

Abstract
Aggregation of disordered amyloidogenic peptides into oligomers is the causative agent of amyloid-related diseases. In solution, disordered protein states are characterized by heterogeneous ensembles. Among these, β-rich conformers self-assemble via a conformational selection mechanism to form energetically-favored cross-β structures, regardless of their precise sequences. These disordered peptides can also penetrate the membrane, and electrophysiological data indicate that they form ion-conducting channels. Based on these and additional data, including imaging and molecular dynamic simulations of a range of amyloid peptides, Alzheimer’s amyloid-β (Aβ) peptide, its disease-related variants with point mutations and N-terminal truncated species, other amyloidogenic peptides, as well as a cytolytic peptide and a synthetic gel-forming peptide, we suggest that disordered amyloidogenic peptides can also present a common motif in the membrane. The motif consists of curved, moon-like β-rich oligomers associated into annular organizations. The motif is favored in the lipid bilayer since it permits hydrophobic side chains to face and interact with the membrane and the charged/polar residues to face the solvated channel pores. Such channels are toxic since their pores allow uncontrolled leakage of ions into/out of the cell, destabilizing cellular ionic homeostasis. Here we detail Aβ, whose aggregation is associated with Alzheimer’s disease (AD) and for which there are the most abundant data. AD is a protein misfolding disease characterized by a build-up of Aβ peptide as senile plaques, neurodegeneration, and memory loss. Excessively produced Aβ peptides may directly induce cellular toxicity, even without the involvement of membrane receptors through Aβ peptide-plasma membrane interactions.
Keywords: conformational disorder, intrinsically disordered peptides, ion-channel, cell membrane, molecular dynamics simulations, oligomers, bilayer, β-sheet, protein aggregation, amyloid, cell homeostasis
1. Introduction
Protein misfolding causes abnormal protein aggregates that link to fatal protein deposition diseases including a number of neurodegenerative diseases, such as Alzheimer’s, Huntington’s, Parkinson’s, familial British dementia (FED), familial Danish dementia (FDD), and prion encephalopathies, type II diabetes and eye cataracts.1–6 Amyloid aggregates are aging-related, symptomatically associated with Alzheimer’s disease (AD), which is characterized by the presence of extracellular plaques, intracellular neurofibrillary tangles, and the loss of synapses and neurons in the brain of AD patients.7,8 Despite the prevalence of amyloid-related diseases, their origins, mechanisms of toxicity, and how to prevent, halt or delay amyloidosis are still open questions. Common view has long held that protein misfolding-induced amyloids result in disease either by disrupting regular protein function or by inducing a gain-of-function, often causing pathophysiologic cell response by destabilizing cellular ionic homeostasis.1–3 In solution, many of the amyloid aggregates form by disordered peptides (or fragments) assembling into a common, regular cross-β structures through conformational selection of preferred β conformers.9
The traditional amyloid hypothesis holds that accumulation of β-amyloid (Aβ) peptide in the brain is the primary cause of AD pathogenesis, leading to synapse loss and neuronal cell death.10–14 The extracellular plaques mainly contain Aβ peptides and the intracellular tangles include aggregates of Tau protein.4,5 Amyloid fibrils with a β-sheet pattern are commonly found in these aggregates, deposited both in the extracellular space and in the cytoplasm.15,16 Early studies pointed to fibrillar deposits of Aβ peptides in the extracellular plaques as directly associated with the cause of the disease.16 However, a long term clinical study revealed that even though an experimental drug (AN1792) could remove the extracellular plaques in AD patients, it failed to prevent progressive neurodegeneration.17 The current amyloid cascade hypothesis in AD points to small Aβ oligomers as the main toxic species,18–22 gradually shifting the research focus to Aβ oligomers rather than fibrils.23,24 This hypothesis suggests that early stage symptoms of AD, including reduced synaptic function as well as impairment of learning and memory formation processes, are associated with oligomeric assemblies.5,25 The interaction of Aβ with the cell membrane is a fundamental mechanistic chemical feature leading to AD pathogenesis.26–29 Here, we suggest that small oligomers of Aβ and other disordered amyloidogenic peptides may insert into the membrane and assemble into common β-sheet rich annular structural motifs and review the literature in this light, focusing on Aβ which has abundant data.
The 39–43 (40 and 42 are the most common) amino acids long Aβ peptide is a fragment of the amyloid precursor protein (APP) (Fig. 1A). APP cleavage is driven by β-secretase (BACE) at position 1 outside the cell and γ-secretase at positions 40 or 42 within the cell membrane (Fig. 1B). While the production of Aβ1-40 is energetically more favorable than Aβ1-42, Aβ1-42 is more toxic to neurons than Aβ1-40.30 In addition to the full-length Aβ1-40/42 peptide, N-terminal truncated fragments are also formed via cleavage of the APP by β′- and γ-secretases producing the Aβ11-40/42 peptide (Fig. 1C), and α - and γ-secretases cleavage yields Aβ17-40/42 peptide (Fig. 1D).11,31 Since these truncated peptides were putatively treated as nonpathogenic species, drugs to inhibit BACE were used to block production of the full-length Aβ peptides, and at the same time, enhance the production of the N-terminal truncated Aβ peptides.32 However, recent studies using complementary techniques of atomic force microscopy (AFM), molecular dynamics (MD) simulations, planar lipid bilayer (PLB), cell calcium imaging, neuritic degeneration, and cell death assays demonstrated that the N-terminal truncated Aβ peptides, Aβ17-42 (p3) and Aβ9-42 (N9), formed toxic ion channels in the lipid bilayers.33–38 In particular, the p3 peptide was reported to induce toxicity in AD and is known to be the main constituent of cerebellear preamyloid lesions in Down Syndrome (DS).39–41
Figure 1.

Productions of β-amyloid (Aβ) via various cleavages. (A) A cartoon representing the cleavage process by α-, β-, β′- and γ-secretases of amyloid precursor protein (APP). The Aβ1-42 domain is shown in gray with the sequence and numbering of the amino acids. In single letter amino acid codes, hydrophobic, polar/Gly, positively charged, and negatively charged residues are colored white, green, blue, and red, respectively. Various Aβ fragments are processed by different secretase combinations. (B) Amyloidogenic fragment of Aβ1-40/42 by β- and γ-secretases, and non-amyloidogenic fragments of (C) Aβ11-40/42 by β′- and γ-secretases and (D) Aβ17-40/42 by α- and γ-secretases. These cartoons were inspired by previous publication.31 (B and D from Jang et al., 2010,37 are reprinted with permission).
In spite of over a century of research, there is still no strategy to prevent or cure the AD.42,43 An important reason for this is the lack of knowledge of a high resolution structure based on x-ray diffraction for the toxic amyloid oligomers, hampering the development of therapeutic drugs.44 Solution and solid state NMR (ssNMR), without and with coordinated metals such as zinc and copper, indicated a range of conformations,45–52 as did other spectroscopic techniques,53,54 and molecular dynamics simulations.55–62 The different amyloid states emerging from these underscore the chemical nature of Aβ: a disordered peptide with energetically fairly similar conformational states separated by low barriers, with the prevailing states highly sensitive to conditions and the chemical environment: solution or bilayer, peptide concentration, presence of ions, membrane composition, cholesterol, metals, presence of other amyloids such as of Tau protein known to co-aggregate with Aβ, and other proteins, and more. Further, slight sequence alterations, such as those involving single point mutations and truncated peptides, taking place under physiological conditions and in disease, can also be expected to shift the free energy landscape of amyloids.63,64 The different conformations may self-assemble into multiple oligomeric cross-β seed states, propagating into a broad range of fibrils, differing in their organizations and dimensions.65–70 It can be expected that the range of currently observed conformations will increase. On a different resolution scale, a substantial body of evidence obtained by AFM techniques illustrated assembled channel-like oligomer structures for a series of different amyloids.21,36,71–78 Electron microscopy (EM) also provided images of amyloid oligomers with doughnut-like structures.79–81 Given these predicaments, the problem of predicting amyloid conformations using MD simulations, coarse grained, implicit, or explicit solvent description- in the solution state, without and with metal ions, on and in the membrane, in the presence and absence of AD-related mutations, and truncated fragments has drawn much attention.82–102 Recently, a series of MD simulations provided insight into the molecular conformations of oligomeric Aβ channel structures at atomic-level resolution,33–38,77,78,103–106 exhibiting that Aβ channels are heterogeneous, consisting of β-sheet-rich subunits with morphologies and dimensions in good agreement with the imaged AFM channels.21,72,73
We propose that heterogeneous, disordered amyloidogenic peptides with different sequences frequently insert into the membrane and assemble into channel structural motifs. Insertion may depend on the membrane composition and net charge, which varies across tissues and organism types. We center on Aβ channel structures derived from modeling and MD simulations for Aβ sequences and monomer morphologies, and relate these to AD. Similar structural motifs were obtained by simulations for other amyloidogenic peptides (a fragment of β2-microglobulin (K3)75 and the human islet amyloid polypeptide (hIAPP)84,107 and by AFM for a still broader range.21 They were also obtained for the cytolytic antimicrobial peptide (AMP), protegrin-1 (PG-1),108,109 and for a synthetic peptide which self-assembles into a hydrogel.110 Unregulated toxic ion channels consisting of β-rich oligomers annularly associated and supported by their bilayer environment may be a preferred state for heterogeneous disordered peptides. In solution, aggregated amyloid states typically present the cross-β structures. In the membrane, small β-sheet rich subunits may insert and if their concentration is sufficiently high, oligomerize to form circular organization. In both, conformational and organizational details vary with the sequence and physical environment. The MD simulations described below were performed by the CHARMM111 program with the NAMD code112 on the Biowulf cluster (http://biowulf.nih.gov) at the NIH.
2. Mechanisms of Aβ toxicity
Pathological amyloid folding alters the three-dimensional conformations from soluble native structures113,114 to insoluble non-native β-sheet-rich aggregates,115,116 ranging from small oligomers to fibers.117 Upon binding to the cell membrane, these conformational changes, catalyzed by the membrane, disrupt cellular function inducing cytotoxicity.4,5,118 Aβ toxicity can be a direct consequence of ion channel formation.119 Amyloid channels consist of small oligomers with a β-sheet motif, self-assembled around an aqueous cavity in the lipid environment. The formation of water cavity provides passage for unregulated ionic currents across the lipid membrane, destabilizing cellular ionic homeostasis. In the early 90’s, Arispe et al.120–124 first reported electrophysiology data of Aβ ion channels in the PLB (or BLM for black lipid membrane) neuron experiments, proposing the amyloid ion channel hypothesis. The measured ionic flux across the reconstituted membrane detected the emergence of stepwise ionic currents over time pointing to ion channels (Fig. 2A). The Aβ channels were cation-selective, voltage-independent and blocked by zinc73,124–127 (Fig. 2B). Unlike typical regulated ion channels, Aβ channels exhibited multiple, large single channel conductances in the range of 0.4–4 nS, inducing an abrupt change in the cellular ionic concentration, leading to significant disruption of the membrane potential and loss of cellular homeostasis. Similar observations were made for other channel-forming amyloids including IAPP,128–133 prion protein fragment,134 polyglutamine,135 β2-microglobulin,136 transthyretin (TTR),137 and serum amyloid A (SAA).138
Figure 2.

An example of stepwise current feature across planar lipid bilayer (PLB, or BLM representing “black lipid membrane”) membrane produced by amyloid channels. (A) The electrophysiological activity of Aβ1-42 ion channels embedded in PLB. (B) Inhibition of channel activity by Zn2+ addition. Time of Zn2+ addition (2 mM) is marked by arrow. (From Capone et al., 2012,103 reprinted with permission).
Indirect large oligomer-induced toxicity effects relate to neuronal oxidative stress, inflammation, or cell membrane-mediated signaling pathways.139–141 An alternative hypothesis to explain disruption of cellular ionic homeostasis suggested that large amyloid oligomers cause mechanical damage to the cell membrane inducing membrane thinning with consequent nonselective ion leakage through the low dielectric barrier in the locally perturbed membrane.142,143 Recently, amyloid fiber growth on the membrane surface was found to produce fragmentation of the cell membrane, inducing non-specific leakages.144–147 All of these Aβ-induced effects, whether via channel formation, receptor-mediated pathways, or membrane thinning destabilized the cellular ionic homeostasis, primarily by increased levels of intracellular calcium.
3. Structures of Aβ peptide
3.1 Aβ1-42 vs. Aβ1-40
Early nuclear magnetic resonance (NMR) data suggested that Aβ monomers were generally disordered in aqueous environments,114,148 but recent studies indicate that they are partially-folded α-helical structures.149–151 When aggregated into oligomers or fibrils, however, the helical intermediates convert into β-sheet-rich structures. Lührs et al.152 reported the Aβ1-42 fibril structure from a combination of hydrogen/deuterium-exchange NMR data, side-chain packing constraints from pairwise mutagenesis, ssNMR and EM (pdb id: 2BEG). They obtained the coordinates for residues 17–42, while the N-terminal coordinates (residues 1–16) were missing due to disorder (Fig. 3A). The monomer conformation in the fibril was U-shaped with a β-strand-turn-β-strand motif and a turn located at Ser26-Ile31 with an intermolecular salt bridge Asp23/Lys28. The U-shaped monomer topology of Aβ peptides has been first introduced for solvated oligomers of Aβ16-35 through modeling and MD simulations.153 The peptides with the β-strand-turn-β-strand motif assembled in register presenting a parallel organization with the intramolecular salt bridge of Asp23/Lys28. Similar U-shaped peptides could be observed in small Aβ1-40 protofibrils (pdb ids: 2LMN and 2LMO) defined by solid state NMR (ssNMR).67 The U-shaped Aβ1-40 peptide had a turn at Asp23-Gly29 and the same salt bridge as the Aβ16-35 peptide (Fig. 3B). The N-terminal coordinates (residues 1–8) were missing due to disorder. Recently, another U-shaped Aβ1-40 peptide154 with a turn at Val24-Ala30, which is similar to the previous Aβ1-40 model,67 was reported. Combined, it appears that the more C-terminal turn at Asp23-Gly29 is an intrinsic turn of Aβ1-40. In contrast, Aβ1-42 preferentially adopted the less C-terminal turn at Ser26-Ile31.152 Variants with additional turns near the C-terminal have also been detected, primary among these are those seeded from the brain extracts of two Alzheimer’s disease patients presenting a triangular shape organization155 reminiscent of earlier triangular organizations.65,70
Figure 3.

Three-dimensional structures of Alzheimer’s amyloids. (A) The pentameric Aβ1-42 fibril structure obtained from a combination of hydrogen/deuterium-exchange nuclear magnetic resonance (NMR) data, side-chain packing constraints from pairwise mutagenesis, solid state NMR (ssNMR) and electron microscopy (EM) (pdb id: 2BEG).152 The coordinates for residues 1–16 were missing due to disorder. (B) The ssNMR model for small Aβ1-40 protofibrils (pdb ids: 2LMN).67 The coordinates for residues 1–8 were missing due to disorder. Residues at both termini are marked.
3.2 Two Aβ1-42 conformers derived from the NMR-based structures
The NMR-derived models of small Aβ1-40/42 protofibrils only provided the N-terminal truncated Aβ coordinates due to conformational disorder.67,152,154 In order to paint a complete picture for Aβ toxicity involving full-length Aβ peptides, structural information of the β-sheet peptide is needed.156–159 Recently, computational modeling provided two U-shaped monomer conformations of Aβ1-42 based on the NMR structures.77,78,103,104 In the MD simulations, the Aβ1-16 coordinates in the absence of Zn2+ (pdb id: 1ZE7)47 were used for the missing N-terminal portions of the peptides.67,152 For each combination of the N-terminal structure with the U-shaped motifs of Aβ17-42 and Aβ9-40, two Aβ1-42 conformers were generated (Fig. 4). Conformer 1 has a turn at Ser26-Ile31, and conformer 2 at Asp23-Gly29. In the latter conformer, two C-terminal residues, Ile41 and Ala42 were added to create Aβ1-42. Both Aβ1-42 conformers retained the U-shaped β-strand-turn-β-strand motif and can be divided into four domains: the N-terminal fragment (residues 1–16 and 1–8 for conformer 1 and 2, respectively), pore-lining β-strand (residues 17–25 and 9–22 for conformer 1 and 2, respectively), turn (residues 26–31 and 23–29 for conformer 1 and 2, respectively), and C-terminal β-strand (residues 32–42 and 30–42 for conformer 1 and 2, respectively).
Figure 4.

A cartoon representing the constructions of the full-length Aβ1-42 peptides. The N-terminal truncated Aβ monomers are U-shape with the β-strand-turn-β-strand motif. The missing N-terminal portions of these NMR-derived Aβ peptides are recovered with the coordinates from the solution structure of Aβ1-16 (pdb id: 1ZE7).47 By covalently connecting the N-terminal to the truncated Aβ peptides, two Aβ1-42 conformers (conformer 1 and 2) with different turns can be generated.
4. Aβ channels in the lipid bilayers
4.1 N-terminal truncated Aβ17-42 and Aβ9-42 channels
APP cleavages by combinations of β′-/γ-secretases and α-/γ-secretases produce Aβ11-42 and Aβ17-42 (p3) peptides, respectively (Fig. 1C,D).11,31 Adding two more residues to the N-terminal of Aβ11-42 peptide obtains the Aβ9-42 (N9) peptide. The pioneering computational studies of amyloid ion channels in the lipid bilayer have begun with these N-terminal truncated Aβ peptides.33–38 The NMR-based U-shaped Aβ peptides67,152 were directly employed in the modeling of the channels. Annular β-sheets in the channel and barrel topologies were initially constructed in a lipid environment as starting points for the explicit MD simulations. To construct the initial channel structure with conventional β-strands arrangement, 12-36 U-shaped peptides were inserted without inclination with respect to the membrane normal, generating the channel topology (Fig. 5A).33–37 To construct the barrel structure, the U-shaped peptides were inclined ~37° relative to the pore axis and then a 12–20 fold rotational symmetry operation was performed with respect to the pore axis, creating the barrel topology (Fig. 5B).38 In both topologies, the polar/charged N-terminal β-strands (residues 17–25 for p3 and 9–22 for N9 topologies) encompassed the water pore, and the hydrophobic C-terminal β-strands (residues 32–42 for p3 and 30–42 for N9 topologies) faced lipids. Other β-sheet-forming peptides also exhibited the channel and barrel topologies (Fig. 5C–E).
Figure 5.

Conventional β-sheet channel vs. β-barrel designs. (A) The conventional β-sheet channel has the β-strands that orient parallel to the membrane normal, (B) while the β-strands that orient obliquely to the membrane normal generate β-barrel structure. Above examples are shown for the p3 (Aβ17-42) channel and barrel. Other examples of the β-sheet channel and barrel formed by the U-shaped K3 (a fragment of β2-microglobulin) peptide and by the PG-1 and MAX β-hairpins. (C) The 24-mer channel embedded in the DOPC bilayer in stereo view.75 (D) The 8-mer PG-1 channels with the antiparallel and parallel β-strand arrangements.108 (E) The 10-mer antiparallel MAX channels and barrels in the NCCN and NCNC packing modes (from Gupta et al., 2013,110 reprinted with permission).
The annular channels/barrels gradually relaxed toward heterogeneous shapes in the lipid bilayer during the simulations (Fig. 6). The simulations illustrated that the Aβ channels and barrels consist of loosely attached β-sheet-rich subunits with the morphologies and dimensions in good agreement with the imaged AFM channels.36 The outer dimensions and the pore diameters for the p3 and N9 channels/barrels from the simulations depended on the number of peptides assembled in the channels/barrels (Tables 1 and 2); however, the number of subunits which were formed during the simulations reflected the fluidic bilayer dynamics. Remarkably, the MD simulations presented optimal toxic ion channel sizes ranging between 16 and 24 monomers.35,37 This range was also found to hold for other toxic β-sheet channels; 24-mer K3 (a fragment of β2-microglobulin) channels with 24 β-strands,75 8-/10-mer protegrin-1 (PG-1) channels with 16–20 β-strands,108,109 18-/24-mer human islet amyloid polypeptide (hIAPP) channels with 18–24 β-strands,107 and 10-mer for the synthetic hydrogel-forming peptide MAX barrels with 20 β-strands.110
Figure 6.

Truncated Aβ channel/barrel conformations in the lipid bilayer. Averaged pore structures calculated by the HOLE program184 embedded in the averaged channel/barrel conformations during the simulations for the p3 (Aβ17-42) (A) channels and (B) barrels, and the N9 (Aβ9-42) (C) channels and (D) barrels. (A from Jang et al., 2010,37 B and D from Jang et al., 2010,38 and C from from Jang et al., 2009,35 reprinted with permission).
Table 1.
Structural features of the p3 (Aβ17-42) channels/barrels embedded in various lipid bilayers.
| p3 (Aβ17-42) | Outer diameter (nm) | Pore diameter (nm) | Pore status | Number of subunits | Embedded lipid | Reference |
|---|---|---|---|---|---|---|
| 12-mer channel | ~6.1 | ~0.8 | collapsed | - | DOPC | 37 |
| 16-mer channel | ~6.8–6.9 | ~1.7 | opened | 4, 5 | DOPC | 36,37 |
| 20-mer channel | ~7.4 | ~1.9 | opened | 5 | DOPC | 37 |
| 24-mer channel | ~8.0 | ~2.5–2.7 | opened | 3,5 | DOPC POPC/POPG=4:1 |
33,34,37 |
| 36-mer channel | ~9.3 | ~3.9 | reduced | 6 | DOPC | 37 |
| 12-mer barrel | ~6.7 | ~1.8 | opened | 3 | DOPC | 38 |
| 16-mer barrel | ~6.8 | ~1.5 | opened | 5 | DOPC | 38 |
| 20-mer barrel | ~7.3–7.9 | ~1.7–2.2 | opened | 4,5 | DOPC | 38 |
Table 2.
Structural features of the N9 (Aβ9-42) channels/barrels embedded in various lipid bilayers.
| N9 (Aβ9-42) | Outer diameter (nm) | Pore diameter (nm) | Pore status | Number of subunits | Embedded lipid | Reference |
|---|---|---|---|---|---|---|
| 12-mer channel | ~6.6 | ~0.6 | collapsed | 2 | DOPC | 35 |
| 16-mer channel | ~7.3–7.4 | ~1.5–1.7 | opened | 5 | DOPC | 35,36 |
| 20-mer channel | ~7.8 | ~1.9 | opened | 3 | DOPC | 35 |
| 24-mer channel | ~7.8–8.3 | ~2.2–2.5 | opened | 4,5 | DOPC POPC/POPG=4:1 |
34,35 |
| 36-mer channel | ~10.3 | ~3.7 | reduced | 6 | DOPC | 35 |
| 12-mer barrel | ~6.6 | ~1.3 | opened | 3 | DOPC | 38 |
| 16-mer barrel | ~7.2 | ~1.6 | opened | 4 | DOPC | 38 |
| 20-mer barrel | ~7.7–8.0 | ~1.9 | opened | 5,6 | DOPC | 38 |
4.2 L- and D-enantiomers Aβ1-42 channels
The indirect mechanism for Aβ-mediated destabilization of ionic homeostasis suggested that Aβ binds to cell membrane receptors via stereospecific interactions, resulting in opening existing ion channels or transporters.115,139 Cell binding studies by Ciccotosto et al.160 showed that although both all L- and all D-amino acids Aβ1-42 peptides (L-Aβ1-42 and D-Aβ1-42 peptides) bound to cultured cortical neurons, only the L-Aβ1-42 peptide was neurotoxic, suggesting stereospecific interactions of L-Aβ1-42 peptide. However, stereospecificity could be studied through comparison of the biological activities of the L- and D-enantiomers, since putative cellular receptors would not bind the D-Aβ due to the lack of conformational fitting. Recent comprehensive studies including AFM, PLB, and MD simulations demonstrated that the D-Aβ isomer formed ion channel in the bilayer with size, shape, and ion conductance behavior indistinguishable from the wild type L-Aβ isomer.77,103 This suggested that Aβ toxicity occurred via a receptor independent, nonstereoselective mechanism. In the computational studies, the L- and D-enantiomers ion channels were modelled using two Aβ1-42 conformers (Fig. 4) with the β-strand-turn-β-strand motif. The D-Aβ1-42 conformers were mirror images of L-Aβ1-42, obtained by reflecting the L-coordinates with respect to the reference plane (Fig. 7). The conformers retained the U-shaped β-strand-turn-β-strand motif similar to their L-Aβ1-42 counterparts, regardless of their chirality. Conformer 1 D-Aβ1-42 had a turn at Ser26-Ile31 and conformer 2 D-Aβ1-42 at Asp23-Gly29, following the wild type ssNMR models. 18-mers, L- and D-enantiomers Aβ1-42 barrels were simulated in an anionic lipid bilayer containing 1,2-dioleoyl-sn-glycero-3-phosphoserine (DOPS) and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE) with a mole ratio DOPS:POPE=1:2, and a zwitterionic lipid bilayer composed of 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC).
Figure 7.

Schematic diagrams representing the all L- and all-D-amino acids Aβ1-42 peptides with different conformers. The coordinates of all D-amino acids Aβ1-42 are mirror-imaged coordinates of all L-amino acids Aβ1-42 for (A) the conformer 1 and (B) the conformer 2. The standard CHARMM111 force can be directly used for D-amino acids. However, the parameters include the dihedral angle cross-term map (CMAP), which, for D-amino acids, needs to be corrected since the map was constructed for L-amino acids. To simulate D-amino acids, corrected CMAP data for D-amino acids should be applied via reflecting the phi-psi CMAP matrix for L-amino acids.77,103,110
During the simulations, the Aβ barrels were gradually relaxed through their interaction with the surrounding lipids, presenting the assembled-subunits channel morphology (Fig. 8A–D). Regardless of the D- and L-amino acids chirality, both Aβ conformers and isomers preserved the barrel conformation in a way such that the membrane-embedded, pore-lining β-strands encompassed the solvated pore, and the C-terminal β-stands interacted with lipid tails. The N-terminal portions were disordered and stayed extramembranous. The computational studies verified that D-Aβ1-42 was able to form ion channels and be active independent of stereospecific receptor interactions, presenting indistinguishable pore structures formed by both isomers as imaged by AFM (Fig. 8E). No differences in the calculated outer and pore dimensions for the Aβ1-42 barrels between different Aβ conformers and/or between different Aβ isomers were observed (Table 3).
Figure 8.

Full-length Aβ1-42 barrel conformations in the lipid bilayer. Simulated barrel structure with an embedded pore structure and highlighted subunits for (A) the conformer 1 and (B) the conformer 2 D-Aβ1-42 barrels, and (C) the conformer 1 and (D) the conformer 2 L-Aβ1-42 barrels. (E) High resolution atomic force microscopy (AFM) images of D- and L-Aβ1-42 reconstituted in the lipid bilayer. The number of subunits is resolved and indicated for each channel. (From Connelly et al., 2012,77 reprinted with permission).
Table 3.
Structural features of the 18-mer Aβ1-42 barrels composed of both Aβ conformer 1 and 2 and/or both L- and D-Aβ isomers in various lipid bilayers.
| 18-mer Aβ1-42 barrel | Outer diameter (nm) | Pore diameter (nm) | Pore status | Number of subunits | Embedded lipid | Reference |
|---|---|---|---|---|---|---|
| Conformer 1 (L-isomer) | ~7.9–8.3 | ~1.8–2.2 | opened | 4 | DOPC DOPS/POPE=1:2 |
77,78,106 |
| Conformer 1 (D-isomer) | ~7.8 | ~1.9 | opened | 4 | DOPS/POPE=1:2 | 77 |
| Conformer 2 (L-isomer) | ~8.0–8.1 | ~1.9–2.2 | opened | 3 | DOPC DOPS/POPE=1:2 |
77,78,106 |
| Conformer 2 (D-isomer) | ~8.2 | ~2.1 | opened | 5 | DOPS/POPE=1:2 | 77 |
4.3 Aβ mutants: F19P, F20C, and Osaka mutant (ΔE22) channels
Several point mutations linked to AD occur naturally in the Aβ peptide, clustered around the central region of the peptide.11,161 These include the Flemish (A21G),162 Artic (E22G),163 Italian (E22K),164 Dutch (E22Q),162,164,165 and Iowa (D23N)166 mutants. Since they affect the salt bridge in the turn region, several studies, experimental and computational, probed these substitutions in a water environment to understand their conformational consequences.55,167–174 In addition, a designed synthetic proline substitution in the central region for a Phe residue is of particular interest, since proline is a β-sheet breaker, preventing the Aβ propagation into β-sheet-rich oligomers or fibrils.175,176 The distinct behavior of the proline substitution in the Aβ channels indicated that substitution of Phe19 with Pro (F19P) in both truncated p3 (Aβ17-42) and full-length Aβ1-42 channel/barrel conformations prevented pore activity and hence cellular toxicity.36,78,104 Computational studies of these F19P mutant channels/barrels verified that kinks at Pro19 destabilized an inner β-sheet formed by the pore-lining β-strands (Fig. 9A). As a result, the F19P substitution induced collapsed pores which prevented ions permeating across the bilayer. However, unlike the collapsed pore induced by the F19P substitution, the F20C substitution preserved the solvated pore with channel activity comparable to the wild type (Fig. 9B). Both F19P and F20C, channels and barrels, presented outer diameters in the wild type range, while the pore sizes significantly decreased for the F19P and slightly reduced for the F20C (Table 4).
Figure 9.
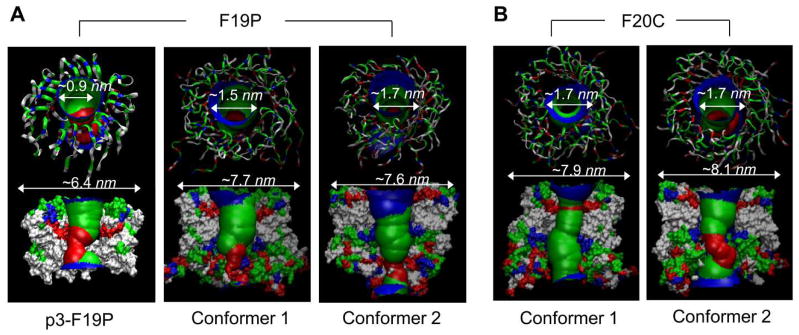
Aβ mutant channels/barrels in the lipid bilayers. (A) Simulated channel/barrel structures with an embedded pore structure for the p3-F19P (from the truncated Aβ17-42) mutant channel (left panel, from Jang et al., 2010,36 reprinted with permission) and for the conformer 1 and the conformer 2 F19P (from the full-length Aβ1-42) mutant barrels (middle and right panels, from Connelly et al., 2012,78 reprinted with permission). (B) Simulated barrel structures with an embedded pore structure for the conformer 1 and the conformer 2 F20C (from the full-length Aβ1-42) mutant barrels (from Connelly et al., 2012,78 reprinted with permission).
Table 4.
Structural features of the p3 (Aβ17-42) F19P mutant channel, the full-length (Aβ1-42) F19P and F20C barrels, and the Osaka mutant (ΔE22) barrels with an absence of Glu22 in various lipid bilayers.
| Aβ mutant channel/barrel | Outer diameter (nm) | Pore diameter (nm) | Pore status | Number of peptides | Embedded lipid | Reference |
|---|---|---|---|---|---|---|
| p3-F19P channel | ~6.4 | ~0.9 | clogged up | 16 | DOPC | 36 |
| F19P barrel (Conformer 1) | ~7.7 | ~1.5 | clogged up | 18 | DOPS/POPE=1:2 | 78,104 |
| F19P barrel (Conformer 2) | ~7.6 | ~1.7 | collapsed | 18 | DOPS/POPE=1:2 | 78,104 |
| F20C barrel (Conformer 1) | ~7.9 | ~1.7 | opened | 18 | DOPS/POPE=1:2 | 78,104 |
| F20C barrel (Conformer 2) | ~8.1 | ~1.7 | partially collapsed | 18 | DOPS/POPE=1:2 | 78,104 |
| ΔE22 barrel (Conformer 1) | ~7.9 | ~2.1 | opened | 18 | DOPC | 106 |
| ΔE22 barrel (Conformer 2) | ~7.6 | ~1.5 | reduced | 18 | DOPC | 106 |
Another mutation in the central region of the Aβ peptide, nicknamed the Osaka mutation, is a deletion of Glu22 (ΔE22).177 It was known that Aβ mutants with a familial Alzheimer’s disease (FAD)-linked point substitutions at Glu22 were toxic species.178 Like these point substitutions, the complete elimination of the Glu22 position, rather than an amino acid substitution, is still linked to FAD. Recent computational studies provided a membrane-bound conformation of the ΔE22 barrel in atomic-level detail,106 demonstrating that the mutant barrels presented similar morphologies and dimensions as those of the wild type Aβ1-42 (Fig. 10 and Table 4). This suggested that the Osaka mutant could directly relate to the Aβ ion channel-mediated mechanism as observed for the wild type Aβ peptide in AD pathology.
Figure 10.

Osaka mutant (ΔE22) barrels in the lipid bilayers. (A) Monomer conformations of the Osaka mutant with different turns at Ser25-Ile30 (conformer 1) and Asp22-Gly28 (conformer 2). (B) The U-shaped monomer conformations are similar to the wild type Aβ1-42 peptides with different turns at Ser26-Ile31 (conformer 1) and Asp23-Gly29 (conformer 2). Simulated barrel structures with an embedded pore structure for (C) the conformer 1 and (D) the conformer 2 Osaka mutant (ΔE22) barrels. (From Jang et al., 2013,106 reprinted with permission).
4.4 AβpE3-42 channels
Pyroglutamate (pE) modified Aβ peptides, in particular, the AβpE3-42 peptide have been increasingly associated with enhanced toxicity, possibly due to its increased stability and higher aggregation propensity.179,180 This peptide is generated post-translationally by cleavage of the first two N-terminal amino acids of Aβ1-42, leaving an exposed glutamate (E) residue in position 3. The pyroglutamate (pE) ring is subsequently generated by intramolecular dehydration catalyzed by the glutaminyl cyclase (QC) enzyme.179 Our preliminary AFM results indicate that the AβpE3-42 peptide is able to form a channel in the lipid bilayer, with similar characteristics and dimensions as the channels observed in previous studies.77,78 Subsequent MD studies also provide a model of AβpE3-42 barrel in atomic details (Fig. 11).
Figure 11.

Computational modelling of pyroglutamate (pE) modified Aβ barrels. (A) Molecular structure of pyroglutamate (pE) at position 3. (B) Monomer conformations of AβpE3-42 (upper) and AβpE3-40 (lower) peptides. (C) Modelled structures of 18-mer AβpE3-42 (left) and AβpE3-40 (right) barrels.
5. Conclusions
Although the molecular mechanisms of Aβ that lead to cellular dysfunction are still unclear, they involve interactions of oligomeric species with the cell membrane.19 Lipids and amyloid peptides can reciprocally affect their respective conformations. Aβ peptides have the potential to affect the structural integrity of the membrane, ultimately leading to cytotoxicity. Conversely, lipid membranes can promote the conversion of amyloid monomers into β-sheet-rich toxic oligomeric species.29,181 Aβ membrane binding and insertion suggested that β-sheet oligomers spontaneously inserted to form membrane-bound aggregates.159 These aggregates in the membrane were cytotoxic, and their presence, validated by electrophysiological recordings, set the amyloid channel hypothesis.120,121 Amyloid channels were stable over time with lifetimes ranging from several minutes to hours. The cationic selective channels were voltage independent and blocked by zinc, presenting multiple interconverting conductance levels, suggesting that Aβ-mediated permeabilization is specifically caused by formation of intrinsic calcium permeable membrane pores.182
Several AFM structural studies have shown doughnut-like amyloid channels in lipid bilayers with outer diameters typically raging between 8–12 nm and inner diameters of ~2 nm.21,72–74 Unlike typical ion channels, which have a well-defined number of subunits, amyloid channels present a varying number of subunits, ranging between trimers and hexamers.77 Subsequent MD simulations provided amyloid channel conformations in atomic-level detail.33–38,75,77,78,103,104,106 These computational channels were modeled using the U-shaped Aβ peptide with the β-strand-turn-β-strand motif. In the simulated channels, the solvated pore was lined by the central β-strands containing polar/charged residues, while the hydrophobic C-terminal strands interacted predominantly with the lipids. The modeled channels exhibited the water pore, wide enough for multiple ions to simultaneously enter and exit.
Given their prevalence, disordered states have been of increasing interest. Amyloidogenic deposits typically arise from disordered peptide species or fragments of amyloidogenic proteins. Disordered states are characterized by broad ensembles with no clear, highly populated state. Within the ensembles there are also β-rich conformers, and these associate through hydrophobic interactions and favorable generic backbone hydrogen bonds. The regular self-assembled structures permit seed formation which can propagate to form long and branched aggregates. The common occurrence of these in solution as a typical organization, irrespective of precise sequence reflects the cross-β stability. Peptides have long been known to insert into membranes. Here we posited that in membrane environments disordered peptides similarly tend to form common favored motifs. While here we focused on Aβ, we detailed their occurrence for Aβ variants – mutational and truncated species, as well as other amyloidogenic peptides, a cytolytic peptide and a synthetic gel-forming peptide. The fact that if the concentration of the disordered peptides is sufficiently high they tend to form annular organizations in membrane environments is not surprising: channels allow outer facing hydrophobic residues to favorably interact with the lipid membrane environment, and charged/polar residues to face a solvated water pore. Certain polar lipid-facing side chains in the β-strand can still be satisfied by some water molecules that permeate the membrane. This dislike of a membrane environment by the charged/polar surfaces already induces curvature to the oligomers, shrinking these exposed surfaces and expanding the membrane-loving hydrophobic, outer-surfaces, preorganizing the oligomers for channel formation. On the down side, while energetically favorable, such channels are toxic, since they permit unregulated passage of ions in their solvated pores, thus disturbing the cellular ionic homeostatis.
Although there has been significant support for the Aβ channel conformation consisting of the U-shape motif, this does not necessarily imply that such conformational species are always the preferred conformational states. The amyloid landscape is highly heterogeneous and different channel conformations may be populated.9,44,63 Highly polymorphic membrane-permeated channels could evolve from different seed formations.105,159 Whether these channels assembled in identical shape in the membrane or not, abundant structural evidence for Aβ channels with the β-sheet morphology by AFM and MD studies strongly suggests that a direct mechanism for the loss of cell ionic homeostasis in AD may also be operable in the cell.126,183
Acknowledgments
This project has been funded in whole or in part with Federal funds from the Frederick National Laboratory for Cancer Research, National Institutes of Health, under contract HHSN261200800001E. This research was supported [in part] by the Intramural Research Program of NIH, Frederick National Lab, Center for Cancer Research. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products or organizations imply endorsement by the US Government. All simulations had been performed using the high-performance computational facilities of the Biowulf PC/Linux cluster at the National Institutes of Health, Bethesda, MD (http://biowulf.nih.gov).
References
- 1.Cohen FE, Kelly JW. Nature. 2003;426:905–909. doi: 10.1038/nature02265. [DOI] [PubMed] [Google Scholar]
- 2.Temussi PA, Masino L, Pastore A. Embo J. 2003;22:355–361. doi: 10.1093/emboj/cdg044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dobson CM. Nature. 2003;426:884–890. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- 4.Chiti F, Dobson CM. Annu Rev Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 5.DeToma AS, Salamekh S, Ramamoorthy A, Lim MH. Chem Soc Rev. 2012;41:608–621. doi: 10.1039/c1cs15112f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eisenberg D, Jucker M. Cell. 2012;148:1188–1203. doi: 10.1016/j.cell.2012.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Monsonego A, Zota V, Karni A, Krieger JI, Bar-Or A, Bitan G, Budson AE, Sperling R, Selkoe DJ, Weiner HL. J Clin Invest. 2003;112:415–422. doi: 10.1172/JCI18104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Di Paolo G, Kim TW. Nature Rev Neurosci. 2011;12:284–296. doi: 10.1038/nrn3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ma B, Nussinov R. Journal of molecular biology. 2012;421:172–184. doi: 10.1016/j.jmb.2011.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tanzi RE, Bertram L. Cell. 2005;120:545–555. doi: 10.1016/j.cell.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 11.Iversen LL, Mortishiresmith RJ, Pollack SJ, Shearman MS. Biochem J. 1995;311:1–16. doi: 10.1042/bj3110001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jakob-Roetne R, Jacobsen H. Angewandte Chemie International Edition. 2009;48:3030–3059. doi: 10.1002/anie.200802808. [DOI] [PubMed] [Google Scholar]
- 13.Hardy Current Alzheimer Research. 2006;3:71–73. doi: 10.2174/156720506775697098. [DOI] [PubMed] [Google Scholar]
- 14.Hardy JA, Higgins GA. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 15.Sunde M, Blake C. Adv Protein Chem. 1997;50:123–159. doi: 10.1016/s0065-3233(08)60320-4. [DOI] [PubMed] [Google Scholar]
- 16.Sipe JD, Cohen AS. J Struct Biol. 2000;130:88–98. doi: 10.1006/jsbi.2000.4221. [DOI] [PubMed] [Google Scholar]
- 17.Holmes C, Boche D, Wilkinson D, Yadegarfar G, Hopkins V, Bayer A, Jones RW, Bullock R, Love S, Neal JW, Zotova E, Nicoll JAR. Lancet. 2008;372:216–223. doi: 10.1016/S0140-6736(08)61075-2. [DOI] [PubMed] [Google Scholar]
- 18.Bernstein SL, Dupuis NF, Lazo ND, Wyttenbach T, Condron MM, Bitan G, Teplow DB, Shea JE, Ruotolo BT, Robinson CV, Bowers MT. Nature Chem. 2009;1:326–331. doi: 10.1038/nchem.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Butterfield SM, Lashuel HA. Angewandte Chemie International Edition. 2010;49:5628–5654. doi: 10.1002/anie.200906670. [DOI] [PubMed] [Google Scholar]
- 20.Glabe CG. J Biol Chem. 2008;283:29639–29643. doi: 10.1074/jbc.R800016200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Quist A, Doudevski I, Lin H, Azimova R, Ng D, Frangione B, Kagan B, Ghiso J, Lal R. Proc Natl Acad Sci USA. 2005;102:10427–10432. doi: 10.1073/pnas.0502066102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matsumura S, Shinoda K, Yamada M, Yokojima S, Inoue M, Ohnishi T, Shimada T, Kikuchi K, Masui D, Hashimoto S, Sato M, Ito A, Akioka M, Takagi S, Nakamura Y, Nemoto K, Hasegawa Y, Takamoto H, Inoue H, Nakamura S, Nabeshima Y, Teplow DB, Kinjo M, Hoshia M. J Biol Chem. 2011;286:11555–11562. doi: 10.1074/jbc.M110.181313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klein WL, Krafft GA, Finch CE. Trends Neurosci. 2001;24:219–224. doi: 10.1016/s0166-2236(00)01749-5. [DOI] [PubMed] [Google Scholar]
- 24.Lambert MP, Viola KL, Chromy BA, Chang L, Morgan TE, Yu JX, Venton DL, Krafft GA, Finch CE, Klein WL. J Neurochem. 2001;79:595–605. doi: 10.1046/j.1471-4159.2001.00592.x. [DOI] [PubMed] [Google Scholar]
- 25.Haass C, Selkoe DJ. Nature Revs Mol Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 26.Chi EY, Ege C, Winans A, Majewski J, Wu GH, Kjaer K, Lee KYC. Proteins. 2008;72:1–24. doi: 10.1002/prot.21887. [DOI] [PubMed] [Google Scholar]
- 27.Relini A, Cavalleri O, Rolandi R, Gliozzi A. Chem Phys Lipids. 2009;158:1–9. doi: 10.1016/j.chemphyslip.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 28.Garner B. Biochim Biophys Acta. 2010;1801:747–749. doi: 10.1016/j.bbalip.2010.06.003. [DOI] [PubMed] [Google Scholar]
- 29.Williams TL, Serpell LC. FEBS J. 2011;278:3905–3917. doi: 10.1111/j.1742-4658.2011.08228.x. [DOI] [PubMed] [Google Scholar]
- 30.Singh R, Barman A, Prabhakar R. J Phys Chem B. 2009;113:2990–2999. doi: 10.1021/jp811154w. [DOI] [PubMed] [Google Scholar]
- 31.Thinakaran G, Koo EH. J Biol Chem. 2008;283:29615–29619. doi: 10.1074/jbc.R800019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Postina R, Schroeder A, Dewachter I, Bohl J, Schmitt U, Kojro E, Prinzen C, Endres K, Hiemke C, Blessing M, Flamez P, Dequenne A, Godaux E, van Leuven F, Fahrenholz F. J Clin Invest. 2004;113:1456–1464. doi: 10.1172/JCI20864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jang H, Zheng J, Nussinov R. Biophys J. 2007;93:1938–1949. doi: 10.1529/biophysj.107.110148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jang H, Zheng J, Lal R, Nussinov R. Trends Biochem Sci. 2008;33:91–100. doi: 10.1016/j.tibs.2007.10.007. [DOI] [PubMed] [Google Scholar]
- 35.Jang H, Arce FT, Capone R, Ramachandran S, Lal R, Nussinov R. Biophys J. 2009;97:3029–3037. doi: 10.1016/j.bpj.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jang H, Arce FT, Ramachandran S, Capone R, Azimova R, Kagan BL, Nussinov R, Lal R. Proc Natl Acad Sci USA. 2010;107:6538–6543. doi: 10.1073/pnas.0914251107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jang H, Arce FT, Ramachandran S, Capone R, Lal R, Nussinov R. J Phys Chem B. 2010;114:9445–9451. doi: 10.1021/jp104073k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jang H, Arce FT, Ramachandran S, Capone R, Lal R, Nussinov R. J Mol Biol. 2010;404:917–934. doi: 10.1016/j.jmb.2010.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gowing E, Roher AE, Woods AS, Cotter RJ, Chaney M, Little SP, Ball MJ. J Biol Chem. 1994;269:10987–10990. [PubMed] [Google Scholar]
- 40.Higgins LS, Murphy GM, Forno LS, Catalano R, Cordell B. Am J Pathol. 1996;149:585–596. [PMC free article] [PubMed] [Google Scholar]
- 41.Lalowski M, Golabek A, Lemere CA, Selkoe DJ, Wisniewski HM, Beavis RC, Frangione B, Wisniewski T. J Biol Chem. 1996;271:33623–33631. doi: 10.1074/jbc.271.52.33623. [DOI] [PubMed] [Google Scholar]
- 42.Lansbury PT, Lashuel HA. Nature. 2006;443:774–779. doi: 10.1038/nature05290. [DOI] [PubMed] [Google Scholar]
- 43.Hardy J. Neuron. 2006;52:3–13. doi: 10.1016/j.neuron.2006.09.016. [DOI] [PubMed] [Google Scholar]
- 44.Jang H, Connelly L, Arce FT, Ramachandran S, Lal R, Kagan BL, Nussinov R. Phys Chem Chem Phys. 2013;15:8868–8877. doi: 10.1039/c3cp00017f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lim KH, Kim YK, Chang YT. Biochemistry. 2007;46:13523–13532. doi: 10.1021/bi701112z. [DOI] [PubMed] [Google Scholar]
- 46.Noy D, Solomonov I, Sinkevich O, Arad T, Kjaer K, Sagi I. Journal of the American Chemical Society. 2008;130:1376–1383. doi: 10.1021/ja076282l. [DOI] [PubMed] [Google Scholar]
- 47.Zirah S, Kozin SA, Mazur AK, Blond A, Cheminant M, Segalas-Milazzo I, Debey P, Rebuffat S. J Biol Chem. 2006;281:2151–2161. doi: 10.1074/jbc.M504454200. [DOI] [PubMed] [Google Scholar]
- 48.Talmard C, Guilloreau L, Coppel Y, Mazarguil H, Faller P. Chembiochem: a European journal of chemical biology. 2007;8:163–165. doi: 10.1002/cbic.200600319. [DOI] [PubMed] [Google Scholar]
- 49.Gaggelli E, Janicka-Klos A, Jankowska E, Kozlowski H, Migliorini C, Molteni E, Valensin D, Valensin G, Wieczerzak E. J Phys Chem B. 2008;112:100–109. doi: 10.1021/jp075168m. [DOI] [PubMed] [Google Scholar]
- 50.Parthasarathy S, Long F, Miller Y, Xiao Y, McElheny D, Thurber K, Ma B, Nussinov R, Ishii Y. Journal of the American Chemical Society. 2011;133:3390–3400. doi: 10.1021/ja1072178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miller Y, Ma B, Nussinov R. Proc Natl Acad Sci U S A. 2010;107:9490–9495. doi: 10.1073/pnas.0913114107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Miller Y, Ma BY, Nussinov R. Coordin Chem Rev. 2012;256:2245–2252. [Google Scholar]
- 53.Siddiqua A, Luo Y, Meyer V, Swanson MA, Yu X, Wei G, Zheng J, Eaton GR, Ma B, Nussinov R, Eaton SS, Margittai M. Journal of the American Chemical Society. 2012;134:10271–10278. doi: 10.1021/ja303498q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Luo Y, Dinkel P, Yu X, Margittai M, Zheng J, Nussinov R, Wei G, Ma B. Chem Commun (Camb) 2013;49:3582–3584. doi: 10.1039/c3cc00241a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rosenman DJ, Connors CR, Chen W, Wang C, Garcia AE. Journal of molecular biology. 2013;425:3338–3359. doi: 10.1016/j.jmb.2013.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ball KA, Phillips AH, Wemmer DE, Head-Gordon T. Biophys J. 2013;104:2714–2724. doi: 10.1016/j.bpj.2013.04.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lin YS, Bowman GR, Beauchamp KA, Pande VS. Biophys J. 2012;102:315–324. doi: 10.1016/j.bpj.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lv Z, Roychaudhuri R, Condron MM, Teplow DB, Lyubchenko YL. Sci Rep. 2013;3:2880. doi: 10.1038/srep02880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Barz B, Urbanc B. PLoS One. 2012;7:e34345. doi: 10.1371/journal.pone.0034345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cote S, Laghaei R, Derreumaux P, Mousseau N. J Phys Chem B. 2012;116:4043–4055. doi: 10.1021/jp2126366. [DOI] [PubMed] [Google Scholar]
- 61.Zhang T, Zhang J, Derreumaux P, Mu Y. J Phys Chem B. 2013;117:3993–4002. doi: 10.1021/jp312573y. [DOI] [PubMed] [Google Scholar]
- 62.Meral D, Urbanc B. Journal of molecular biology. 2013;425:2260–2275. doi: 10.1016/j.jmb.2013.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Miller Y, Ma B, Nussinov R. Chem Rev. 2010;110:4820–4838. doi: 10.1021/cr900377t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ndlovu H, Ashcroft AE, Radford SE, Harris SA. Biophys J. 2012;102:587–596. doi: 10.1016/j.bpj.2011.12.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Miller Y, Ma B, Tsai CJ, Nussinov R. Proc Natl Acad Sci U S A. 2010;107:14128–14133. doi: 10.1073/pnas.1004704107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Miller Y, Ma B, Nussinov R. Journal of the American Chemical Society. 2011;133:2742–2748. doi: 10.1021/ja1100273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Petkova AT, Yau WM, Tycko R. Biochemistry. 2006;45:498–512. doi: 10.1021/bi051952q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Paravastu AK, Leapman RD, Yau WM, Tycko R. Proc Natl Acad Sci U S A. 2008;105:18349–18354. doi: 10.1073/pnas.0806270105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Meinhardt J, Sachse C, Hortschansky P, Grigorieff N, Fandrich M. Journal of molecular biology. 2009;386:869–877. doi: 10.1016/j.jmb.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang R, Hu X, Khant H, Ludtke SJ, Chiu W, Schmid MF, Frieden C, Lee JM. Proc Natl Acad Sci U S A. 2009;106:4653–4658. doi: 10.1073/pnas.0901085106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rhee SK, Quist AP, Lal R. J Biol Chem. 1998;273:13379–13382. doi: 10.1074/jbc.273.22.13379. [DOI] [PubMed] [Google Scholar]
- 72.Bhatia R, Lin H, Lal R. FASEB J. 2000;14:1233–1243. doi: 10.1096/fasebj.14.9.1233. [DOI] [PubMed] [Google Scholar]
- 73.Lin H, Bhatia R, Lal R. FASEB J. 2001;15:2433–2444. doi: 10.1096/fj.01-0377com. [DOI] [PubMed] [Google Scholar]
- 74.Lal R, Lin H, Quist AP. Biochim Biophys Acta-Biomembr. 2007;1768:1966–1975. doi: 10.1016/j.bbamem.2007.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mustata M, Capone R, Jang H, Arce FT, Ramachandran S, Lal R, Nussinov R. J Am Chem Soc. 2009;131:14938–14945. doi: 10.1021/ja9049299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Arce FT, Jang H, Ramachandran S, Landon PB, Nussinov R, Lal R. Soft Matter. 2011;7:5267–5273. doi: 10.1039/C1SM05162H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Connelly L, Arce FT, Jang H, Capone R, Kotler SA, Ramachandran S, Kagan BL, Nussinov R, Lal R. J Phys Chem B. 2012;116:1728–1735. doi: 10.1021/jp2108126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Connelly L, Jang H, Arce FT, Ramachandran S, Kagan BL, Nussinov R, Lal R. Biochemistry. 2012;51:3031–3038. doi: 10.1021/bi300257e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lashuel HA, Hartley D, Petre BM, Walz T, Lansbury PT., Jr Nature. 2002;418:291. doi: 10.1038/418291a. [DOI] [PubMed] [Google Scholar]
- 80.Lashuel HA, Hartley DM, Petre BM, Wall JS, Simon MN, Walz T, Lansbury PT., Jr J Mol Biol. 2003;332:795–808. doi: 10.1016/s0022-2836(03)00927-6. [DOI] [PubMed] [Google Scholar]
- 81.Lashuel HA, Lansbury PT., Jr Q Rev Biophys. 2006;39:167–201. doi: 10.1017/S0033583506004422. [DOI] [PubMed] [Google Scholar]
- 82.Qi R, Luo Y, Ma B, Nussinov R, Wei G. Biomacromolecules. 2014;15:122–131. doi: 10.1021/bm401406e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Olubiyi OO, Frenzel D, Bartnik D, Gluck JM, Brener O, Nagel-Steger L, Funke SA, Willbold D, Strodel B. Curr Med Chem. 2013 doi: 10.2174/0929867321666131129122247. [DOI] [PubMed] [Google Scholar]
- 84.Poojari C, Xiao D, Batista VS, Strodel B. Biophys J. 2013;105:2323–2332. doi: 10.1016/j.bpj.2013.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Smith MD, Cruz L. J Phys Chem B. 2013;117:14907–14915. doi: 10.1021/jp408579v. [DOI] [PubMed] [Google Scholar]
- 86.Miller C, Zerze GH, Mittal J. J Phys Chem B. 2013;117:16066–16075. doi: 10.1021/jp409755y. [DOI] [PubMed] [Google Scholar]
- 87.Buchanan LE, Dunkelberger EB, Tran HQ, Cheng PN, Chiu CC, Cao P, Raleigh DP, de Pablo JJ, Nowick JS, Zanni MT. Proc Natl Acad Sci U S A. 2013;110:19285–19290. doi: 10.1073/pnas.1314481110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Guo J, Zhang Y, Ning L, Jiao P, Liu H, Yao X. Biochim Biophys Acta. 2013;1840:357–366. doi: 10.1016/j.bbagen.2013.09.012. [DOI] [PubMed] [Google Scholar]
- 89.Friedman R, Caflisch A. Proteins. 2014;82:399–404. doi: 10.1002/prot.24402. [DOI] [PubMed] [Google Scholar]
- 90.Xu L, Wang X, Shan S. J Comput Chem. 2013;34:2524–2536. doi: 10.1002/jcc.23416. [DOI] [PubMed] [Google Scholar]
- 91.Chiu CC, Singh S, de Pablo JJ. Biophys J. 2013;105:1227–1235. doi: 10.1016/j.bpj.2013.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wu C, Shea JE. PLoS Comput Biol. 2013;9:e1003211. doi: 10.1371/journal.pcbi.1003211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ndlovu H, Ashcroft AE, Radford SE, Harris SA. Beilstein J Nanotechnol. 2013;4:429–440. doi: 10.3762/bjnano.4.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Maiorana A, Marino T, Minicozzi V, Morante S, Russo N. Biophys Chem. 2013;182:86–93. doi: 10.1016/j.bpc.2013.07.002. [DOI] [PubMed] [Google Scholar]
- 95.Di Scala C, Troadec JD, Lelievre C, Garmy N, Fantini J, Chahinian H. J Neurochem. 2014;128:186–195. doi: 10.1111/jnc.12390. [DOI] [PubMed] [Google Scholar]
- 96.Hernandez-Rodriguez M, Correa-Basurto J, Benitez-Cardoza CG, Resendiz-Albor AA, Rosales-Hernandez MC. Protein Sci. 2013;22:1320–1335. doi: 10.1002/pro.2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Skeby KK, Sorensen J, Schiott B. Journal of the American Chemical Society. 2013;135:15114–15128. doi: 10.1021/ja405530p. [DOI] [PubMed] [Google Scholar]
- 98.Lemkul JA, Bevan DR. Biochemistry. 2013;52:4971–4980. doi: 10.1021/bi400562x. [DOI] [PubMed] [Google Scholar]
- 99.Shea JE, Urbanc B. Curr Top Med Chem. 2012;12:2596–2610. doi: 10.2174/1568026611212220012. [DOI] [PubMed] [Google Scholar]
- 100.Gessel MM, Wu C, Li H, Bitan G, Shea JE, Bowers MT. Biochemistry. 2012;51:108–117. doi: 10.1021/bi201520b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yu X, Wang Q, Pan Q, Zhou F, Zheng J. Phys Chem Chem Phys. 2013;15:8878–8889. doi: 10.1039/c3cp44448a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Liang G, Zhao J, Yu X, Zheng J. Biochemistry. 2013;52:1089–1100. doi: 10.1021/bi301525e. [DOI] [PubMed] [Google Scholar]
- 103.Capone R, Jang H, Kotler SA, Connelly L, Arce FT, Ramachandran S, Kagan BL, Nussinov R, Lal R. J Chem Theory Comput. 2012;8:1143–1152. doi: 10.1021/ct200885r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Capone R, Jang H, Kotler SA, Kagan BL, Nussinov R, Lal R. Biochemistry. 2012;51:776–785. doi: 10.1021/bi2017427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tofoleanu F, Buchete NV. Prion. 2012;6:339–345. doi: 10.4161/pri.21022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jang H, Teran Arce F, Ramachandran S, Kagan BL, Lal R, Nussinov R. J Phys Chem B. 2013;117:11518–11529. doi: 10.1021/jp405389n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhao J, Luo Y, Jang H, Yu X, Wei G, Nussinov R, Zheng J. Biochim Biophys Acta. 2012;1818:3121–3130. doi: 10.1016/j.bbamem.2012.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Jang H, Ma B, Lal R, Nussinov R. Biophys J. 2008;95:4631–4642. doi: 10.1529/biophysj.108.134551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Capone R, Mustata M, Jang H, Arce FT, Nussinov R, Lal R. Biophys J. 2010;98:2644–2652. doi: 10.1016/j.bpj.2010.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gupta K, Jang H, Harlen K, Puri A, Nussinov R, Schneider JP, Blumenthal R. Biophys J. 2013;105:2093–2103. doi: 10.1016/j.bpj.2013.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. J Comp Chem. 1983;4:187–217. [Google Scholar]
- 112.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. J Comp Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Shao H, Jao S, Ma K, Zagorski MG. Journal of molecular biology. 1999;285:755–773. doi: 10.1006/jmbi.1998.2348. [DOI] [PubMed] [Google Scholar]
- 114.Hou LM, Shao HY, Zhang YB, Li H, Menon NK, Neuhaus EB, Brewer JM, Byeon IJL, Ray DG, Vitek MP, Iwashita T, Makula RA, Przybyla AB, Zagorski MG. Journal of the American Chemical Society. 2004;126:1992–2005. doi: 10.1021/ja036813f. [DOI] [PubMed] [Google Scholar]
- 115.Verdier Y, Zarandi M, Penke B. J Pept Sci. 2004;10:229–248. doi: 10.1002/psc.573. [DOI] [PubMed] [Google Scholar]
- 116.Shirwany NA, Payette D, Xie J, Guo Q. Neuropsychiatr Dis Treat. 2007;3:597–612. [PMC free article] [PubMed] [Google Scholar]
- 117.Buchete NV. Biophys J. 2012;103:1411–1413. doi: 10.1016/j.bpj.2012.08.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 119.Kagan BL. Prog Mol Biol Transl Sci. 2012;107:295–325. doi: 10.1016/B978-0-12-385883-2.00001-1. [DOI] [PubMed] [Google Scholar]
- 120.Arispe N, Rojas E, Pollard HB. Proc Natl Acad Sci USA. 1993;90:567–571. doi: 10.1073/pnas.90.2.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Arispe N, Pollard HB, Rojas E. Proc Natl Acad Sci USA. 1993;90:10573–10577. doi: 10.1073/pnas.90.22.10573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Pollard HB, Rojas E, Arispe N. Ann N Y Acad Sci. 1993;695:165–168. doi: 10.1111/j.1749-6632.1993.tb23046.x. [DOI] [PubMed] [Google Scholar]
- 123.Arispe N, Pollard HB, Rojas E. Calcium Hypothesis of Aging and Dementia. 1994;747:256–266. doi: 10.1111/j.1749-6632.1994.tb44414.x. [DOI] [PubMed] [Google Scholar]
- 124.Arispe N, Pollard HB, Rojas E. Proc Natl Acad Sci U S A. 1996;93:1710–1715. doi: 10.1073/pnas.93.4.1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hirakura Y, Lin MC, Kagan BL. Journal of Neuroscience Research. 1999;57:458–466. [PubMed] [Google Scholar]
- 126.Kagan BL, Azimov R, Azimova R. J Membr Biol. 2004;202:1–10. doi: 10.1007/s00232-004-0709-4. [DOI] [PubMed] [Google Scholar]
- 127.Kagan BL, Hirakura Y, Azimov R, Azimova R, Lin MC. Peptides. 2002;23:1311–1315. doi: 10.1016/s0196-9781(02)00067-0. [DOI] [PubMed] [Google Scholar]
- 128.Mirzabekov TA, Lin MC, Kagan BL. J Biol Chem. 1996;271:1988–1992. doi: 10.1074/jbc.271.4.1988. [DOI] [PubMed] [Google Scholar]
- 129.Brender JR, Heyl DL, Samisetti S, Kotler SA, Osborne JM, Pesaru RR, Ramamoorthy A. Phys Chem Chem Phys. 2013;15:8908–8915. doi: 10.1039/c3cp44696d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Sciacca MF, Milardi D, Messina GM, Marletta G, Brender JR, Ramamoorthy A, La Rosa C. Biophys J. 2013;104:173–184. doi: 10.1016/j.bpj.2012.11.3811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Sciacca MF, Brender JR, Lee DK, Ramamoorthy A. Biochemistry. 2012;51:7676–7684. doi: 10.1021/bi3009888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Brender JR, Hartman K, Reid KR, Kennedy RT, Ramamoorthy A. Biochemistry. 2008;47:12680–12688. doi: 10.1021/bi801427c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Brender JR, Durr UH, Heyl D, Budarapu MB, Ramamoorthy A. Biochim Biophys Acta. 2007;1768:2026–2029. doi: 10.1016/j.bbamem.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Lin MC, Mirzabekov T, Kagan BL. J Biol Chem. 1997;272:44–47. doi: 10.1074/jbc.272.1.44. [DOI] [PubMed] [Google Scholar]
- 135.Hirakura Y, Azimov R, Azimova R, Kagan BL. Journal of neuroscience research. 2000;60:490–494. doi: 10.1002/(sici)1097-4547(20000515)60:4<490::aid-jnr7>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 136.Hirakura Y, Kagan BL. Amyloid-J Protein Fold Disord. 2001;8:94–100. doi: 10.3109/13506120109007350. [DOI] [PubMed] [Google Scholar]
- 137.Hirakura Y, Azimova R, Azimov R, Kagan B. Biophys J. 2001;80:129a–129a. [Google Scholar]
- 138.Hirakura Y, Carreras I, Sipe JD, Kagan BL. Amyloid. 2002;9:13–23. doi: 10.3109/13506120209072440. [DOI] [PubMed] [Google Scholar]
- 139.Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, Viola KL, Klein WL. J Neurosci. 2007;27:796–807. doi: 10.1523/JNEUROSCI.3501-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.De Felice FG, Velasco PT, Lambert MP, Viola K, Fernandez SJ, Ferreira ST, Klein WL. J Biol Chem. 2007;282:11590–11601. doi: 10.1074/jbc.M607483200. [DOI] [PubMed] [Google Scholar]
- 141.De Felice FG, Wu D, Lambert MP, Fernandez SJ, Velasco PT, Lacor PN, Bigio EH, Jerecic J, Acton PJ, Shughrue PJ, Chen-Dodson E, Kinney GG, Klein WL. Neurobiol Aging. 2008;29:1334–1347. doi: 10.1016/j.neurobiolaging.2007.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kayed R, Sokolov Y, Edmonds B, McIntire TM, Milton SC, Hall JE, Glabe CG. J Biol Chem. 2004;279:46363–46366. doi: 10.1074/jbc.C400260200. [DOI] [PubMed] [Google Scholar]
- 143.Sokolov Y, Kozak JA, Kayed R, Chanturiya A, Glabe C, Hall JE. J Gen Physiol. 2006;128:637–647. doi: 10.1085/jgp.200609533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Schauerte JA, Wong PT, Wisser KC, Ding H, Steel DG, Gafni A. Biochemistry. 2010;49:3031–3039. doi: 10.1021/bi901444w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Sciacca MFM, Kotler SA, Brender JR, Chen J, Lee DK, Ramamoorthy A. Biophys J. 2012;103:702–710. doi: 10.1016/j.bpj.2012.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Sciacca MFM, Brender JR, Lee DK, Ramamoorthy A. Biochemistry. 2012;51:7676–7684. doi: 10.1021/bi3009888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Milanesi L, Sheynis T, Xue WF, Orlova EV, Hellewell AL, Jelinek R, Hewitt EW, Radford SE, Saibil HR. Proc Natl Acad Sci USA. 2012;109:20455–20460. doi: 10.1073/pnas.1206325109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Lazo ND, Grant MA, Condron MC, Rigby AC, Teplow DB. Protein Science. 2005;14:1581–1596. doi: 10.1110/ps.041292205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Crescenzi O, Tomaselli S, Guerrini R, Salvadori S, D’Ursi AM, Temussi PA, Picone D. Eur J Biochem. 2002;269:5642–5648. doi: 10.1046/j.1432-1033.2002.03271.x. [DOI] [PubMed] [Google Scholar]
- 150.Tomaselli S, Esposito V, Vangone P, van Nuland NAJ, Bonvin AMJJ, Guerrini R, Tancredi T, Temussi PA, Picone D. Chembiochem. 2006;7:257–267. doi: 10.1002/cbic.200500223. [DOI] [PubMed] [Google Scholar]
- 151.Vivekanandan S, Brender JR, Lee SY, Ramamoorthy A. Biochem Biophys Res Commun. 2011;411:312–316. doi: 10.1016/j.bbrc.2011.06.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Lührs T, Ritter C, Adrian M, Riek-Loher D, Bohrmann B, Doeli H, Schubert D, Riek R. Proc Natl Acad Sci USA. 2005;102:17342–17347. doi: 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ma B, Nussinov R. Proc Natl Acad Sci U S A. 2002;99:14126–14131. doi: 10.1073/pnas.212206899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Bertini I, Gonnelli L, Luchinat C, Mao J, Nesi A. Journal of the American Chemical Society. 2011;133:16013–16022. doi: 10.1021/ja2035859. [DOI] [PubMed] [Google Scholar]
- 155.Lu JX, Qiang W, Yau WM, Schwieters CD, Meredith SC, Tycko R. Cell. 2013;154:1257–1268. doi: 10.1016/j.cell.2013.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Chimon S, Shaibat MA, Jones CR, Calero DC, Aizezi B, Ishii Y. Nat Struct Mol Biol. 2007;14:1157–1164. doi: 10.1038/nsmb1345. [DOI] [PubMed] [Google Scholar]
- 157.Wu JW, Breydo L, Isas JM, Lee J, Kuznetsov YG, Langen R, Glabe C. J Biol Chem. 2010;285:6071–6079. doi: 10.1074/jbc.M109.069542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Ladiwala AR, Litt J, Kane RS, Aucoin DS, Smith SO, Ranjan S, Davis J, Vannostrand WE, Tessier PM. J Biol Chem. 2012;287:24765–24773. doi: 10.1074/jbc.M111.329763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Jang H, Connelly L, Arce FT, Ramachandran S, Kagan BL, Lal R, Nussinov R. J Chem Theory Comput. 2013;9:822–833. doi: 10.1021/ct300916f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Ciccotosto GD, Tew DJ, Drew SC, Smith DG, Johanssen T, Lal V, Lau TL, Perez K, Curtain CC, Wade JD, Separovic F, Masters CL, Smith JP, Barnham KJ, Cappai R. Neurobiol Aging. 2011;32:235–248. doi: 10.1016/j.neurobiolaging.2009.02.018. [DOI] [PubMed] [Google Scholar]
- 161.de Groot NS, Aviles FX, Vendrell J, Ventura S. FEBS J. 2006;273:658–668. doi: 10.1111/j.1742-4658.2005.05102.x. [DOI] [PubMed] [Google Scholar]
- 162.Wang Z, Natte R, Berliner JA, van Duinen SG, Vinters HV. Stroke. 2000;31:534–538. doi: 10.1161/01.str.31.2.534. [DOI] [PubMed] [Google Scholar]
- 163.Nilsberth C, Westlind-Danielsson A, Eckman CB, Condron MM, Axelman K, Forsell C, Stenh C, Luthman J, Teplow DB, Younkin SG, Naslund J, Lannfelt L. Nat Neurosci. 2001;4:887–893. doi: 10.1038/nn0901-887. [DOI] [PubMed] [Google Scholar]
- 164.Miravalle L, Tokuda T, Chiarle R, Giaccone G, Bugiani O, Tagliavini F, Frangione B, Ghiso J. J Biol Chem. 2000;275:27110–27116. doi: 10.1074/jbc.M003154200. [DOI] [PubMed] [Google Scholar]
- 165.Levy E, Carman MD, Fernandez-Madrid IJ, Power MD, Lieberburg I, van Duinen SG, Bots GT, Luyendijk W, Frangione B. Science. 1990;248:1124–1126. doi: 10.1126/science.2111584. [DOI] [PubMed] [Google Scholar]
- 166.Grabowski TJ, Cho HS, Vonsattel JP, Rebeck GW, Greenberg SM. Ann Neurol. 2001;49:697–705. doi: 10.1002/ana.1009. [DOI] [PubMed] [Google Scholar]
- 167.Viet MH, Li MS. J Chem Phys. 2012;136:245105. doi: 10.1063/1.4730410. [DOI] [PubMed] [Google Scholar]
- 168.Wise-Scira O, Xu L, Kitahara T, Perry G, Coskuner O. J Chem Phys. 2011;135:205101. doi: 10.1063/1.3662490. [DOI] [PubMed] [Google Scholar]
- 169.Thirumalai D, Reddy G, Straub JE. Acc Chem Res. 2012;45:83–92. doi: 10.1021/ar2000869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Paparcone R, Pires MA, Buehler MJ. Biochemistry. 2010;49:8967–8977. doi: 10.1021/bi100953t. [DOI] [PubMed] [Google Scholar]
- 171.Davis CH, Berkowitz ML. J Phys Chem B. 2009;113:14480–14486. doi: 10.1021/jp905889z. [DOI] [PubMed] [Google Scholar]
- 172.Reddy G, Straub JE, Thirumalai D. J Phys Chem B. 2009;113:1162–1172. doi: 10.1021/jp808914c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Krone MG, Baumketner A, Bernstein SL, Wyttenbach T, Lazo ND, Teplow DB, Bowers MT, Shea JE. Journal of molecular biology. 2008;381:221–228. doi: 10.1016/j.jmb.2008.05.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Fawzi NL, Phillips AH, Ruscio JZ, Doucleff M, Wemmer DE, Head-Gordon T. Journal of the American Chemical Society. 2008;130:6145–6158. doi: 10.1021/ja710366c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Walsh DM, Lomakin A, Benedek GB, Condron MM, Teplow DB. J Biol Chem. 1997;272:22364–22372. doi: 10.1074/jbc.272.35.22364. [DOI] [PubMed] [Google Scholar]
- 176.Williams AD, Portelius E, Kheterpal I, Guo JT, Cook KD, Xu Y, Wetzel R. Journal of molecular biology. 2004;335:833–842. doi: 10.1016/j.jmb.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 177.Tomiyama T, Nagata T, Shimada H, Teraoka R, Fukushima A, Kanemitsu H, Takuma H, Kuwano R, Imagawa M, Ataka S, Wada Y, Yoshioka E, Nishizaki T, Watanabe Y, Mori H. Ann Neurol. 2008;63:377–387. doi: 10.1002/ana.21321. [DOI] [PubMed] [Google Scholar]
- 178.Murakami K, Irie K, Morimoto A, Ohigashi H, Shindo M, Nagao M, Shimizu T, Shirasawa T. J Biol Chem. 2003;278:46179–46187. doi: 10.1074/jbc.M301874200. [DOI] [PubMed] [Google Scholar]
- 179.Jawhar S, Wirths O, Bayer TA. J Biol Chem. 2011;286:38825–38832. doi: 10.1074/jbc.R111.288308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Saido TC, Iwatsubo T, Mann DM, Shimada H, Ihara Y, Kawashima S. Neuron. 1995;14:457–466. doi: 10.1016/0896-6273(95)90301-1. [DOI] [PubMed] [Google Scholar]
- 181.Poojari C, Kukol A, Strodel B. Biochimica et Biophysica Acta (BBA) - Biomembranes. 2012;1828:327–339. doi: 10.1016/j.bbamem.2012.09.001. [DOI] [PubMed] [Google Scholar]
- 182.DeMuro A, Smith M, Parker I. J Cell Biol. 2011;195:515–524. doi: 10.1083/jcb.201104133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Kagan BL, Thundimadathil J. Adv Exp Med Biol. 2010;677:150–167. doi: 10.1007/978-1-4419-6327-7_13. [DOI] [PubMed] [Google Scholar]
- 184.Smart OS, Goodfellow JM, Wallace BA. Biophys J. 1993;65:2455–2460. doi: 10.1016/S0006-3495(93)81293-1. [DOI] [PMC free article] [PubMed] [Google Scholar]