

Abstract
Logistic regression is usually applied to investigate the association between inherited genetic variants and a binary disease phenotype. A limitation of standard methods used to estimate the parameters of logistic regression models is their strong dependence on a few observations deviating from the majority of the data.
We used data from the Genetic Analysis Workshop 18 to explore the possible benefit of robust logistic regression to estimate the genetic risk of hypertension. The comparison between standard and robust methods relied on the influence of departing hypertension profiles (outliers) on the estimated odds ratios, areas under the receiver operating characteristic curves, and clinical net benefit.
Our results confirmed that single outliers may substantially affect the estimated genotype relative risks. The ranking of variants by probability values was different in standard and in robust logistic regression. For cutoff probabilities between 0.2 and 0.6, the clinical net benefit estimated by leave-one-out cross-validation in the investigated sample was slightly larger under robust regression, but the overall area under the receiver operating characteristic curve was larger for standard logistic regression. The potential advantage of robust statistics in the context of genetic association studies should be investigated in future analyses based on real and simulated data.
Background
Hypertension is a common chronic medical condition characterized by elevated arterial blood pressure. High blood pressure is associated with an increased risk of stroke, heart attack, and other serious diseases. Age, gender, tobacco smoking, alcohol consumption, and high body mass index constitute established risk factors for hypertension [1]. A genetic component has also been postulated. It has been shown that individuals with a family history of hypertension have on average a higher blood pressure than individuals without a family history. Yanek et al found a 44% higher prevalence of hypertension in siblings of affected persons than in the general reference population [2]. In a Canadian study, standardized risk ratios of hypertension were higher for first-degree relatives than for spouses of probands with hypertension [3]. In genetic studies, a large number of polymorphisms has been associated with hypertension and validated in independent collectives; 14 loci have been identified (as of 2010) and many genetic studies are currently in progress [4-8].
The relationship between inherited genetic polymorphisms and a binary response variable (with/without hypertension) can be investigated using logistic regression models that simultaneously consider the effects of multiple risk factors. Standard methods used to estimate the parameters of logistic regression models--for example, iteratively reweighted least squares--are limited by their dependence on a few observations departing from the majority of the data. This contrasts with the purpose of genetic risk models that aim to predict a particular health outcome that holds for the bulk of individuals, and to identify persons with a deviating high risk of disease. We use data from the Genetic Analysis Workshop (GAW18) to explore the possible benefit of robust parameter estimates in logistic regression models for the genetic prediction of hypertension risk.
Methods
The analysed data (real phenotypes) were derived from 142 unrelated individuals who participated in the San Antonio Family Heart or Family Diabetes/Gallbladder studies. Longitudinal information on hypertension, age, gender, and current tobacco smoking was measured up to 4 times per individual; the present analyses relied on the first available measurement. Further information is provided in several articles [9-12].
The original data was filtered according to the following criteria: (a) at least 1 measurement with complete information on hypertension and age, (b) monomorphisms were excluded and each polymorphism had to be represented by at least 2 individuals, (c) individuals with more than 5% missing genotypes were excluded, and, finally, (d) variants with missing data in any individual were removed.
The relationship between hypertension and age, gender, and current tobacco smoking was first investigated by χ2 tests. Covariates significantly associated at the 5% confidence level entered the intercept-only model to build the baseline model. Subsequently, standard logistic regression (iteratively reweighted least squares) was used to identify possible hypertension-associated single-nucleotide polymorphisms (SNPs) with minimal deviance, taking into account associated covariates. The deviance is defined as minus twice the logarithm of the likelihood. Genotypes were coded according to an additive penetrance model; that is, 0, 1, and 2. Departing observations (outliers) according to standard logistic regression were identified based on the Cook's distance in the baseline model. The Cook's distance for observation i is defined as
where denotes the full regression model prediction for observation j, represents the regression model prediction for observation j estimated omitting observation i, and MSE indicates the mean square error of the regression model with q explanatory variables.
To investigate the possible benefit of robust parameter estimates in logistic regression, model coefficients were also estimated by solving
where with the Pearson residuals and the Huber function
with the ith diagonal element of the matrix , and .
This estimator is based on a quasi-likelihood, asymptotically normally distributed and Fisher consistent [13]. The objective of the Huber function is to downweight the influence of outliers and to assign inliers the usual weight. Variable selection under robust logistic regression relied on the minimal quasideviance as described by Cantoni and Ronchetti, which is a robust test statistic for model selection [13]. The quasideviance between 2 nested models is defined as
where with such that and such that and the estimated linear predictor is associated to the estimate of β and is associated to which is the estimate of . Linkage disequilibrium was not accounted for during variant selection neither for standard logistic regression nor for robust logistic regression.
Our comparison of the performance of standard and robust logistic regression was based on different statistics. First, standard and robust estimates of age effects were used to exemplify the potential influence of departing observations. Because of a different handling of outliers, it was expected that different age-genotype models were selected under standard and robust logistic regression. Consequently, the areas under the receiver operating characteristic curves (AUCs) were subsequently compared in order to investigate the discriminative performance of the selected models. Comparisons were conducted for the complete data set and after exclusion of potential outliers.
In addition, concordance, sensitivity, specificity, clinical net benefit, and AUCs were estimated for age-genotype models using a leave-one-out cross-validation approach [14]. Concordance was defined as the proportion of correctly estimated hypertension statuses using several cutoff values for the predicted affection probability. The clinical net benefit (NB) was defined by
where c is the chosen threshold for allocating an individual to the cases based on the logistic regression probability estimate. Note that the net benefit depends on the hypertension prevalence in the study population. The standard and robust logistic regression models were also compared based on the integrated discrimination index (IDI) estimated by cross-validation
where , , , and denote the probability estimates from the robust and standard logistic regression models for cases and controls [15]. This index represents the difference in the discrimination slopes of the 2 compared models. A positive IDI indicates that the robust model discriminates better between hypertensive and normotensive individuals than the standard model. Statistical analyses were carried out using the statistical language R, version 2.15.1 [16].
Results
χ2 tests revealed no influence of gender (p = 0.95) and tobacco smoking (p = 1.00) on hypertension risk. Hence, only age was included in the logistic regression models as covariate. Filter criteria resulted in 130 individuals (43 cases and 87 controls) with complete genotype and phenotype information. The age of the individuals ranged between 20 and 95 years with a median age of 52 years. The total number of measured SNPs on chromosome 3 in the investigated GAW18 data set was 35,045.
A plot of Cook's distances under the age-only standard logistic regression model revealed several observations (Figure 1) that departed from the majority of the sample. Considering a threshold of 0.05 for the Cook's distance, 4 observations could be defined as outliers. Information on disease status and age of deviating individuals is shown in Table 1. Individuals 62, 58, and 24 were older than 80 years and normotensive. Individual number 60 was affected by the condition early in life, at 38 years of age. Table 1 shows the influence of the 4 identified outliers on standard and robust parameter estimates of age effects. For example, the exclusion of individual 62 resulted in an 11.2% increase of the excess risk of hypertension per year according to standard logistic regression, compared to a 7.8% increase for robust logistic regression. Table 2 shows the odds of hypertension by age interval.
Figure 1.
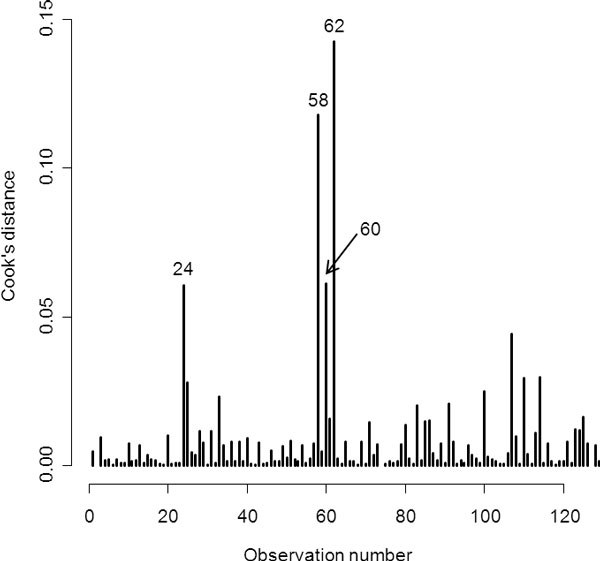
Cook's distances from the age-only standard logistic regression model. The 4 most prominent outliers are indicated by their observation number.
Table 1.
Estimated odds ratios per year of age
| Excluded individuals |
HTN | Age | Standard logistic regression | Robust logistic regression | ||
|---|---|---|---|---|---|---|
| OR-Age (95% CI) | % Change | OR-Age (95% CI) | % Change | |||
| None | 1.085 (1.050, 1.121) | ref. | 1.084 (1.048, 1.122) | ref. | ||
| 62 | 0 | 90.23 | 1.095 (1.057, 1.133) | +11.2% | 1.091 (1.052, 1.131) | +7.8% |
| 58 | 0 | 87.66 | 1.094 (1.056, 1.132) | +10.0% | 1.091 (1.052, 1.131) | +7.9% |
| 60 | 1 | 38.44 | 1.091 (1.054, 1.128) | +6.5% | 1.089 (1.051, 1.128) | +5.1% |
| 24 | 0 | 80.27 | 1.091 (1.054, 1.128) | +6.6% | 1.091 (1.052, 1.131) | +7.6% |
Odds ratios (ORs) were estimated based on standard and robust logistic regression models for the complete set of individuals and after exclusion of the 4 most remarkable outliers.
HTN: Hypertension.
Table 2.
Overall odds of hypertension per age interval
| Age interval (number of cases-to-controls) | |||
|---|---|---|---|
| <39.0 (1:22) | [39.0, 46.0) (2:20) | [46.0, 56.2) (9:23) | ≥56.2 (31:22) |
| 0.05 | 0.10 | 0.39 | 1.41 |
Age intervals were defined by the age quartiles in controls.
Standard logistic regression identified SNP rs3934103 located in the ULK4 gene as the variant that most improved the model fit. Robust logistic regression identified SNP rs11918360 in RP11-408H1.3 as the variant with the strongest association signal. Under both standard and robust regression, model selection clearly favored the 2 identified SNPs as represented in Figure 2. The pairwise r2 between SNP rs3934103 and SNP rs11918360 was 0.003.
Figure 2.
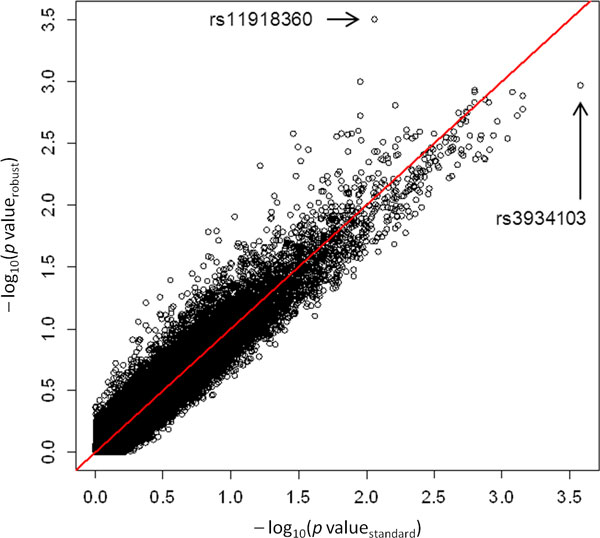
Quantile-quantile plots from the age-genotype standard and robust logistic regression models. The 2 selected SNPs are indicated by their reference SNP ID number.
Table 3 shows the influence of the 4 outliers on the AUCs from the standard and robust logistic regression models. Robust and standard AUCs for the age-only models were identical. For the age-genotype models, the AUCs were slightly smaller and also slightly less outlier-dependent for robust logistic regression than for standard logistic regression.
Table 3.
Area under the receiver operating characteristic curve (AUC)
| Excluded individuals |
Standard logistic regression | Robust logistic regression | ||||||
|---|---|---|---|---|---|---|---|---|
| AUC-Age (% Change) |
AUC-Age + SNP (% Change) |
AUC-Age (% Change) |
AUC-Age + SNP (% Change) |
|||||
| None | 0.811 | (ref.) | 0.852 | (ref.) | 0.811 | (ref.) | 0.843 | (ref.) |
| 62 | 0.820 | +1.1% | 0.861 | +1.1% | 0.820 | +1.1% | 0.852 | +1.0% |
| 58 | 0.820 | +1.1% | 0.861 | +1.1% | 0.820 | +1.1% | 0.853 | +1.2% |
| 60 | 0.825 | +1.7% | 0.859 | +0.9% | 0.825 | +1.7% | 0.851 | +0.9% |
| 24 | 0.819 | +1.0% | 0.859 | +0.9% | 0.819 | +1.0% | 0.844 | +0.0% |
AUCs were calculated for the complete set of individuals and after exclusion of the 4 most remarkable outliers. The relative contributions of the variables age and SNP (rs3934103 and rs11918360, respectively) are also shown.
Table 4 summarizes the results from the leave-one-out cross-validation. The concordance was better for the robust logistic regression model at every cutoff probability. Both models allocated best at probability 0.5 and almost identically at probability 0.3 (the investigated population included 43 cases and 87 controls; that is 33% hypertension prevalence). At a probability of 0.3, sensitivities were identical and the specificity was slightly higher under robust regression. Standard and robust estimates showed similar discriminative performances supported by an IDI of −0.07 at every cutoff probability. AUCs were also almost identical. The clinical net benefit was slightly larger for the robust logistic regression model in the probability range between 0.2 and 0.6.
Table 4.
Concordance, sensitivity, specificity, clinical net benefit, and overall AUCs.
| Probability cutoff | Standard logistic regression | Robust logistic regression | ||||||
|---|---|---|---|---|---|---|---|---|
| Concordance N (%) |
Sensitivity | Specificity | Clinical net benefit | Concordance N (%) |
Sensitivity | Specificity | Clinical net benefit | |
| 0.0 | 43 (33.1) | 1.00 | 0.00 | 0.33 | 43 (33.1) | 1.00 | 0.00 | 0.33 |
| 0.1 | 79 (60.8) | 0.95 | 0.44 | 0.27 | 82 (63.1) | 0.88 | 0.51 | 0.26 |
| 0.2 | 90 (69.2) | 0.86 | 0.61 | 0.22 | 97 (74.6) | 0.86 | 0.69 | 0.23 |
| 0.3 | 98 (75.4) | 0.81 | 0.72 | 0.19 | 99 (76.2) | 0.81 | 0.74 | 0.19 |
| 0.4 | 98 (75.4) | 0.70 | 0.78 | 0.13 | 102 (78.5) | 0.72 | 0.82 | 0.16 |
| 0.5 | 101 (77.7) | 0.60 | 0.86 | 0.11 | 107 (82.3) | 0.67 | 0.90 | 0.15 |
| 0.6 | 97 (74.6) | 0.40 | 0.92 | 0.05 | 102 (78.5) | 0.51 | 0.92 | 0.09 |
| 0.7 | 99 (76.2) | 0.35 | 0.97 | 0.06 | 100 (76.9) | 0.42 | 0.94 | 0.05 |
| 0.8 | 93 (71.5) | 0.19 | 0.98 | 0.00 | 97 (74.6) | 0.30 | 0.97 | 0.01 |
| 0.9 | 91 (70.0) | 0.12 | 0.99 | −0.03 | 93 (71.5) | 0.19 | 0.98 | −0.08 |
| 1.0 | 87 (66.9) | 0.00 | 1.00 | - | 87 (66.9) | 0.00 | 1.00 | - |
| AUC | 0.835 | 0.830 | ||||||
These characteristics rely on the age-genotype models for standard and robust logistic regression estimated based on leave-one-out cross-validation.
Discussion
Present results confirmed that single individuals (1/130 = 0.8% of the observations) with a departing risk of hypertension may substantially affect the overall risk estimates in the baseline model, causing up to an 11.2% change in the estimated excess risk of hypertension per year according to standard logistic regression in the present exercise.
The identification of outliers is relatively straightforward using routine diagnostic plots, but outlier management is extremely challenging. For example, the specification of thresholds for outlier definition is often arbitrary. Robust statistics aim to generate estimates that hold for the majority of the population using complete data. The unequal weighting of outliers by standard and robust regression resulted in prediction models that included different genetic variants.
Although robust estimates of age effects and AUCs for age-genotype models were less sensitive to outliers than standard estimates in the investigated sample, cross-validation AUCs based on standard and robust logistic regression, as well as IDI, were almost identical. The other investigated performance characteristics (concordance, sensitivity, specificity, and clinical net benefit) were equal or better for robust logistic regression around the probability that reflects the case-control ratio.
The standard logistic regression model selected 1 variant in the ULK4 gene. It was previously shown that variants in this gene are associated with hypertension [4,17]. Among others, 4 variants (rs2272007, rs3774372, rs1716975, rs1052501) mentioned in the 2 publications were also genotyped in the GAW18 collective, and we found them to be in linkage disequilibrium (r2 values 0.83, 0.73, 0.83, and 0.83, respectively) with the associated SNP rs3934103.
Conclusions
Preliminary findings suggest some advantage of robust statistics in the context of genetic association studies. However, present results were limited to a given sample size, as well as to particular genetic effect sizes and proportions of outliers. Additional analyses based on both real data and more general simulated scenarios should be conducted to validate initial findings.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
MK analysed and interpreted the data, created figures and tables, searched literature, and drafted the manuscript. CL identified relevant literature, supported data analysis and interpretation, and reviewed the manuscript. BP and MKa supported data analysis and interpretation and reviewed the manuscript. CF and UH supported interpretation and reviewed the manuscript. JLB formulated study goals, supported data analysis and interpretation, and reviewed the manuscript. All authors read and approved the final version.
Contributor Information
Miriam Kesselmeier, Email: miriam.kesselmeier@med.uni-jena.de.
Carine Legrand, Email: legrand@imbi.uni-heidelberg.de.
Barbara Peil, Email: peil@imbi.uni-heidelberg.de.
Maria Kabisch, Email: m.kabisch@dkfz-heidelberg.de.
Christine Fischer, Email: christine.fischer@med.uni-heidelberg.de.
Ute Hamann, Email: u.hamann@dkfz-heidelberg.de.
Justo Lorenzo Bermejo, Email: lorenzo@imbi.uni-heidelberg.de.
Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (DFG) grant SFB/TRR77 (Project Z2).
The GAW18 whole genome sequence data were provided by the T2D-GENES Consortium, which is supported by NIH grants U01 DK085524, U01 DK085584, U01 DK085501, U01 DK085526, and U01 DK085545. The other genetic and phenotypic data for GAW18 were provided by the San Antonio Family Heart Study and San Antonio Family Diabetes/Gallbladder Study, which are supported by NIH grants P01 HL045222, R01 DK047482, and R01 DK053889. The Genetic Analysis Workshop is supported by NIH grant R01 GM031575.
This article has been published as part of BMC Proceedings Volume 8 Supplement 1, 2014: Genetic Analysis Workshop 18. The full contents of the supplement are available online at http://www.biomedcentral.com/bmcproc/supplements/8/S1. Publication charges for this supplement were funded by the Texas Biomedical Research Institute.
References
- Jonas BS, Franks P, Ingram DD. Are symptoms of anxiety and depression risk factors for hypertension? Longitudinal evidence from the National Health and Nutrition Examination Survey I Epidemiologic Follow-up Study. Arch Fam Med. 1997;6:43–49. doi: 10.1001/archfami.6.1.43. [DOI] [PubMed] [Google Scholar]
- Yanek LR, Moy TF, Blumenthal RS, Raqueño JV, Yook RM, Hill MN, Becker LC, Becker DM. Hypertension among siblings of persons with premature coronary heart disease. Hypertension. 1998;32:123–128. doi: 10.1161/01.HYP.32.1.123. [DOI] [PubMed] [Google Scholar]
- Katzmarzyk PT, Rankinen T, Pérusse L, Rao DC, Bouchard C. Familial risk of high blood pressure in the Canadian population. Am J Hum Biol. 2001;13:620–625. doi: 10.1002/ajhb.1100. [DOI] [PubMed] [Google Scholar]
- Levy D, Ehret GB, Rice K, Verwoert GC, Launer LJ, Dehghan A, Glazer NL, Morrison AC, Johnson AD, Aspelund T. et al. Genome-wide association study of blood pressure and hypertension. Nat Genet. 2009;41:677–687. doi: 10.1038/ng.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton-Cheh C, Johnson T, Gateva V, Tobin MD, Bochud M, Coin L, Najjar SS, Zhao JH, Heath SC, Eyheramendy S. et al. Genome-wide association study identifies eight loci associated with blood pressure. Nat Genet. 2009;41:666–676. doi: 10.1038/ng.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, O'Connell JR, McArdle PF, Wade JB, Dorff SE, Shah SJ, Shi X, Pan L, Rampersaud E, Shen H. et al. Whole-genome association study identifies STK39 as a hypertension susceptibility gene. Proc Natl Acad Sci U S A. 2009;106:226–231. doi: 10.1073/pnas.0808358106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehret GB. Genome-wide association studies: contribution of genomics to understanding blood pressure and essential hypertension. Curr Hypertens Rep. 2010;12:17–25. doi: 10.1007/s11906-009-0086-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmanabhan S, Newton-Cheh C, Dominiczak AF. Genetic basis of blood pressure and hypertension. Trends Genet. 2012;28:397–408. doi: 10.1016/j.tig.2012.04.001. [DOI] [PubMed] [Google Scholar]
- Mitchell BD, Kammerer CM, Blangero J, Mahaney MC, Rainwater DL, Dyke B, Hixson JE, Henkel RD, Sharp RM, Comuzzie AG. et al. Genetic and environmental contributions to cardiovascular risk factors in Mexican Americans: The San Antonio Family Heart Study. Circulation. 1996;94:2159–2170. doi: 10.1161/01.CIR.94.9.2159. [DOI] [PubMed] [Google Scholar]
- Duggirala R, Blangero J, Almasy L, Dyer TD, Williams KL, Leach RJ, O'Connell P, Stern MP. Linkage of type 2 diabetes mellitus and of age at onset to a genetic location on chromosome 10q in Mexican Americans. Am J Hum Genet. 1999;64:1127–1140. doi: 10.1086/302316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt KJ, Lehman DM, Arya R, Fowler S, Leach RJ, Göring HH, Almasy L, Blangero J, Dyer TD, Duggirala R. et al. Genome-wide linkage analyses of type 2 diabetes in Mexican Americans: the San Antonio Family Diabetes/Gallbladder Study. Diabetes. 2005;54:2655–2662. doi: 10.2337/diabetes.54.9.2655. [DOI] [PubMed] [Google Scholar]
- Almasy L, Dyer TD, Peralta JM, Jun G, Fuchsberger C, Almeida MA, Kent JW Jr, Fowler S, Duggirala R, Blangero J. Data for Genetic Analysis Workshop 18: human whole genome sequence, blood pressure, and simulated phenotypes in extended pedigrees. BMC Proc. 2014;8(suppl 2):S2. doi: 10.1186/1753-6561-8-S1-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantoni E, Ronchetti E. Robust inference for generalized linear models. J Am Stat Assoc. 2001;96:1022–1030. doi: 10.1198/016214501753209004. [DOI] [Google Scholar]
- Vickers AJ, Elkin EB. Decision curve analysis: a novel method for evaluating prediction models. Med Decis Making. 2006;26:565–574. doi: 10.1177/0272989X06295361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pencina MJ, D'Agostino RB Sr, D'Agostino RB Jr, Vasan RS. Evaluating the added predictive ability of a new marker: from area under the ROC curve to reclassification and beyond. Stat Med. 2008;27:157–172. doi: 10.1002/sim.2929. [DOI] [PubMed] [Google Scholar]
- R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. 2012. http://www.R-project.org/
- Ho JE, Levy D, Rose L, Johnson AD, Ridker PM, Chasman DI. Discovery and replication of novel blood pressure genetic loci in the Women's Genome Health Study. J Hypertens. 2011;29:62–69. doi: 10.1097/HJH.0b013e3283406927. [DOI] [PMC free article] [PubMed] [Google Scholar]