordinate, and the pixel y-coordinate shows the size of the small window (the higher the value, the larger the window size). We next reduced the domainograms to a plot in which the x-axis represented the genomic coordinate and the y-axis represented the –log10 of the most significant P value among all the small windows of different sizes centered on a given genomic coordinate (Fig. 1B).
Next, we explored the correlations between the 4C signal and the density of various genomic features. The genomic locations of the CGIs were obtained from the cpgIslandEx table for the galGal4 genome from the UCSC genome browser.72 Putative transcription factor binding sites were identified by matching a position weight matrix (PWM) for the factor of interest to the galGal4 genome (R packages Biostrings and BSgenome). The PWMs were downloaded using the MotifDb R package. To account for the nucleotide content of a TF binding motif, the order of positions in the PWMs was shuffled, which generated control motifs with different sequences but similar nucleotide compositions. The 4C signals were correlated with the numbers of occurrences of the genomic features in 100 Kb bins; the region surrounding the anchor (NPRL3 anchor - chr14: 11500000–12800000) was not taken into account. The correlations were separately calculated for the whole genome, for the bait-containing chr14 and for other chromosomes. The corresponding scatter plots were plotted with an overlaid linear regression line. The putative G-quadruplex sequences following the pattern d(G3+N1–7G3+N1–7G3+N1–7G3+) in the galGal4 genome were identified using the quadparser software.73
To measure average 4C signal in the vicinity of various genomic features (e.g., CGIs, CTCF binding sites, TSS), the following procedure was applied. 40 consecutive bins of equal length (1Kb) surrounding every occurrence of a genomic feature were taken, 20 upstream and 20 downstream. 4C signal coverage was measured in each bin and then averaged over all bins positioned in the same way relative to a genomic feature, yielding the averaged 4C profile around genomic features. To estimate the variance of the averages, a bootstrapping procedure was applied 10 times. Every time, a set of features with the same size as the original set was generated by choosing random elements from the original set (thus, an element could be chosen more than once or never), then for each bootstrapped feature set an averaged signal profile was calculated. To plot averaged 4C signal profiles between CGIs, regions between consecutive CGIs were split into 40 consecutive bins of equal length (thus, bins for different loci could differ in length), then the same procedure was applied.
Mapping the CTCF deposition sites by ChIP-Seq
Chromatin immunoprecipitation was performed as described previously,74 but with modifications according to the Life Technologies protocol (http://tools.lifetechnologies.com/content/sfs/manuals/DynabeadsProteinG_man.pdf). Approximately 1 × 108 logarithmically growing cells were fixed with 1% v/v formaldehyde in DMEM/F-12 (1:1) medium (Invitrogen, 42400–010) at room temperature for 8 min. The fixed cells were pelleted at 700 g for 4 min at 4 °C, washed with PBS containing 1 mM AEBSF and 2 μl/ml of a protease inhibitor cocktail (Sigma, P8340), pelleted again, and re-suspended in 500 μl of lysis buffer (50 mM TRIS-HCl pH 8.0, 1% SDS, 10 mM EDTA). The lysate was incubated for 10 min on ice and sonicated with a Cole-Parmer CP750 ultrasonic processor (30% amplitude, 40 cycles for 3 s each with 10 s intervals). The cell debris was removed using a microcentrifuge (10 min, 13 000 rpm, 4 °C), and the supernatant was diluted 10-fold with 16.7 mM TRIS-HCl, pH 8.0, 16.7 mM NaCl, 1.2 mM EDTA, 1% Triton X-100, 0.01% SDS, 1 mM AEBSF, and 1 μl/ml of the protease inhibitor cocktail. Aliquots were taken at this stage for subsequent use as the input control. The cell lysates were pre-cleared by incubation with Dynabeads Protein G magnetic beads (Life Technologies) and then incubated with 30 μg of anti-CTCF antibodies overnight at 4 °C with rotation. The rabbit custom anti-CTCF antibodies were prepared against the N-terminal region of the chicken CTCF protein comprising amino acids 86–233. The specificity of these antibodies was confirmed by western blot analysis.75 After incubation with antibodies, the DNA-protein complexes were collected with the Dynabeads Protein G magnetic beads, washed according to the manufacturer’s recommendations (Life Technologies), and eluted by two incubations for 15 min in elution buffer (1% SDS, 0.1 M NaHCO3) at room temperature. The DNA samples were purified using the QIAquick Gel Extraction Kit according to the manufacturer’s recommendations (Qiagen). The immunoprecipitated and input DNA probes (10 ng each) were sequenced at Evrogen (http://www.evrogen.ru) using an Illumina HiSeq platform, and 41–50 million reads were sequenced per sample. The reads were mapped to the galGal4 genome using the Bowtie2 software.76 ChIP peak calling was performed as described77 with a P value threshold parameter of 10−5. To verify the validity of the CTCF deposition sites mapped, we have inspected the upstream area of the c-myc gene in HD3 cells and found a strong peak in the same place where the CTCF binding/deposition sites were originally mapped by Lobanenkov et al.78,79 (Fig. S5).
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by the Presidium of the Russian Academy of Sciences (grants from the Program on Molecular and Cellular Biology to A.A.G. and S.V.R.) by the Russian Foundation for Basic Research (grants 12-04-93109, 12-04-33040, 12-04-00036-а, 13-04-40086-Н, 13-04-40089-Н, 13-04-93105, 12-04-00313) and by Dmitri Zimin's foundation “Dynasty.”
Glossary
Abbreviations:
- CGI
CpG island
- 3C
chromosome conformation capture
- 4C
chromosome conformation capture on chip
- ACH
active chromatin hub
- TSS
transcription start site
- PWM
position weight matrix
References
- 1.Holwerda S, de Laat W. Chromatin loops, gene positioning, and gene expression. Front Genet. 2012;3:217. doi: 10.3389/fgene.2012.00217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.de Wit E, de Laat W. A decade of 3C technologies: insights into nuclear organization. Genes Dev. 2012;26:11–24. doi: 10.1101/gad.179804.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Palstra RJ. Close encounters of the 3C kind: long-range chromatin interactions and transcriptional regulation. Brief Funct Genomic Proteomic. 2009;8:297–309. doi: 10.1093/bfgp/elp016. [DOI] [PubMed] [Google Scholar]
- 4.de Laat W, Grosveld F. Spatial organization of gene expression: the active chromatin hub. Chromosome Res. 2003;11:447–59. doi: 10.1023/A:1024922626726. [DOI] [PubMed] [Google Scholar]
- 5.de Laat W, Klous P, Kooren J, Noordermeer D, Palstra RJ, Simonis M, Splinter E, Grosveld F. Three-dimensional organization of gene expression in erythroid cells. Curr Top Dev Biol. 2008;82:117–39. doi: 10.1016/S0070-2153(07)00005-1. [DOI] [PubMed] [Google Scholar]
- 6.Noordermeer D, de Laat W. Joining the loops: beta-globin gene regulation. IUBMB Life. 2008;60:824–33. doi: 10.1002/iub.129. [DOI] [PubMed] [Google Scholar]
- 7.Gavrilov AA, Razin SV. Spatial configuration of the chicken alpha-globin gene domain: immature and active chromatin hubs. Nucleic Acids Res. 2008;36:4629–40. doi: 10.1093/nar/gkn429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou GL, Xin L, Song W, Di LJ, Liu G, Wu XS, Liu DP, Liang CC. Active chromatin hub of the mouse alpha-globin locus forms in a transcription factory of clustered housekeeping genes. Mol Cell Biol. 2006;26:5096–105. doi: 10.1128/MCB.02454-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vernimmen D, De Gobbi M, Sloane-Stanley JA, Wood WG, Higgs DR. Long-range chromosomal interactions regulate the timing of the transition between poised and active gene expression. EMBO J. 2007;26:2041–51. doi: 10.1038/sj.emboj.7601654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vernimmen D, Marques-Kranc F, Sharpe JA, Sloane-Stanley JA, Wood WG, Wallace HA, Smith AJ, Higgs DR. Chromosome looping at the human alpha-globin locus is mediated via the major upstream regulatory element (HS -40) Blood. 2009;114:4253–60. doi: 10.1182/blood-2009-03-213439. [DOI] [PubMed] [Google Scholar]
- 11.Ulianov SV, Gavrilov AA, Razin SV. Spatial organization of the chicken beta-globin gene domain in erythroid cells of embryonic and adult lineages. Epigenetics Chromatin. 2012;5:16. doi: 10.1186/1756-8935-5-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gavrilov AA, Gushchanskaya ES, Strelkova O, Zhironkina O, Kireev II, Iarovaia OV, Razin SV. Disclosure of a structural milieu for the proximity ligation reveals the elusive nature of an active chromatin hub. Nucleic Acids Res. 2013;41:3563–75. doi: 10.1093/nar/gkt067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Razin SV, Gavrilov AA, Ioudinkova ES, Iarovaia OV. Communication of genome regulatory elements in a folded chromosome. FEBS Lett. 2013;587:1840–7. doi: 10.1016/j.febslet.2013.04.027. [DOI] [PubMed] [Google Scholar]
- 14.Misteli T. Concepts in nuclear architecture. Bioessays. 2005;27:477–87. doi: 10.1002/bies.20226. [DOI] [PubMed] [Google Scholar]
- 15.Misteli T. Beyond the sequence: cellular organization of genome function. Cell. 2007;128:787–800. doi: 10.1016/j.cell.2007.01.028. [DOI] [PubMed] [Google Scholar]
- 16.Osborne CS, Chakalova L, Brown KE, Carter D, Horton A, Debrand E, Goyenechea B, Mitchell JA, Lopes S, Reik W, et al. Active genes dynamically colocalize to shared sites of ongoing transcription. Nat Genet. 2004;36:1065–71. doi: 10.1038/ng1423. [DOI] [PubMed] [Google Scholar]
- 17.Faro-Trindade I, Cook PR. Transcription factories: structures conserved during differentiation and evolution. Biochem Soc Trans. 2006;34:1133–7. doi: 10.1042/BST0341133. [DOI] [PubMed] [Google Scholar]
- 18.Carter DR, Eskiw C, Cook PR. Transcription factories. Biochem Soc Trans. 2008;36:585–9. doi: 10.1042/BST0360585. [DOI] [PubMed] [Google Scholar]
- 19.Sutherland H, Bickmore WA. Transcription factories: gene expression in unions? Nat Rev Genet. 2009;10:457–66. doi: 10.1038/nrg2592. [DOI] [PubMed] [Google Scholar]
- 20.Razin SV, Gavrilov AA, Pichugin A, Lipinski M, Iarovaia OV, Vassetzky YS. Transcription factories in the context of the nuclear and genome organization. Nucleic Acids Res. 2011;39:9085–92. doi: 10.1093/nar/gkr683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cook PR. A model for all genomes: the role of transcription factories. J Mol Biol. 2010;395:1–10. doi: 10.1016/j.jmb.2009.10.031. [DOI] [PubMed] [Google Scholar]
- 22.Schoenfelder S, Clay I, Fraser P. The transcriptional interactome: gene expression in 3D. Curr Opin Genet Dev. 2010;20:127–33. doi: 10.1016/j.gde.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 23.Schoenfelder S, Sexton T, Chakalova L, Cope NF, Horton A, Andrews S, Kurukuti S, Mitchell JA, Umlauf D, Dimitrova DS, et al. Preferential associations between co-regulated genes reveal a transcriptional interactome in erythroid cells. Nat Genet. 2010;42:53–61. doi: 10.1038/ng.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bartlett J, Blagojevic J, Carter D, Eskiw C, Fromaget M, Job C, Shamsher M, Trindade IF, Xu M, Cook PR. Specialized transcription factories. Biochem Soc Symp. 2006:67–75. doi: 10.1042/bss0730067. [DOI] [PubMed] [Google Scholar]
- 25.Osborne CS, Chakalova L, Mitchell JA, Horton A, Wood AL, Bolland DJ, Corcoran AE, Fraser P. Myc dynamically and preferentially relocates to a transcription factory occupied by Igh. PLoS Biol. 2007;5:e192. doi: 10.1371/journal.pbio.0050192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Philonenko ES, Klochkov DB, Borunova VV, Gavrilov AA, Razin SV, Iarovaia OV. TMEM8 - a non-globin gene entrapped in the globin web. Nucleic Acids Res. 2009;37:7394–406. doi: 10.1093/nar/gkp838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kowalczyk MS, Hughes JR, Babbs C, Sanchez-Pulido L, Szumska D, Sharpe JA, Sloane-Stanley JA, Morriss-Kay GM, Smoot LB, Roberts AE, et al. Nprl3 is required for normal development of the cardiovascular system. Mamm Genome. 2012;23:404–15. doi: 10.1007/s00335-012-9398-y. [DOI] [PubMed] [Google Scholar]
- 28.Flint J, Tufarelli C, Peden J, Clark K, Daniels RJ, Hardison R, Miller W, Philipsen S, Tan-Un KC, McMorrow T, et al. Comparative genome analysis delimits a chromosomal domain and identifies key regulatory elements in the alpha globin cluster. Hum Mol Genet. 2001;10:371–82. doi: 10.1093/hmg/10.4.371. [DOI] [PubMed] [Google Scholar]
- 29.Tufarelli C, Hardison R, Miller W, Hughes J, Clark K, Ventress N, Frischauf AM, Higgs DR. Comparative analysis of the alpha-like globin clusters in mouse, rat, and human chromosomes indicates a mechanism underlying breaks in conserved synteny. Genome Res. 2004;14:623–30. doi: 10.1101/gr.2143604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Klochkov D, Rincón-Arano H, Ioudinkova ES, Valadez-Graham V, Gavrilov A, Recillas-Targa F, Razin SV. A CTCF-dependent silencer located in the differentially methylated area may regulate expression of a housekeeping gene overlapping a tissue-specific gene domain. Mol Cell Biol. 2006;26:1589–97. doi: 10.1128/MCB.26.5.1589-1597.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Razin SV, Kekelidze MG, Lukanidin EM, Scherrer K, Georgiev GP. Replication origins are attached to the nuclear skeleton. Nucleic Acids Res. 1986;14:8189–207. doi: 10.1093/nar/14.20.8189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Simonis M, Klous P, Splinter E, Moshkin Y, Willemsen R, de Wit E, van Steensel B, de Laat W. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C) Nat Genet. 2006;38:1348–54. doi: 10.1038/ng1896. [DOI] [PubMed] [Google Scholar]
- 33.Tolhuis B, Blom M, Kerkhoven RM, Pagie L, Teunissen H, Nieuwland M, Simonis M, de Laat W, van Lohuizen M, van Steensel B. Interactions among Polycomb domains are guided by chromosome architecture. PLoS Genet. 2011;7:e1001343. doi: 10.1371/journal.pgen.1001343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Phillips-Cremins JE, Sauria ME, Sanyal A, Gerasimova TI, Lajoie BR, Bell JS, Ong CT, Hookway TA, Guo C, Sun Y, et al. Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell. 2013;153:1281–95. doi: 10.1016/j.cell.2013.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Splinter E, Heath H, Kooren J, Palstra RJ, Klous P, Grosveld F, Galjart N, de Laat W. CTCF mediates long-range chromatin looping and local histone modification in the beta-globin locus. Genes Dev. 2006;20:2349–54. doi: 10.1101/gad.399506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim TH, Abdullaev ZK, Smith AD, Ching KA, Loukinov DI, Green RD, Zhang MQ, Lobanenkov VV, Ren B. Analysis of the vertebrate insulator protein CTCF-binding sites in the human genome. Cell. 2007;128:1231–45. doi: 10.1016/j.cell.2006.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Essien K, Vigneau S, Apreleva S, Singh LN, Bartolomei MS, Hannenhalli S. CTCF binding site classes exhibit distinct evolutionary, genomic, epigenomic and transcriptomic features. Genome Biol. 2009;10:R131. doi: 10.1186/gb-2009-10-11-r131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nakahashi H, Kwon KR, Resch W, Vian L, Dose M, Stavreva D, Hakim O, Pruett N, Nelson S, Yamane A, et al. A genome-wide map of CTCF multivalency redefines the CTCF code. Cell Rep. 2013;3:1678–89. doi: 10.1016/j.celrep.2013.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cayrou C, Coulombe P, Puy A, Rialle S, Kaplan N, Segal E, Méchali M. New insights into replication origin characteristics in metazoans. Cell Cycle. 2012;11:658–67. doi: 10.4161/cc.11.4.19097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Besnard E, Babled A, Lapasset L, Milhavet O, Parrinello H, Dantec C, Marin JM, Lemaitre JM. Unraveling cell type-specific and reprogrammable human replication origin signatures associated with G-quadruplex consensus motifs. Nat Struct Mol Biol. 2012;19:837–44. doi: 10.1038/nsmb.2339. [DOI] [PubMed] [Google Scholar]
- 41.Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–93. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sanyal A, Lajoie BR, Jain G, Dekker J. The long-range interaction landscape of gene promoters. Nature. 2012;489:109–13. doi: 10.1038/nature11279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Habermann FA, Cremer M, Walter J, Kreth G, von Hase J, Bauer K, Wienberg J, Cremer C, Cremer T, Solovei I. Arrangements of macro- and microchromosomes in chicken cells. Chromosome Res. 2001;9:569–84. doi: 10.1023/A:1012447318535. [DOI] [PubMed] [Google Scholar]
- 44.Barbieri M, Fraser J, Lavitas LM, Chotalia M, Dostie J, Pombo A, Nicodemi M. A polymer model explains the complexity of large-scale chromatin folding. Nucleus. 2013;4:267–73. doi: 10.4161/nucl.25432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nagano T, Lubling Y, Stevens TJ, Schoenfelder S, Yaffe E, Dean W, Laue ED, Tanay A, Fraser P. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature. 2013;502:59–64. doi: 10.1038/nature12593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sexton T, Umlauf D, Kurukuti S, Fraser P. The role of transcription factories in large-scale structure and dynamics of interphase chromatin. Semin Cell Dev Biol. 2007;18:691–7. doi: 10.1016/j.semcdb.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 47.Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes Dev. 2011;25:1010–22. doi: 10.1101/gad.2037511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Martin S, Pombo A. Transcription factories: quantitative studies of nanostructures in the mammalian nucleus. Chromosome Res. 2003;11:461–70. doi: 10.1023/A:1024926710797. [DOI] [PubMed] [Google Scholar]
- 49.Iborra FJ, Pombo A, Jackson DA, Cook PR. Active RNA polymerases are localized within discrete transcription “factories’ in human nuclei. J Cell Sci. 1996;109:1427–36. doi: 10.1242/jcs.109.6.1427. [DOI] [PubMed] [Google Scholar]
- 50.Jackson DA, Iborra FJ, Manders EM, Cook PR. Numbers and organization of RNA polymerases, nascent transcripts, and transcription units in HeLa nuclei. Mol Biol Cell. 1998;9:1523–36. doi: 10.1091/mbc.9.6.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gavrilov AA, Zukher IS, Philonenko ES, Razin SV, Iarovaia OV. Mapping of the nuclear matrix-bound chromatin hubs by a new M3C experimental procedure. Nucleic Acids Res. 2010;38:8051–60. doi: 10.1093/nar/gkq712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cayrou C, Coulombe P, Vigneron A, Stanojcic S, Ganier O, Peiffer I, Rivals E, Puy A, Laurent-Chabalier S, Desprat R, et al. Genome-scale analysis of metazoan replication origins reveals their organization in specific but flexible sites defined by conserved features. Genome Res. 2011;21:1438–49. doi: 10.1101/gr.121830.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sequeira-Mendes J, Díaz-Uriarte R, Apedaile A, Huntley D, Brockdorff N, Gómez M. Transcription initiation activity sets replication origin efficiency in mammalian cells. PLoS Genet. 2009;5:e1000446. doi: 10.1371/journal.pgen.1000446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fujita M, Ishimi Y, Nakamura H, Kiyono T, Tsurumi T. Nuclear organization of DNA replication initiation proteins in mammalian cells. J Biol Chem. 2002;277:10354–61. doi: 10.1074/jbc.M111398200. [DOI] [PubMed] [Google Scholar]
- 55.Hozák P, Cook PR. Replication factories. Trends Cell Biol. 1994;4:48–52. doi: 10.1016/0962-8924(94)90009-4. [DOI] [PubMed] [Google Scholar]
- 56.Cseresnyes Z, Schwarz U, Green CM. Analysis of replication factories in human cells by super-resolution light microscopy. BMC Cell Biol. 2009;10:88. doi: 10.1186/1471-2121-10-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ellis RJ. Macromolecular crowding: obvious but underappreciated. Trends Biochem Sci. 2001;26:597–604. doi: 10.1016/S0968-0004(01)01938-7. [DOI] [PubMed] [Google Scholar]
- 58.Hancock R. A role for macromolecular crowding effects in the assembly and function of compartments in the nucleus. J Struct Biol. 2004;146:281–90. doi: 10.1016/j.jsb.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 59.Marenduzzo D, Micheletti C, Cook PR. Entropy-driven genome organization. Biophys J. 2006;90:3712–21. doi: 10.1529/biophysj.105.077685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nolis IK, McKay DJ, Mantouvalou E, Lomvardas S, Merika M, Thanos D. Transcription factors mediate long-range enhancer-promoter interactions. Proc Natl Acad Sci U S A. 2009;106:20222–7. doi: 10.1073/pnas.0902454106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Botta M, Haider S, Leung IX, Lio P, Mozziconacci J. Intra- and inter-chromosomal interactions correlate with CTCF binding genome wide. Mol Syst Biol. 2010;6:426. doi: 10.1038/msb.2010.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zlatanova J, Caiafa P. CCCTC-binding factor: to loop or to bridge. Cell Mol Life Sci. 2009;66:1647–60. doi: 10.1007/s00018-009-8647-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Phillips JE, Corces VG. CTCF: master weaver of the genome. Cell. 2009;137:1194–211. doi: 10.1016/j.cell.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ohlsson R, Lobanenkov V, Klenova E. Does CTCF mediate between nuclear organization and gene expression? Bioessays. 2010;32:37–50. doi: 10.1002/bies.200900118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ong CT, Corces VG. CTCF: an architectural protein bridging genome topology and function. Nat Rev Genet. 2014;15:234–46. doi: 10.1038/nrg3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lee BK, Iyer VR. Genome-wide studies of CCCTC-binding factor (CTCF) and cohesin provide insight into chromatin structure and regulation. J Biol Chem. 2012;287:30906–13. doi: 10.1074/jbc.R111.324962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ruiz-Narváez EA, Campos H. Evolutionary rate heterogeneity of Alu repeats upstream of the APOA5 gene: do they regulate APOA5 expression? J Hum Genet. 2008;53:247–53. doi: 10.1007/s10038-008-0245-7. [DOI] [PubMed] [Google Scholar]
- 68.Beug H, Doederlein G, Freudenstein C, Graf T. Erythroblast cell lines transformed by a temperature-sensitive mutant of avian erythroblastosis virus: a model system to study erythroid differentiation in vitro. J Cell Physiol Suppl. 1982;1:195–207. doi: 10.1002/jcp.1041130427. [DOI] [PubMed] [Google Scholar]
- 69.Beug H, von Kirchbach A, Döderlein G, Conscience JF, Graf T. Chicken hematopoietic cells transformed by seven strains of defective avian leukemia viruses display three distinct phenotypes of differentiation. Cell. 1979;18:375–90. doi: 10.1016/0092-8674(79)90057-6. [DOI] [PubMed] [Google Scholar]
- 70.Splinter E, de Wit E, van de Werken HJ, Klous P, de Laat W. Determining long-range chromatin interactions for selected genomic sites using 4C-seq technology: from fixation to computation. Methods. 2012;58:221–30. doi: 10.1016/j.ymeth.2012.04.009. [DOI] [PubMed] [Google Scholar]
- 71.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gardiner-Garden M, Frommer M. CpG islands in vertebrate genomes. J Mol Biol. 1987;196:261–82. doi: 10.1016/0022-2836(87)90689-9. [DOI] [PubMed] [Google Scholar]
- 73.Huppert JL, Balasubramanian S. G-quadruplexes in promoters throughout the human genome. Nucleic Acids Res. 2007;35:406–13. doi: 10.1093/nar/gkl1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Orlando V. Mapping chromosomal proteins in vivo by formaldehyde-crosslinked-chromatin immunoprecipitation. Trends Biochem Sci. 2000;25:99–104. doi: 10.1016/S0968-0004(99)01535-2. [DOI] [PubMed] [Google Scholar]
- 75.Kotova ES, Sorokina IV, Akopov SB, Nikolaev LG, Sverdlov ED. Expression of chicken CTCF gene in COS-1 cells and partial purification of CTCF protein. Biochemistry (Mosc) 2013;78:879–83. doi: 10.1134/S0006297913080038. [DOI] [PubMed] [Google Scholar]
- 76.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lobanenkov VV, Nicolas RH, Plumb MA, Wright CA, Goodwin GH. Sequence-specific DNA-binding proteins which interact with (G + C)-rich sequences flanking the chicken c-myc gene. Eur J Biochem. 1986;159:181–8. doi: 10.1111/j.1432-1033.1986.tb09850.x. [DOI] [PubMed] [Google Scholar]
- 79.Klenova EM, Nicolas RH, Paterson HF, Carne AF, Heath CM, Goodwin GH, Neiman PE, Lobanenkov VV. CTCF, a conserved nuclear factor required for optimal transcriptional activity of the chicken c-myc gene, is an 11-Zn-finger protein differentially expressed in multiple forms. Mol Cell Biol. 1993;13:7612–24. doi: 10.1128/mcb.13.12.7612. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.