

Abstract
Background
Human epidermal growth factor receptor 2 (HER2) has an important role in cancer aggressiveness and poor prognosis. HER2 has been used as a drug target for cancers. In particular, to effectively treat HER2-positive cancer, small molecule inhibitors were developed to target HER2 kinase. Knowing that curcumin has been used as food to inhibit cancer activity, this study evaluated the efficacy of natural curcumins and curcumin analogs as HER2 inhibitors using in vitro and in silico studies. The curcumin analogs considered in this study composed of 4 groups classified by their core structure, β-diketone, monoketone, pyrazole, and isoxazole.
Results
In the present study, both computational and experimental studies were performed. The specificity of curcumin analogs selected from the docked results was examined against human breast cancer cell lines. The screened curcumin compounds were then subjected to molecular dynamics simulation study. By modifying curcumin analogs, we found that protein-ligand affinity increases. The benzene ring with a hydroxyl group could enhance affinity by forming hydrophobic interactions and the hydrogen bond with the hydrophobic pocket. Hydroxyl, carbonyl or methoxy group also formed hydrogen bonds with residues in the adenine pocket and sugar pocket of HER2-TK. These modifications could suggest the new drug design for potentially effective HER2-TK inhibitors. Two outstanding compounds, bisdemethylcurcumin (AS-KTC006) and 3,5-bis((E)-3,4-dimethoxystyryl)isoxazole (AS-KTC021 ),were well oriented in the binding pocket almost in the simulation time, 30 ns. This evidence confirmed the results of cell-based assays and the docking studies. They possessed more distinguished interactions than known HER2-TK inhibitors, considering them as a promising drug in the near future.
Conclusions
The series of curcumin compounds were screened using a computational molecular docking and followed by human breast cancer cell lines assay. Both AS-KTC006 and AS-KTC021 could inhibit breast cancer cell lines though inhibiting of HER2-TK. The intermolecular interactions were confirmed by molecular dynamics simulation studies. This information would explore more understanding of curcuminoid structures and HER2-TK.
Electronic supplementary material
The online version of this article (doi:10.1186/1471-2105-15-261) contains supplementary material, which is available to authorized users.
Keywords: HER2, Tyrosine kinase, Curcuminoid analogs, Docking, Molecular dynamics simulation
Background
Human Epidermal Growth Factor Receptor 2 (HER2) is one of the tyrosine kinase receptors in EGFR family, which includes EGFR/ErbB1, HER2/ErbB2, HER3/ErbB3 and HER4/ErbB4 [1]. Since there is no natural ligand specific to HER2, HER2 tends to form heterodimer with other ligand-induced members [2]. After dimerization, the complex can trigger downstream pathways such as Ras/Raf/MAPK and PI3K/AKT pathways to increase cell growth, cell survival and cell differentiation [3, 4]. Considerable evidencesshowed that HER2 over expression was involved in many types of cancer such as breast, ovarian, gastric and prostate cancers [5]. Therefore, HER2 is considered as a drug target for cancer therapy focusing on inhibiting HER2 to reduce tumor growth.
At present, there are two main approaches used to inhibit HER2, namely; monoclonal antibodies such as Trastuzumab, and small molecule inhibitors such as Lapatinib [6] and SYR127063 (called SYR for short) [7] targetingon tyrosine kinase domain (HER2-TK). Although Trastuzumab can downregulate HER expression, cardiotoxicity and drug resistance can be found in Trastuzumab-treated patients [8, 9]. Moreover, side effects such as diarrhea, rash or nausea can be observed in Lapatinib treatment [10]. Hence, new inhibitors are urgently required for HER2-overexpressed cancer treatment.
Recently, in 2011, the first HER2-TK structure complex with pyrimidine compound SYR was released (PDB access 3PP0), providing the new understanding of the kinase structure [7]. Unlike the active- or inactive-conformations of EGFR-TK, HER2-TK configuration was somewhat in the middle of these typical conformations. It was named “the active-like conformation”, due to, the orientation of the helix-αC-out, the DFG-in and unformed secondary structure of the activation loop. The second crystal of HER2-TK complex with TAK-285 (PDB access 3RCD) adopted the similar conformation as mentioned above [11].
Curcumin (also known as diferuloylmethane) is generally found as the major compound in rhizomes of turmeric plants; Curcuma longa Linaeusas yellow residue. It has been used as spice and ingredients in folk medicinal remedies in many Asian countries. The curcumin and its three natural analogs, curcumin II (demethoxycurcumin), curcumin III (bisdemethoxycurcumin) and cyclocurcuminpossess the remarkable pharmacological effects for centuries, such as anti-inflammatory [12, 13], antioxidant [14], anti-carcinogenesis [15–18]. Moreover, curcumins is safe to use in high dose with non-toxic report [19, 20]. Despite many advantages of curcumins, the poor stability and bioavailability profiles of curcumins are questionable when it comes to directly using crude curcumin as the potent and selective cancer drug. Many researchers have been focusing on the developing the curcumin analogs to enhance their stability and bioavailability. In particular, the novel series of curcumin-analog compounds have been synthesized and studied their effect in various cell targets [21–26]. They possess several properties, potent activity against parasite in Trypanosoma and Leishmania species [21], antimycobacterial activity [22], inhibiting nitric oxide production from Lps-activated microglial cells [25] and estrogenic properties [23, 24, 26]. Thus, in this paper, we aimed to investigate the effect of this set of curcumin analogs on the HER2-TK activity using both experimental and computational methods.
Curcumin has been shown to inhibit cancer growth by means of inhibiting several tyrosine kinases including EGFR, HER2, MAPK, phosphorylase kinase, pp60c-src tyrosine kinase, protein kinase C, and protein kinase A [18, 27–34]. Furthermore, the curcumins can inhibit various types of cancer including breast cancer cells [15, 28] and also induce the internalization of HER2 from cell surface [35]. Recently, curcumin analog cyclohexanone has shown to selectively inhibit tyrosine kinase domain of EGFR, in vitro, in vivo and in silico studies [36] which reveals an opportunity for direct binding between curcumins and tyrosine kinase domains of other EGFR family members. Furthermore, the in silico screening of the natural database against HER2 kinase showed that such curcumins could interact with kinase through benzene rings for hydrophobic interactions and carboxyl groups for hydrogen bond formation [37].
In this study, we investigated interactions between curcumin analogs and HER2-TK by using virtual screening based on molecular docking in order to find potential compounds against HER2-TK. The hit compounds have been validated by different inhibitions between two types of breast cancer cell-lines with both HER2-overexpression and HER2-non-overexpression. Such findings might be useful for further development of curcumins as a new HER2 inhibitor in the future.
Methods
Computational procedures
The preparation of ligand
The two dimensional (2D) structure of 143 curcuminoid analogs were collected from the previous studies [21–26] (Additional file 1: Table S1). The ionization states, tautomers, stereochemistries, and ring conformations of all curcuminoid structures were calculated and OPLS-2005 force field was applied using LigPrep module in Schrödinger package. These structures were used as an initial material during computational docking procedure to study interactions with the binding site of the HER2 tyrosine kinase domain.
The preparation of protein
The atomic coordinate of HER2 tyrosine kinase domain (HER2-TK) was obtained from the crystallographic structure, accession no. 3PP0 in Protein Data Bank (PDB) [7]. This structure contains asymmetric dimer of HER2-TK complex with selective inhibitor HER2-TK, pyrrolo[3,2-d]pyrimidine-based potent, SYR. In order to perform the docking calculations, only chain A was selected as the target template. Another chain of HER2-TK as well as the co-crystalized ligand(s) and crystal water molecules were removed. Hydrogen atoms were assigned and parameterized with Optimized Potential for Liquid Simulation version 2005 (OPLS-2005 force field) using the protein preparation wizard, which continuously minimized the whole structure by the Impref module in the Schrödinger package.
Docking procedure using Glide standard precision mode (SP mode)
The structures of protein and ligands were prepared as previously described. The OPLS-2005 force field was applied to both protein and ligands. The complexes of HER2-TK and each curcuminoid, including the co-crystal ligand were generated with molecular docking approach using Grid-based Ligand Docking with Energetics (Glide)with standard precision mode (SP mode) [38, 39]. The grid map was generated in Receptor Grid Generation by setting the center of the grid map around the catalytic site. Self-docking between HER2-TK and SYR was performed to validate all parameters before being applied to the study of interactions between HER2 and curcumins.
Post-docking analysis
In order to handle considerable number of docking results, the sub-groups of modified core structure of curcuminoids were classified. Top ranks docking score of each sub-group were selected to further test in cell-based assay. In addition, the poor scores of each curcumin sub-groups were also chosen to be the control set in breast cancer cell-line assay.
Molecular interaction and stability in binding pocket
All simulation steps were performed using the SANDER module of the AMBER 12 package and AMBER FF03 force-field parameters [40]. The partial atomic charges of ligand were computed by using AM1-BCC method as implemented in the Antechamber module of the AMBER package. Their atom types and missing force field parameters were assigned based on the general AMBER force field (GAFF). Each complex was immersed in an isomeric truncated-octahedron box of TIP3P water molecules (10 Å from the solute surface) and neutralized by additional Cl- anions. The system was then minimized with the five-step procedure (Additional file 1: Table S2). All steps included 5,000 steepest-descent minimization cycles and 5,000 conjugate-gradient minimization cycles with different restraints on the protein structure. For the first step, harmonic restraints with a force constant of 5 kcal/(mol · Å2) were used to immobilize the heavy atoms of protein coordinates, excluding hydrogen atoms, at the starting positions, while solvent molecules were allowed to relax the unfavorable contacts with other solvent and solute molecules. For the second, third and fourth steps, harmonic restraints with force constants of 5, 1 and 0.5 kcal/(mol · Å2), respectively, were used to restrain the backbone of the protein. In the last step, the entire system was minimized with no positional restraints.
With weak positional restraints on the protein (force constant of 5 kcal/(mol · Å2)), all systems were heated from 0 to 300 K during a 200 ps MD simulations. After removing the restraints from the protein, we equilibrated the system with constant volume and set the constant temperature at 300 K for 500 ps. Note that we observed the equilibrium of energy (potential, kinetic and total energy), temperature, pressure, volume, density and RMSD before moving on to the production runs. The production MD simulations were performed from 30 ns while maintaining constant pressure and temperature. With a collision frequency of 1 ps-1, the temperature in all simulations was controlled by Langevin dynamics. Using an isotropic position scaling algorithm with a relaxation time of 2 ps, the pressure in NPT simulations was maintained at an average pressure of 1 atm. The random number generator was reseeded [41] for every simulation. A cut-off of 10 Å and the particle mesh Ewald method were employed with the default parameters to calculate long-range non bonded interactions. With the tolerance parameter of 10-5 Å, SHAKE constraints [42] were used to eliminate bond-stretching freedom for all bonds involving hydrogen, thereby allowing the use of a 2 fs time step. To monitor the stabilities of all systems, the Cα root-mean-square deviations (RMSD) were calculated. The RMSD of binding residues within 5 Å of the inhibitor were examined. The ptraj modules in the AMBER software were used to calculate the hydrogen bond occupancy and hydrogen bond distance between inhibitors and proteins [43, 44]. All MD simulations were calculated on 22-node Linux High Performance Computer Cluster with 32 cores of AMD 2.2 GHz.
The energy calculations were done as implemented in the MMPBSA.py script in AmberTools. The MM-PBSA approach is an acceptable method to compute the free energies of binding of ligands to proteins or to estimate the absolute free energies of molecules [43, 44]. One hundred frames from the last 5 ns of each 30 ns MD studies were selected for the analysis of ligand binding energies, sampled at 50 ps intervals. Binding free energy was estimated from each energy terms as following equations (equation 1–4),
| 1 |
| 2 |
| 3 |
| 4 |
‹∆GMMPBSA› is referred to final calculated MM-PBSA binding energy. It is described by the difference of ΔGcomplex by the summation of ΔGprotein and ΔGligand (1). The free energy of each molecular system is given by the expression in the terms of equation (2). ‹∆EMM› is the total molecular mechanics energy in the gas phase, ‹∆Gsolv› is a correction term (solvation free energy) of each system surrounded by solvent, and ‹T∆S› is the entropy. ‹∆EMM› includes electrostatic ‹∆Eele›, and van der Waals ‹∆Evdw› energies, while ‹∆Gsolv› is the sum of electrostatic solvation energy ‹∆Gpb›, and the non-electrostatic solvation component ‹∆Gnp› (3–4). The polar contribution is calculated using PB model, while the non-polar energy is estimated by solvent accessible surface area (SASA). In this study, ‹T∆S› term was excluded.
Experimental procedures
Proliferation and viability assay
Cell proliferation and viability were measured by tetrazolium 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) assay. The reaction was catalyzed by mitochondrial succinate dehydrogenease and requires NADH, which must be supplied by living cells. SKBR3 and MCF7 cell line were seeded in flat-bottomed 96-well tissue culture plates as 1 × 104 cells/well/100 μL and cultured overnight. Pure curcumin extracts and its analogs with different concentrations were dissolved in 100 μL 10% FBS-RPMI 1640 medium and added into the cells and incubated for 48 h. Then 100 μL of medium was removed and 10 μL of MTT dye (Sigma-Aldrich; USA) was added, followed by 4hourof incubation. Subsequently, the supernatant from each well was aspirated off, leaving the purple form azan crystals. Optical density was measured by an ELISA micro plate reader at 540 nm with a reference wavelength of 630 nm. Percentage of cell survival was calculated by the formula below. Each assay was done in triplicate and the standard deviations were calculated.
Absorbance of vehicle control well.
All curcumin analog compounds were synthesized and published [21–26] by the laboratory of Prof. Dr. Apichart Suksamran, Department of Chemistry and Center of Excellence for Innovation in Chemistry, Faculty of Science, Ramkhamhaeng University.
Results and discussion
Selection of the curcumin analogs and structure analyses
Considering to the core structures of all 143 curcuminoid compounds, the middle-linear seven carbon linkage between two phenyl rings can be classified into four sub-groups,β-diketone, monoketone, pyrazole (N-N heterocyclic), and isoxazole (N-O hetorocyclic). Schematic diagram of the workflow is shown in Figure 1. All 143 curcumin analogs were docked against tyrosine kinase of HER2 by Glide SP docking. A few compounds of each sub-group in the docking top ranks were selected. All 24 chosen compounds were classified into four groups based on their core structures. From Table 1, compounds AS-KTC001 to AS-KTC011 were classified into β-diketone group. Monoketone composed of two compounds, AS-KTC012 and AS-KTC013. For AS-KTC014 to AS-KTC017 were categorized as pyrazole curcumin analogs. The last group, isoxazole curcumin analogs consisted of seven molecules, AS-KTC018 to AS-KTC024. The docked conformations of all curcumin compounds were well oriented in the ATP-binding pocket of HER2-TK (Figure 2C and 2D). One phenyl-end of curcuminoids compounds oriented well in deep hydrophobic pocket, while another phenyl-end exposed to the open gate. They could form interactions either with Met801 (adenine region) or with Cys805 (hydrophobic pocket II), depending on each configuration of analog (Figure 2D). According to the revealed three dimensional structure of HER2-TK adopted the active-like conformation, the binding cave stayed in the tunnel shape rather than the opened cave as presented in EGFR-TK. This pocket possesses approximately the volume of 475 Å3. As the binding pocket adopted the tunnel-like shape, the curcuminoid structures are also in the linear cylinder shape that can fit into this pocket quite well. The hydrophobic-I site merged with the phosphate binding region (Ser783, Arg784, Leu785, Leu769, Gly770, Ala771, Met774 and Phe864) as the deep semi-closed site of ATP-binding pocket. This pocket is occupied by the trifluoromethyl-phenoxy fragment of SYR, the co-crystal ligand (Figure 2A and 2B). Furthermore, one of the phenyl rings and its hydrophobic substituent groups of curcuminoids compounds were perfectly fit to this deep hydrophobic pocket. The functional groups of modified middle-linear seven carbon linkage of curcurmin analogs pointed to the DFG motif, especially the adjacent residue Thr862. In addition, another phenyl-end and its hydrophobic substituent groups of curcumin analogs could span around adenine region to hydrophobic-II site (Figure 2C and 2D). This observation was found as the common interaction among curcuminoid compounds, conformed to the pyridine and amine fragments of SYR pointed to the same residues (Figure 2A and 2B). These occupancy and interactions were considered important to increase selectivity and affinity for HER2 inhibitors [45–47]. However, the curcumin analogs could not interact with Met801 as any other EGFR/HER2-TK inhibitors, which contain 4-anilinoquinazoline, pyrrolopyrimidine, pyrrolotriazine and cyanoquinoline cores. The carbonyl group of the linkage chain, from β-diketone or monoketoncurcumins, could form a hydrogen bond directly with one of the key residues Thr862 (Thr862 OH---OC curcumins). This interaction differed from that of SYR127063 and other HER2 inhibitors that was a water-mediated hydrogen bond between N3 of quinazoline and Thr862 [48]. As the direct hydrogen bond formation at this position was crucial to enhance binding affinity, N3 of quinazoline was modified to nitrile group to form the direct hydrogen bond [48]. These occupancy and interactions mimicked those of a heterocyclic core of HER2 inhibitor in an adenine ring of ATP [49]. All 24 selected compounds have been processed in further experiments, to investigate the bioavailability profiles of curcumin analogs on two types of human breast cancer cell lines.
Figure 1.
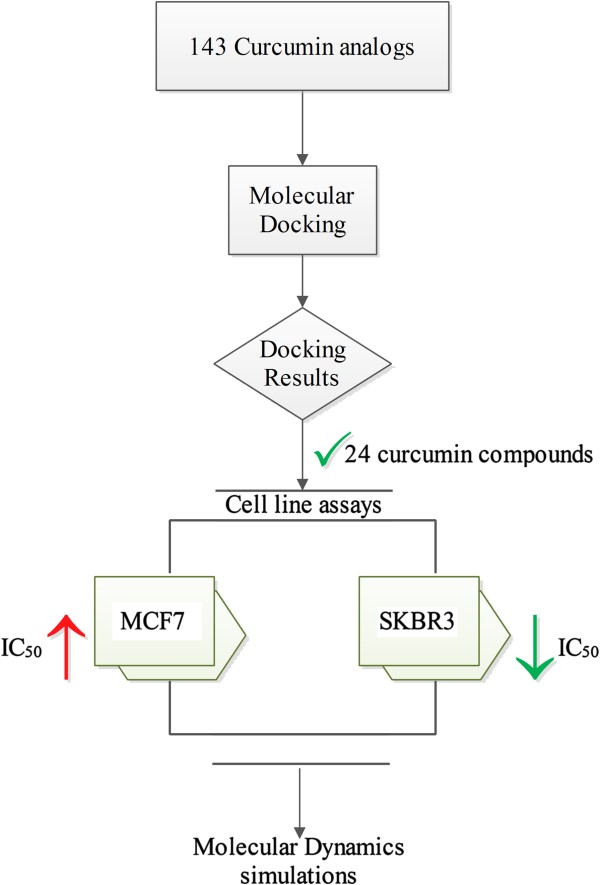
The rational workflow of this study, starting from in silico screening based on molecular docking and validated with cell line assays (SKBR3:a breast cancer cell line which over-expresses the HER2 gene product, MCF-7: a breast cancer cell line that absence of HER2 protein overexpression) and deep interaction study by molecular dynamic simulation.
Table 1.
Two dimensional structures of curcumin analogs and its Gscore
| Code | Structure | Dock score | Ref |
|---|---|---|---|
| AS-KTC001 |
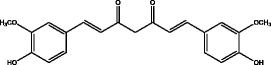
|
-8.35 | [21, 22, 25] |
| AS-KTC002 |
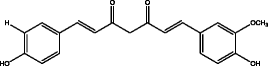
|
-8.17 | [21, 22, 25] |
| AS-KTC003 |
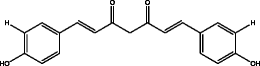
|
-8.79 | [21, 22, 25] |
| AS-KTC004 |
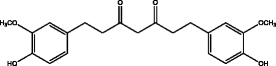
|
-8.24 | [21, 22, 25] |
| AS-KTC005 |
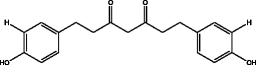
|
-8.65 | [21, 22, 25] |
| AS-KTC006 |
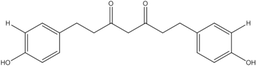
|
-8.18 | [21, 22, 25] |
| AS-KTC007 |
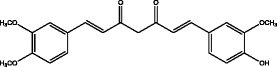
|
-8.20 | [21, 22] |
| AS-KTC008 |
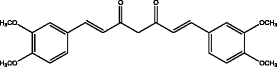
|
-6.40 | [21, 22] |
| AS-KTC009 |
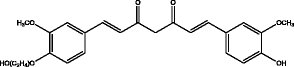
|
-8.66 | [21, 22] |
| AS-KTC010 |
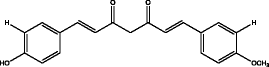
|
-9.03 | [21, 22] |
| AS-KTC011 |
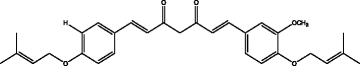
|
-9.26 | [21, 22] |
| AS-KTC012 |
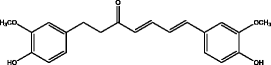
|
-8.56 | [21] |
| AS-KTC013 |
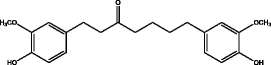
|
-8.24 | [22] |
| AS-KTC014 |
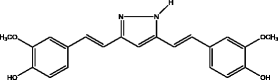
|
-8.10 | [22] |
| AS-KTC015 |
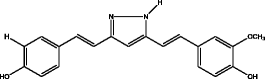
|
-7.71 | [22] |
| AS-KTC016 |
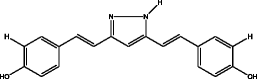
|
-7.82 | [22] |
| AS-KTC017 |
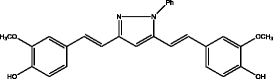
|
-8.61 | [22] |
| AS-KTC018 |
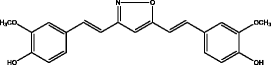
|
-8.04 | [22] |
| AS-KTC019 |
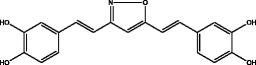
|
-8.67 | [22] |
| AS-KTC020 |
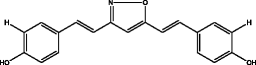
|
-7.76 | [22] |
| AS-KTC021 |
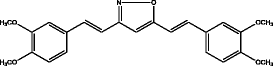
|
-7.12 | [22] |
| AS-KTC022 |
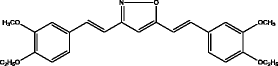
|
-7.14 | [22] |
| AS-KTC023 |
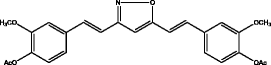
|
-7.35 | [22] |
| AS-KTC024 |
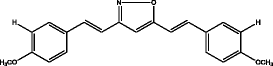
|
-7.33 | [22] |
Figure 2.
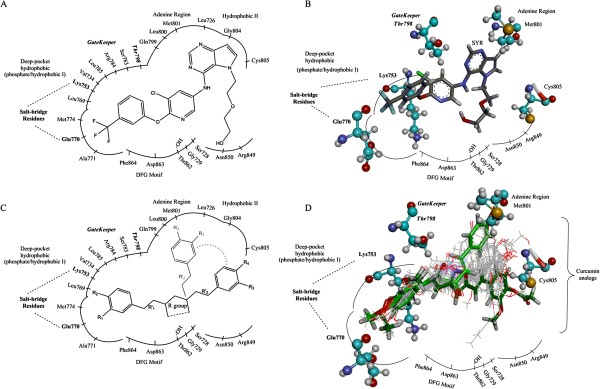
The illustration of A) two dimensional (2D) structure and B) three dimensional (3D) structure of SYR from the x-ray structure 3PP0, while panel C) and D) present 2D and 3D structures of curcumin analogs in the binding pocket of HER2-TK from the docking results.
The bioavailability profiles of curcumin analogs on two types of human breast cancer cell lines
The HER2-TK inhibitory assay was performed using a commercialized HER2-TK, but unfortunately, activity of the HER2-TK could not be detected (data not shown). Moreover curcumins were stable and soluble in very low pH which is not suitable for HER2-TK to stay active. Therefore, the cell-based assay was chosen instead of the purified protein based assay to investigate the inhibitory effect of curcumin analogs to HER2-TK. In order to identify the curcumin analogs against HER2-TK, two different types of human breast cancer cell lines, MCF-7 and SKBR3 were performed. The SKBR3 is a breast cancer cell line which over-expresses the HER2 gene product while the MCF-7 is in absence of HER2 protein overexpression [50–54]. Therefore, such compounds should be more effective against SKBR3 than MCF-7. The lapatinib which was tested on both breast cancer cell lines showed the IC50 on SKBR3 lower than MCF-7 [55]. Twenty-four selected compounds were determined IC50 on both breast cancer cell lines by MTT assay as shown in Table 2. Although most of curcumin analogs have very similar structures, each analog showed different activities on both cells. In the β-diketone group from Table 1, only AS-KTC006 showed the effective inhibiting to HER2-positive cancer cell-line (SKBR3), vice versa, lack of inhibiting to HER2-negative cancer cell-line (MCF-7). The core structure of this group composes of β-diketone which is modified substituent from natural curcumins. On the other hand none of curcumin analogs in monoketone and pyrazole groups showed selectively inhibiting activity among both cancer cell-lines. In the last group, AS-KTC021 also presented the outstanding suppress on SKBR3 but not MCF7 among isoxazole analogs. The IC50 for AS-KTC006 and AS-KTC021 in SKBR3 were 15.4 and 16.9 μM, respectively, and IC50 for both compounds in MCF7 were higher than 100 μM. Since both AS-KTC006 and AS-KTC021 were selected from docking results, they could inhibit the breast cancer cells through blocking of HER2-TK activities. Therefore, the AS-KTC006 and AS-KTC021 were chosen for further investigation the interaction mechanisms by molecular dynamics simulations.
Table 2.
The inhibitory activity profiles of curcumin analogs on MCF7 and SKBR3 cells
| SKBR3 | MCF7 | SKBR3 | MCF7 | ||
|---|---|---|---|---|---|
| AS-KTC | IC50 | IC50 | AS-KTC | IC50 | IC50 |
| (μM) | (μM) | (μM) | (μM) | ||
| 001 | 8.3 ± 0.6 | 41.9 ± 12.3 | 013 | > 100 | > 100 |
| 002 | 13.0 ± 1.8 | 44.4 ± 10.8 | 014 | 10.8 ± 5.5 | 9.9 ± 3.5 |
| 003 | 24.9 ± 2.3 | 81.6 ± 26.0 | 015 | 30.9 ± 4.5 | 15.4 ± 3.8 |
| 004 | > 100 | 79.4 ± 9.8 | 016 | 42.6 ± 5.5 | 33.8 ± 5.8 |
| 005 | > 100 | > 100 | 017 | 21.3 ± 3.8 | 14.3 ± 1.9 |
| 006 | 15.4 ± 3.9 | > 100 | 018 | 33.8 ± 7.2 | 22.4 ± 7.7 |
| 007 | 7.9 ± 2.5 | 17.5 ± 4.5 | 019 | > 100 | 38.9 ± 9.1 |
| 008 | 7.9 ± 2.5 | 36.7 ± 5.8 | 020 | 25 ± 5.5 | 44.1 ± 9.0 |
| 009 | 8.2 ± 0.4 | 22.1 ± 0.1 | 021 | 16.9 ± 3.4 | > 100 |
| 010 | > 100 | > 100 | 022 | > 100 | > 100 |
| 011 | > 100 | > 100 | 023 | > 100 | 24.3 ± 8.7 |
| 012 | 9.9 ± 1.0 | 14.3 ± 1.6 | 024 | > 100 | > 100 |
Molecular interaction, stability binding free energy via MM-PBSA
The molecular dynamics (MD) simulations were performed to examine the molecular interaction of the bothAS-KTC006 and AS-KTC021 curcuminoids in the ATP-binding pocket of HER2-TK. As mention in the previous section, the x-ray crystal structure of HER2-TK, 3PP0 has been used as the model reference template of this study. The complexes of HER2-TK-AS-KTC006 and HER2-TK-AS-KTC021 which were constructed using molecular docking procedure have been used as starting coordinates for MD calculations. The root mean square deviations (RMSD) of all systems (backbone atoms, ligand atoms and binding site atoms) seemed to fit nicely in the binding pocket of HER2-TK (Figure 3, Additional file 1: Table S3). The structure of curcuminoid distinguishes from other known tyrosine kinase inhibitors, which generally containing either quinazoline or pyrrolopyrimidine based structures [56]. The structures of curcumin analogs adopted the long thread with two knots at each end, resulting in freely flexible structure in the tunnel-like binding pocket. Interestingly, the MD results revealed that the hydrogen bonds between H-N atom of Met801 (located on adenine region) and N-atom at position N11 of SYR existed about 94.73% along the entire 30 ns of simulation time. On the other hand, the curcumin analogs possess the interesting hydrogen bond pairs in both AS-KTC006 and AS-KTC021. Rather than forming hydrogen bond with Met801, the β-diketone curcumin analog (AS-KTC006) formed hydrogen bonds to Thr862, Cys805 and Asp863, in the binding pocket of HER2-TK. The formation with these three amino acids existed approximately 34.67%, 10.17% and 9.17%, respectively along the entire MD simulations (Table 3). Distances chromatograms of each pairwise atom from H-bond analyses were shown in Figure 4. One of these three residues is DFG-motif (Asp863), and another is adjacent residue of DFG-motif (Thr862). For AS-KTC021 (isoxazole curcumin analog), the hydrogen boding with Cys805 and Thr862 existed about 35.97% and 5.37%, respectively, along the entire MD simulations. As illustrated in Figure 2C and 2D, one of the phenyl-end of curcumin compounds were well oriented in the ATP-binding pocket of HER2-TK, while the other-end exposed to solvent giving a chance to interact with Cys805, while the linker in the middle of cucurmin structure could interact with Thr862. The binding residues of SYR-HER2-TK systems appear to be stable along the MD simulations (Figure 3A-1 and 3A-2). The system started to converge since 15 ns of simulations time (Figure 3A). In addition, the simulation systems of AS-KTC006-HER2TK and AS-KTC021-HER2TK seem to converge after 25 ns of simulations time (Figure 3). From the molecular dynamic results suggested that AS-KTC006 had better binding affinity with HER2-TK than that of AS-KTC021. Considering at the curcuminoids structures, AS-KTC006 is more flexible than AS-KTC021. In particular, AS-KTC0006 possesses β-diketonemoiety on both sides providing the flexibility of the molecule and allowing O-atom of ketone to interact with Thr862. The binding energy calculations were performed to further understand the interactions of each system. It is to be noted that protein–ligand entropy contributions were not included in these free energy values since the present MM-PBSA are typically time consuming and unreliable. Considering the intermolecular interaction of the ligands with HER2-TK in contribution terms (Table 4), non-polar contributions, the summation of ΔEVDW and ΔGnp, are significant with all ligand(s)-HER2TK systems. Interestingly, ligand AS-KTC006 shows the most favorable electrostatic interaction with the binding pocket of HER2TK. There are the agreement between MM-PBSA binding energy calculations, docking and MD simulations. The consistent observations were presented in all three different computational approaches, namely, focusing on O-atom of ketone to interact with Thr862 of the system AS-KTC006-HER2TK complex. Recently, the computational model of anti-HER2 ligands, the analogous of 4-anilinoquinazoline were reported [57, 58]. These works showed that the vdW term could be a major factor of the ligand-protein interactions; hence, the deep hydrophobic pocket would be the selectivity pocket of HER2-TK [57, 58]. Rather than focusing on the 4-anilinoquinazoline core structure (the known HER2-TK inhibitors), we focused on the curcuminoid core structure in this study. Both selected curcumin analogs form the distinguish interaction moiety from the known inhibitors of HER2-TK. Furthermore, they occupied well in the deep hydrophobic pocket of HER2-TK.
Figure 3.
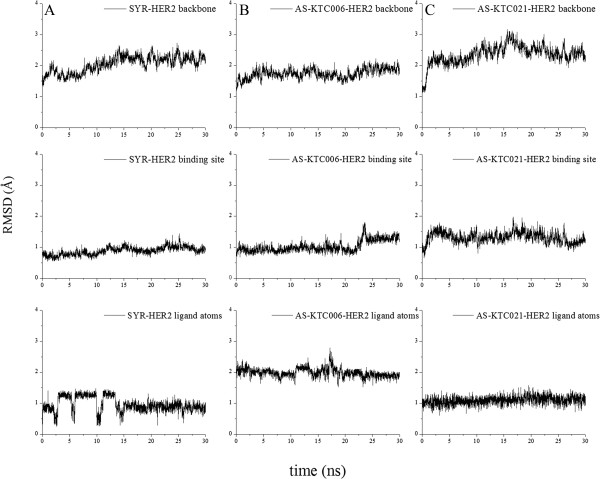
RMSD plots of each MD simulations, presenting the backbone (N, O, C and Cα atoms of HER2-TK, the binding residues atoms of HER2-TK and ligand atoms in upper, middle and lower rows, respectively, in the panel of A) SYR B) AS-KTC006 and C) AS-KTC021.
Table 3.
Conclusion of H-bonds between compounds and tyrosine kinase of HER2
| System | Donor | Acceptor | % occupied | distance (Å) | |||
|---|---|---|---|---|---|---|---|
| SYR-HER2TK | Met801 | N | H | SYR | N11 | 94.73 | 3.128 ± 0.15 |
| SYR | O1 | H1 | Asp863 | OD1 | 10.67 | 2.761 ± 0.17 | |
| AS-KTC006-HER2TK | Thr862 | OG1 | HG1 | AS-KTC006 | O24 | 34.67 | 2.851 ± 0.18 |
| Cys805 | N | H | AS-KTC006 | O25 | 10.17 | 3.191 ± 0.18 | |
| AS-KTC006 | O21 | H39 | Asp863 | O | 9.17 | 2.758 ± 0.16 | |
| AS-KTC021-HER2TK | Cys805 | N | H | AS-KTC021 | O22 | 35.97 | 3.142 ± 0.18 |
| Thr862 | OG1 | HG1 | AS-KTC021 | O21 | 5.37 | 3.100 ± 0.20 | |
Figure 4.
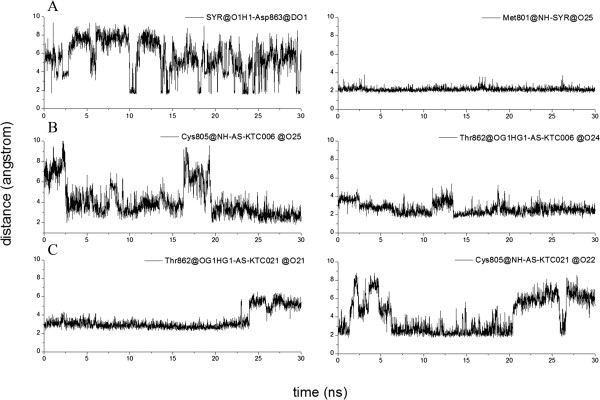
Distance between the pairwise atoms of H-bond analyses, A) SYR-HER2TK, B) AS-KTC006-HER2TK, and C) AS-KTC02-HER2TK.
Table 4.
Individual terms of MM-PBSA binding energy (kcal mol -1 ), entropy term excluded
| System | ‹∆E vdw› | ‹∆E ele› | ‹∆G pb› | ‹∆G np› | ‹∆G solv› | ‹∆G MMPBSA› | Nonpolar/hydrophobic | Polar/electrostatic | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SYR-HER2TK | -67.85 | (0.32) | -16.38 | (0.53) | 45.48 | (0.44) | -43.74 | (0.13) | 1.73 | (0.46) | -82.50 | (0.45) | -111.59 | (0.45) | 29.10 | (0.97) |
| AS-KTC006-HER2TK | -42.81 | (0.28) | -57.70 | (0.46) | 76.27 | (0.37) | -34.02 | (0.08) | 42.25 | (0.37) | -58.26 | (0.39) | -76.83 | (0.36) | 18.57 | (0.83) |
| AS-KTC021-HER2TK | -51.24 | (0.30) | -18.15 | (0.63) | 37.15 | (0.63) | -34.75 | (0.16) | 2.41 | ( 0.60) | -66.98 | (0.38) | -85.99 | (0.46) | 19.00 | (1.26) |
‹∆Evdw› and ‹∆Eele› - van der Waals and electrostatic contributions to binding energy.
‹∆Gpb› and ‹∆Gnp› - electrostatic and nonpolar contributions to the solvation free energy.
‹∆GMMPBSA› - final calculated MM-PBSA binding energy.
Nonpolar contribution = ∆Evdw + ∆Gnp; polar contribution = ∆Eele + ∆Gpb.
Conclusion
In the present study, we screened a series of curcumin compounds using a computational molecular docking. Then, the bioavailability assay of curcumin analogs, were conducted on two types of human breast cancer cell lines to select the specific active HER2 kinase inhibitors. The results suggested that bisdemethylcurcumin compound (AS-KTC006, CAS no. 60831-46-1) and 3,5-bis((E)-3,4-dimethoxystyryl)isoxazole (AS-KTC021, CAS no. 1118765-46-0) could inhibit breast cancer cell lines though HER2-TK. In addition, the intermolecular studies from MD simulation suggested that both selected curcumin analogs form the distinguish interaction moiety from the known inhibitors of HER2-TK. MM-PBSA binding calculation suggested that non-polar contributions are not only significant with all ligand(s)-HER2TK systems but also a major factor of the ligand-protein interactions.
Electronic supplementary material
Additional file 1: Table S1-S3. Table S1. List of 143 curcuminoids compounds, which used in this present study. Table S2. Explanation of each simulation steps of minimization and molecular dynamics simulations. Table S3. List of binding residues of each system, which mentioned in Figure 3. (DOCX 493 KB)
Acknowledgements
This work was supported by Faculty of Science and Grad School Research Fund, Kasetsart University, Kasetsart University Research and Development Institute, and National Research University Project of Thailand, the Office of the Higher Education Commission. We would like to thank Prof. Peter Wolschann, Institute of Theoretical Chemistry, University of Vienna, Austria for providing the Schrödinger programs and the National Center for Genetic Engineering and Biotechnology (BIOTEC) and the National Science and Technology Development Agency (NSTDA) for the use of high performance computer clusters.
Abbreviations
- HER2
Human Epidermal Growth Factor Receptor 2
- EGFR
Epidermal Growth Factor Receptor
- HER2-TK
Tyrosine kinase domain of HER2
- RMSD
Root-mean-square deviations
- SYR127063
2-{2-[4-({5-chloro-6-[3-(trifluoromethyl)phenoxy]pyridin-3-yl}amino)-5H-pyrrolo[3,2-d]pyrimidin-5- yl]ethoxy}ethanol
- TAK-285
N-{2-[4-({3-chloro-4-[3-(trifluoromethyl)phenoxy]phenyl}amino)-5H-pyrrolo[3,2-d]pyrimidin-5-yl]ethyl}-3- hydroxy-3-methylbutanamide
- Glide
Grid-based Ligand Docking with Energetics
- SP mode
Standard precision mode
- AS-KTC006
Bisdemethylcurcumin, also called Di-O-demethylcurcumin, Bis(3,4-Dihydroxy-trans-cinnamoyl)methane (CAS no. 60831-46-1)
- AS-KTC021
3,5-bis((E)-3,4-dimethoxystyryl)isoxazole (CAS no.1118765-46-0)
- SKBR3
A breast cancer cell line which over-expresses the HER2 gene product
- MCF-7
A breast cancer cell line that absence of HER2 protein overexpression.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
WY and OS carried out the molecular docking and drafted the manuscript. SS and NH carried out the cell study and performed the analysis. WM, ST and OS participated in molecular dynamic simulations and drafted the manuscript. AP prepared and provided all curcumin derivatives for the tests. OS and KC conceived the initial concept of the study, and participated in its design and coordination of drafting the manuscript. All authors read and approved the final manuscript.
Contributor Information
Wannarat Yim-im, Email: wanna_kim@hotmail.com.
Orathai Sawatdichaikul, Email: ifrots@ku.ac.th.
Suwanna Semsri, Email: ssemsri@gmail.com.
Natharinee Horata, Email: h_natharinee@hotmail.com.
Wanwimon Mokmak, Email: wanwimon.mok@biotec.or.th.
Sissades Tongsima, Email: sissades@biotec.or.th.
Apichart Suksamrarn, Email: s_apichart@ru.ac.th.
Kiattawee Choowongkomon, Email: fsciktc@ku.ac.th.
References
- 1.Holbro T, Civenni G, Hynes NE. The ErbB receptors and their role in cancer progression. Exp Cell Res. 2003;284(1):99–110. doi: 10.1016/S0014-4827(02)00099-X. [DOI] [PubMed] [Google Scholar]
- 2.Pinkas-Kramarski R, Eilam R, Alroy I, Levkowitz G, Lonai P, Yarden Y. Differential expression of NDF/neuregulin receptors ErbB-3 and ErbB-4 and involvement in inhibition of neuronal differentiation. Oncogene. 1997;15(23):2803–2815. doi: 10.1038/sj.onc.1201466. [DOI] [PubMed] [Google Scholar]
- 3.Pinkas-Kramarski R, Shelly M, Guarino BC, Wang LM, Lyass L, Alroy I, Alimandi M, Kuo A, Moyer JD, Lavi S, Eisenstein M, Ratzkin BJ, Seger R, Bacus SS, Pierce JH, Andrews GC, Yarden Y. ErbB tyrosine kinases and the two neuregulin families constitute a ligand-receptor network. Mol Cell Biol. 1998;18(10):6090–6101. doi: 10.1128/mcb.18.10.6090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tai W, Mahato R, Cheng K. The role of HER2 in cancer therapy and targeted drug delivery. J Control Release. 2010;146(3):264–275. doi: 10.1016/j.jconrel.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Klapper LN, Kirschbaum MH, Sela M, Yarden Y. Biochemical and clinical implications of the ErbB/HER signaling network of growth factor receptors. Adv Cancer Res. 2000;77:25–79. doi: 10.1016/S0065-230X(08)60784-8. [DOI] [PubMed] [Google Scholar]
- 6.Baselga J, Bradbury I, Eidtmann H, Di Cosimo S, de Azambuja E, Aura C, Gómez H, Dinh P, Fauria K, Van Dooren V, Aktan G, Goldhirsch A, Chang TW, Horváth Z, Coccia-Portugal M, Domont J, Tseng L, Kunz G, Sohn JH, Semiglazov V, Lerzo G, Palacova M, Probachai V, Pusztai L, Untch M, Gelber RD, Piccart-Gebhart M. Lapatinib with trastuzumab for HER2-positive early breast cancer (NeoALTTO): a randomised, open-label, multicentre, phase 3 trial. The Lancet. 2012;379(9816):633–640. doi: 10.1016/S0140-6736(11)61847-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aertgeerts K, Skene R, Yano J, Sang B-C, Zou H, Snell G, Jennings A, Iwamoto K, Habuka N, Hirokawa A, Ishikawa T, Tanaka T, Miki H, Ohta Y, Sogabe S. Structural analysis of the mechanism of inhibition and allosteric activation of the kinase domain of HER2 protein. J Biol Chem. 2011;286(21):18756–18765. doi: 10.1074/jbc.M110.206193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guarneri V, Lenihan DJ, Valero V, Durand JB, Broglio K, Hess KR, Michaud LB, Gonzalez-Angulo AM, Hortobagyi GN, Esteva FJ. Long-term cardiac tolerability of trastuzumab in metastatic breast cancer: the M.D. Anderson cancer center experience. J Clin Oncol. 2006;24(25):4107–4115. doi: 10.1200/JCO.2005.04.9551. [DOI] [PubMed] [Google Scholar]
- 9.Scaltriti M, Baselga J. The epidermal growth factor receptor pathway: A model for targeted therapy. Clin Cancer Res. 2006;12(18):5268–5272. doi: 10.1158/1078-0432.CCR-05-1554. [DOI] [PubMed] [Google Scholar]
- 10.Slamon D, Clark G, Wong S, Levin W, Ullrich A, McGuire W. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235:177–182. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- 11.Ishikawa T, Seto M, Banno H, Kawakita Y, Oorui M, Taniguchi T, Ohta Y, Tamura T, Nakayama A, Miki H, Kamiguchi H, Tanaka T, Habuka N, Sogabe S, Yano J, Aertgeerts K, Kamiyama K. Design and synthesis of novel human epidermal growth factor receptor 2 (HER2)/epidermal growth factor receptor (EGFR) dual inhibitors bearing a pyrrolo[3,2-d]pyrimidine scaffold. J Med Chem. 2011;54(23):8030–8050. doi: 10.1021/jm2008634. [DOI] [PubMed] [Google Scholar]
- 12.Ammon HP, Wahl MA. Pharmacology of Curcuma longa. Planta medica. 1991;57(1):1–7. doi: 10.1055/s-2006-960004. [DOI] [PubMed] [Google Scholar]
- 13.Aggarwal BB, Kumar A, Bharti AC. Anticancer potential of curcumin: preclinical and clinical studies. Anticancer Res. 2003;23(1A):363–398. [PubMed] [Google Scholar]
- 14.Sreejayan, Rao MN. Nitric oxide scavenging by curcuminoids. J Pharm Pharmacol. 1997;49(1):105–107. doi: 10.1111/j.2042-7158.1997.tb06761.x. [DOI] [PubMed] [Google Scholar]
- 15.Rao CV, Rivenson A, Simi B, Reddy BS. Chemoprevention of colon carcinogenesis by dietary curcumin, a naturally occurring plant phenolic compound. Cancer Res. 1995;55(2):259–266. [PubMed] [Google Scholar]
- 16.Kuttan R, Bhanumathy P, Nirmala K, George MC. Potential anticancer activity of turmeric (Curcuma longa) Canc Lett. 1985;29(2):197–202. doi: 10.1016/0304-3835(85)90159-4. [DOI] [PubMed] [Google Scholar]
- 17.Anuchapreeda S, Thanarattanakorn P, Sittipreechacharn S, Chanarat P, Limtrakul P. Curcumin inhibits WT1 gene expression in human leukemic K562 cells. Acta Pharmacol Sin. 2006;27(3):360–366. doi: 10.1111/j.1745-7254.2006.00291.x. [DOI] [PubMed] [Google Scholar]
- 18.Dorai T, Gehani N, Katz A. Therapeutic potential of curcumin in human prostate cancer. II. Curcumin inhibits tyrosine kinase activity of epidermal growth factor receptor and depletes the protein. Mol Urol. 2000;4(1):1–6. [PubMed] [Google Scholar]
- 19.Senft C, Polacin M, Priester M, Seifert V, Kogel D, Weissenberger J. The nontoxic natural compound Curcumin exerts anti-proliferative, anti-migratory, and anti-invasive properties against malignant gliomas. BMC cancer. 2010;10:491. doi: 10.1186/1471-2407-10-491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bengmark S. Curcumin, an atoxic antioxidant and natural NFkappaB, cyclooxygenase-2, lipooxygenase, and inducible nitric oxide synthase inhibitor: a shield against acute and chronic diseases. JPEN Journal of parenteral and enteral nutrition. 2006;30(1):45–51. doi: 10.1177/014860710603000145. [DOI] [PubMed] [Google Scholar]
- 21.Changtam C, de Koning HP, Ibrahim H, Sajid MS, Gould MK, Suksamrarn A. Curcuminoid analogs with potent activity against Trypanosoma and Leishmania species. Eur J Med Chem. 2010;45(3):941–956. doi: 10.1016/j.ejmech.2009.11.035. [DOI] [PubMed] [Google Scholar]
- 22.Changtam C, Hongmanee P, Suksamrarn A. Isoxazole analogs of curcuminoids with highly potent multidrug-resistant antimycobacterial activity. Eur J Med Chem. 2010;45(10):4446–4457. doi: 10.1016/j.ejmech.2010.07.003. [DOI] [PubMed] [Google Scholar]
- 23.Su J, Sripanidkulchai K, Suksamrarn A, Hu Y, Piyachuturawat P, Sripanidkulchai B. Pharmacokinetics and organ distribution of diarylheptanoid phytoestrogens from Curcuma comosa in rats. J Nat Med. 2012;66(3):468–475. doi: 10.1007/s11418-011-0607-x. [DOI] [PubMed] [Google Scholar]
- 24.Suksamrarn A, Ponglikitmongkol M, Wongkrajang K, Chindaduang A, Kittidanairak S, Jankam A, Yingyongnarongkul BE, Kittipanumat N, Chokchaisiri R, Khetkam P, Piyachaturawat P. Diarylheptanoids, new phytoestrogens from the rhizomes of Curcuma comosa: Isolation, chemical modification and estrogenic activity evaluation. Bioorg Med Chem. 2008;16(14):6891–6902. doi: 10.1016/j.bmc.2008.05.051. [DOI] [PubMed] [Google Scholar]
- 25.Tocharus J, Jamsuwan S, Tocharus C, Changtam C, Suksamrarn A. Curcuminoid analogs inhibit nitric oxide production from LPS-activated microglial cells. J Nat Med. 2012;66(2):400–405. doi: 10.1007/s11418-011-0599-6. [DOI] [PubMed] [Google Scholar]
- 26.Winuthayanon W, Suksen K, Boonchird C, Chuncharunee A, Ponglikitmongkol M, Suksamrarn A, Piyachaturawat P. Estrogenic activity of diarylheptanoids from Curcuma comosa Roxb. requires metabolic activation. J Agric Food Chem. 2009;57(3):840–845. doi: 10.1021/jf802702c. [DOI] [PubMed] [Google Scholar]
- 27.Korutla L, Kumar R. Inhibitory effect of curcumin on epidermal growth factor receptor kinase activity in A431 cells. Biochim Biophys Acta. 1994;1224(3):597–600. doi: 10.1016/0167-4889(94)90299-2. [DOI] [PubMed] [Google Scholar]
- 28.Korutla L, Cheung JY, Mendelsohn J, Kumar R. Inhibition of ligand-induced activation of epidermal growth factor receptor tyrosine phosphorylation by curcumin. Carcinogenesis. 1995;16(8):1741–1745. doi: 10.1093/carcin/16.8.1741. [DOI] [PubMed] [Google Scholar]
- 29.Hong RL, Spohn WH, Hung MC. Curcumin inhibits tyrosine kinase activity of p185neu and also depletes p185neu. Clin Cancer Res. 1999;5(7):1884–1891. [PubMed] [Google Scholar]
- 30.Tikhomirov O, Carpenter G. Identification of ErbB-2 kinase domain motifs required for geldanamycin-induced degradation. Cancer Res. 2003;63(1):39–43. [PubMed] [Google Scholar]
- 31.Cowley S, Paterson H, Kemp P, Marshall CJ. Activation of MAP kinase kinase is necessary and sufficient for PC12 differentiation and for transformation of NIH 3T3 cells. Cell. 1994;77(6):841–852. doi: 10.1016/0092-8674(94)90133-3. [DOI] [PubMed] [Google Scholar]
- 32.Minden A, Lin A, McMahon M, Lange-Carter C, Derijard B, Davis RJ, Johnson GL, Karin M. Differential activation of ERK and JNK mitogen-activated protein kinases by Raf-1 and MEKK. Science. 1994;266(5191):1719–1723. doi: 10.1126/science.7992057. [DOI] [PubMed] [Google Scholar]
- 33.Reddy S, Aggarwal BB. Curcumin is a non-competitive and selective inhibitor of phosphorylase kinase. FEBS letters. 1994;341(1):19–22. doi: 10.1016/0014-5793(94)80232-7. [DOI] [PubMed] [Google Scholar]
- 34.Liu JY, Lin SJ, Lin JK. Inhibitory effects of curcumin on protein kinase C activity induced by 12-O-tetradecanoyl-phorbol-13-acetate in NIH 3T3 cells. Carcinogenesis. 1993;14(5):857–861. doi: 10.1093/carcin/14.5.857. [DOI] [PubMed] [Google Scholar]
- 35.Catania A, Barrajon-Catalan E, Nicolosi S, Cicirata F, Micol V. Immunoliposome encapsulation increases cytotoxic activity and selectivity of curcumin and resveratrol against HER2 overexpressing human breast cancer cells. Breast Canc Res Treat. 2013;141(1):55–65. doi: 10.1007/s10549-013-2667-y. [DOI] [PubMed] [Google Scholar]
- 36.Xu YY, Cao Y, Ma H, Li HQ, Ao GZ. Design, synthesis and molecular docking of alpha, beta-unsaturated cyclohexanone analogous of curcumin as potent EGFR inhibitors with antiproliferative activity. Bioorg Med Chem. 2013;21(2):388–394. doi: 10.1016/j.bmc.2012.11.031. [DOI] [PubMed] [Google Scholar]
- 37.Yang SC, Chang SS, Chen CY. Identifying HER2 inhibitors from natural products database. PLoS ONE. 2011;6(12):e28793. doi: 10.1371/journal.pone.0028793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47(7):1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 39.Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, Banks JL. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem. 2004;47(7):1750–1759. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- 40.Duan Y, Wu C, Chowdhury S, Lee MC, Xiong G, Zhang W, Yang R, Cieplak P, Luo R, Lee T. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J Comput Chem. 2003;24(16):1999–2012. doi: 10.1002/jcc.10349. [DOI] [PubMed] [Google Scholar]
- 41.Cerutti DS, Duke R, Freddolino PL, Fan H, Lybrand TP. A vulnerability in popular molecular dynamics packages concerning Langevin and Andersen dynamics. J Chem Theor Comput. 2008;4(10):1669–1680. doi: 10.1021/ct8002173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ryckaert J-P, Ciccotti G, Berendsen HJC. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys. 1977;23(3):327–341. doi: 10.1016/0021-9991(77)90098-5. [DOI] [Google Scholar]
- 43.Swanson JMJ, Henchman RH, McCammon JA. Revisiting free energy calculations: a theoretical connection to MM/PBSA and direct calculation of the association free energy. Biophys J. 2004;86(1):67–74. doi: 10.1016/S0006-3495(04)74084-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wittayanarakul K, Hannongbua S, Feig M. Accurate prediction of protonation state as a prerequisite for reliable MM‒PB (GB) SA binding free energy calculations of HIV‒1 protease inhibitors. J Comput Chem. 2007;29(5):673–685. doi: 10.1002/jcc.20821. [DOI] [PubMed] [Google Scholar]
- 45.Jani JP, Finn RS, Campbell M, Coleman KG, Connell RD, Currier N, Emerson EO, Floyd E, Harriman S, Kath JC, Morris J, Moyer JD, Pustilnik LR, Rafidi K, Ralston S, Rossi AM, Steyn SJ, Wagner L, Winter SM, Bhattacharya SK. Discovery and pharmacologic characterization of CP-724,714, a selective ErbB2 tyrosine kinase inhibitor. Cancer Res. 2007;67(20):9887–9893. doi: 10.1158/0008-5472.CAN-06-3559. [DOI] [PubMed] [Google Scholar]
- 46.Rusnak DW, Affleck K, Cockerill SG, Stubberfield C, Harris R, Page M, Smith KJ, Guntrip SB, Carter MC, Shaw RJ, Jowett A, Stables J, Topley P, Wood ER, Brignola PS, Kadwell SH, Reep BR, Mullin RJ, Alligood KJ, Keith BR, Crosby RM, Murray DM, Knight WB, Gilmer TM, Lackey K. The characterization of novel, dual ErbB-2/EGFR, tyrosine kinase inhibitors: potential therapy for cancer. Cancer Res. 2001;61(19):7196–7203. [PubMed] [Google Scholar]
- 47.Wood ER, Truesdale AT, McDonald OB, Yuan D, Hassell A, Dickerson SH, Ellis B, Pennisi C, Horne E, Lackey K, Alligood KJ, Rusnak DW, Gilmer TM, Shewchuk L. A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Res. 2004;64(18):6652–6659. doi: 10.1158/0008-5472.CAN-04-1168. [DOI] [PubMed] [Google Scholar]
- 48.Wissner A, Berger DM, Boschelli DH, Floyd MB, Jr, Greenberger LM, Gruber BC, Johnson BD, Mamuya N, Nilakantan R, Reich MF, Bertucci F. 4-Anilino-6,7-dialkoxyquinoline-3-carbonitrile inhibitors of epidermal growth factor receptor kinase and their bioisosteric relationship to the 4-anilino-6,7-dialkoxyquinazoline inhibitors. J Med Chem. 2000;43(17):3244–3256. doi: 10.1021/jm000206a. [DOI] [PubMed] [Google Scholar]
- 49.Arkin M, Moasser MM. HER-2-directed, small-molecule antagonists. Curr Opin Investig Drugs. 2008;9(12):1264–1276. [PMC free article] [PubMed] [Google Scholar]
- 50.Charafe-Jauffret E, Ginestier C, Monville F, Finetti P, Adelaide J, Cervera N, Fekairi S, Xerri L, Jacquemier J, Birnbaum D, Bertucci F. Gene expression profiling of breast cell lines identifies potential new basal markers. Oncogene. 2006;25(15):2273–2284. doi: 10.1038/sj.onc.1209254. [DOI] [PubMed] [Google Scholar]
- 51.Lacroix M, Leclercq G. Relevance of breast cancer cell lines as models for breast tumours: an update. Breast Canc Res Treat. 2004;83(3):249–289. doi: 10.1023/B:BREA.0000014042.54925.cc. [DOI] [PubMed] [Google Scholar]
- 52.Lacroix M, Toillon RA, Leclercq G. p53 and breast cancer, an update. Endocr Relat Canc. 2006;13(2):293–325. doi: 10.1677/erc.1.01172. [DOI] [PubMed] [Google Scholar]
- 53.Levenson AS, Jordan VC. MCF-7: the first hormone-responsive breast cancer cell line. Cancer Res. 1997;57(15):3071–3078. [PubMed] [Google Scholar]
- 54.Ross DT, Perou CM. A comparison of gene expression signatures from breast tumors and breast tissue derived cell lines. Dis Markers. 2001;17(2):99–109. doi: 10.1155/2001/850531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cai X, Zhai H-X, Wang J, Forrester J, Qu H, Yin L, Lai C-J, Bao R, Qian C. Discovery of 7-(4-(3-Ethynylphenylamino)-7-methoxyquinazolin-6-yloxy)-N-hydroxyheptanamide (CUDC-101) as a Potent Multi-Acting HDAC, EGFR, and HER2 Inhibitor for the Treatment of Cancer. J Med Chem. 2010;53(5):2000–2009. doi: 10.1021/jm901453q. [DOI] [PubMed] [Google Scholar]
- 56.Spector N, Xia W, El-Hariry I, Yarden Y, Bacus S. HER2 therapy. Small molecule HER-2 tyrosine kinase inhibitors. Breast Cancer Res. 2007;9(2):1–8. doi: 10.1186/bcr1652. [DOI] [Google Scholar]
- 57.Ahmed M, Sadek MM, Abouzid KA, Wang F. In silico design: extended molecular dynamic simulations of a new series of dually acting inhibitors against EGFR and HER2. J Mol Graph Model. 2013;44:220–231. doi: 10.1016/j.jmgm.2013.06.004. [DOI] [PubMed] [Google Scholar]
- 58.Ahmed M, Sadek MM, Serrya RA, Kafafy A-HN, Abouzid KA, Wang F. Assessment of new anti-HER2 ligands using combined docking, QM/MM scoring and MD simulation. J Mol Graph Model. 2013;40:91–98. doi: 10.1016/j.jmgm.2012.12.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1-S3. Table S1. List of 143 curcuminoids compounds, which used in this present study. Table S2. Explanation of each simulation steps of minimization and molecular dynamics simulations. Table S3. List of binding residues of each system, which mentioned in Figure 3. (DOCX 493 KB)