

Abstract
We applied a gene-based haplotype approach for the genome-wide association analysis on hypertension using Genetic Analysis Workshop 18 data for unrelated individuals. Association of single-nucleotide polymorphisms and clinical outcome were first assessed and haplotypes were then constructed based on the gene information and the linkage disequilibrium plot. Extensive haplotype analysis was also conducted for the whole chromosome 3. We found 1 block from the ULK4 gene and 2 blocks from the LOC64690 gene that were significantly associated with hypertension.
Background
Hypertension is a major risk factor for many diseases, including stroke and heart failure. Various genetic studies have been done and a number of genes have been identified as having strong associations with hypertension or high blood pressure [1]. In our study, we proposed a haplotype approach to identify blocks on the gene that have strong associations with hypertension. Focusing on a block of the gene instead of looking only at a particular point may better capture the disease pattern and take the potential interactions between markers into account [2]. In addition, because the number of tests is reduced compared with the single-nucleotide polymorphism (SNP) tests, there is less penalty from multiple testing [3]. We report significant haplotypes from association analysis.
Methods
Definition of outcome and predictors
Hypertension was defined as systolic blood pressure >140 mm Hg and diastolic blood pressure > 90 mm Hg, or as being on antihypertensive medications at a specific examination. For this study, we defined our outcome as "ever-hypertension" if an individual was hypertensive in any of the 4 examinations, and "never-hypertension" if hypertension criteria were never met in those 4 examinations. In this way, we created a single hypertension outcome based on the longitudinal structure of the data. The genetic analysis was focused on unrelated individuals.
Gender, smoking habits, and age were selected as the main clinical predictors based on exploratory data analysis. Similar to the definition of outcome, smoking was defined as "ever-smokers" and "nonsmokers" based on multiple examinations. We first treated age as a continuous variable and detected its significant association with hypertension (odds ratio [OR] = 1.034; 95% confidence interval [CI]: 1.009, 1.059; p value = 0.0075). Then we examined the possible nonlinear relation between age and the defined hypertension outcome based on restricted cubic spline method [4] and found that the pattern of OR changed as age changed. Finally, based on the cubic splines plot (Figure 1), we dichotomized age at 55 years.
Figure 1.
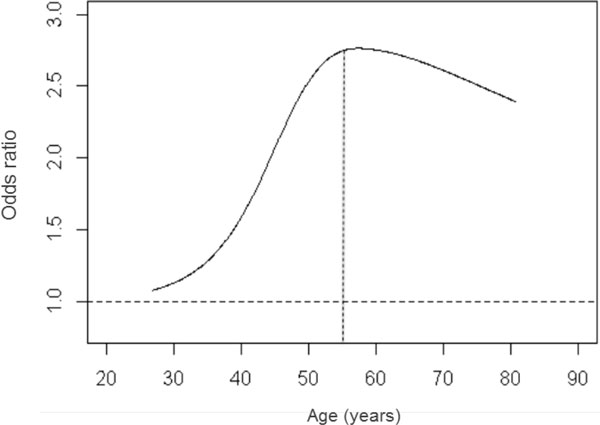
Cubic splines plot for age
Quality control of genotype data
We focused on genome-wide association studies data of chromosome 3, and conducted quality control of genotype data using PLINK [5]. Thresholds for data quality control steps were set as follows: individual genotyping missing rate at 0.05, minor allele frequency at 0.1, missing rate per SNP at 0.05, and Hardy-Weinberg equilibrium at 1 × 10−6. Heterozygosity rate was assessed for potential outliers. We merged our data set with HapMap [6] data and generated a multidimensional scaling plot (Figure 2). To adjust for population stratification effect, we used EIGENSTRAT [7,8] to conduct principal components analysis to explicitly model ancestry differences between individuals and obtained a principal component for each subject.
Figure 2.
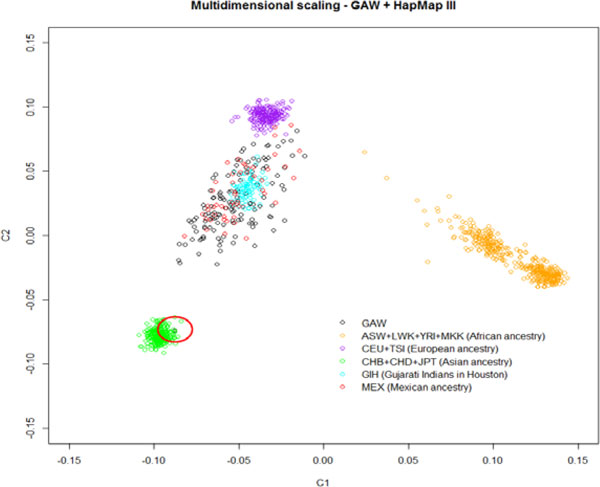
Multidimensional scaling plot (outlier in red circle)
Preliminary analysis and gene-based haplotype construction
A logistic regression model was applied on association analysis for SNPs and the defined hypertension outcome with adjustment for covariates as well as principal component vectors obtained from the population stratification procedure. We first found some nominally significant SNPs (p <5 × 10−4) from this preliminary model, and then located the genes corresponding to such SNPs based on the annotation information (T. Nalpathamkalam et al., unpublished data, 2012). For each gene, we defined the haplotype block based on a high linkage disequilibrium (LD) region containing the significant SNPs we found from the preliminary model. The blocks were defined by CI algorithm [9] as well as the 4-gamete rule algorithm [10]. Then for each block, we estimated the haplotype frequencies and the probability of having each haplotype for all individuals. The estimations of the LD blocks and haplotype frequencies were applied using HAPLOVIEW [11] and PHASE [12-14].
Haplotype analysis
First, omnibus tests on haplotypes were performed for each block of interest. Similar to the preliminary association analysis, logistic regression models were used and then likelihood ratio tests were conducted to see if haplotypes should be included in the model:
| (1) |
| (2) |
where Y represents outcome (Yi = 1 if individual i is defined as "ever-hypertension"), X1 the design matrix representing haplotypes in a particular block, X2 age, X3 gender, X4 smoking habit, and X5 principal component. Difference of log-likelihood between model (2) and model (1) were calculated and a chi-square test was performed. The entries in the design matrix X1 were the inferred conditional probabilities of haplotypes given the genotype [15]. Specifically, for haplotypes hm and hn, the conditional probability of the pair (hm, hn) for the ith individual with genotype Gi is:
| (3) |
where phu and phv denote haplotype frequencies estimated from PHASE. If the omnibus test was significant, which means at least 1 haplotype should be kept in the model, we then conducted haplotype-specific tests for each haplotype in the block and identified the specific haplotype strongly associated with the outcome.
Results
Summary of phenotypes and genotypes
We started with 65,460 SNPs of 142 unrelated individuals. First, we checked missing rate per individual at the 0.05 level and dropped 9 individuals. Second, we excluded SNPs with a minor allele frequency less than 0.1, leaving 46,205 SNPs in the sample. Following that, we excluded SNPs with missing rate greater than 0.05, leaving 46,103 SNPs. Finally, we checked the Hardy-Weinberg equilibrium at 1 × 10−6 level, and all 46,103 SNPs passed the test. Heterozygosity rate was checked for all individuals and none were located outside ±3 SD from the mean heterozygosity rate. We then combined the cleaned data set with HapMap data on common SNPs and obtained the multidimensional scaling plot (see Figure 2). One outlier was identified from family 9 (T2DG0901244), who probably belonged to an Asian population. After quality control, we excluded this individual from the samples and ended up with 42,727 SNPs and 132 individuals. For the 132 individuals left in our sample, 81 were classified as "ever-hypertension" and 51 as "never-hypertension." Table 1 summarizes the distributions of covariates.
Table 1.
Summary of phenotype data
| Characteristics | Count (%) | |
|---|---|---|
| Hypertension | Ever | 81 (61.4) |
| Never | 51 (38.6) | |
| Gender | Male | 57 (43.2) |
| Female | 75 (56.8) | |
| Smoking | Ever | 32 (24.2) |
| Never | 100 (75.8) | |
| Age | <55 years | 75 (56.8) |
| ≥55 years | 57 (43.2) | |
Preliminary association analysis and haplotype construction
The preliminary model had limited power to detect SNPs that strongly associated with hypertension after multiple testing was adjusted. We used QUANTO [16] to conduct power analysis. We needed 433 individuals to have an 80% power to detect the marginal effect of OR = 2.0. Table 2 lists the top 8 SNPs from the preliminary model. They were from 5 genes that may have potential associations with hypertension. Haplotypes were constructed on these genes based on results from the LD plot generated by HAPLOVIEW, and then sample haplotype frequencies were estimated.
Table 2.
Significant SNPs from preliminary model and corresponding genes
| SNP | Gene | OR (CI) | p Value |
|---|---|---|---|
| rs2700464 | ULK4 | 0.29 (0.15, 0.56) | 2 × 10−4 |
| rs2470696 | CBLB | 0.31 (0.18, 0.55) | 7 × 10−5 |
| rs2953768 | ALG1L2 | 0.18 (0.08, 0.39) | 2 × 10−5 |
| rs6785346 | LOC64690 | 3.53 (1.87, 6.64) | 9 × 10−5 |
| rs9857853 | LOC64690 | 3.19 (1.74, 5.87) | 2 × 10−4 |
| rs9848025 | LOC64690 | 3.52 (1.86, 6.66) | 1 × 10−4 |
| rs2129379 | LOC64690 | 3.59 (1.77, 7.28) | 4 × 10−4 |
| rs16862964 | LPP-AS2 | 4.95 (2.06, 11.89) | 3 × 10−4 |
Haplotype analysis
One haplotype from a candidate block of gene ULK4 had significant association with hypertension in the main effect model. Haplotypes from 2 blocks of gene LOC64690 were also significant in the main effect model. We took multiple testing into consideration and determined the significance threshold as 0.05/number of haplotypes being tested in the candidate block. Table 3 summarizes the results from the haplotype analysis. Age was significant in both models, but gender and smoking habit were not.
Table 3.
Significant haplotypes from model 1 in "Methods: Haplotype analysis" section
| Gene (SNP) | Covariate | OR (CI) | p Value | Haplotype Frequency |
|---|---|---|---|---|
| ULK4 (rs2700464) | TAAC | 2.7215 (1.3998, 5.2912) | 0.0032 | 0.3147 |
| Age | 2.7489 (1.2476, 6.0569) | 0.0121 | ||
| LOC64690 (rs6785346, rs9857853) | CC | 0.2430 (0.1202, 0.4913) | 1 × 10−4 | 0.6170 |
| Age | 3.3028 (1.4293, 7.6320) | 0.0052 | ||
| LOC64690 (rs9848025) | GCGTG | 3.8169 (1.7371, 8.3867) | 9 × 10−4 | 0.2477 |
| Age | 3.6333 (1.5983, 8.2590) | 0.0021 | ||
Adding the interactive effect of haplotype and age did not improve the model. Power analysis showed that for gene ULK4, we needed at least 258 individuals to have an 80% power to detect interaction effect with ratio of OR = 2.0, but only 92 individuals were required for the main effects model. For gene LOC64690, 514 individuals were required to gain 80% power for the interaction model (given ratio of OR = 2.0), but only 100 individuals were required for the main effects model to achieve the same level of power.
We also conducted haplotype analysis on whole chromosome 3 in PLINK. In PLINK, haplotype blocks are estimated following the default procedure in HAPLOVIEW and pairwise LD is calculated only for SNPs within 100 kilobases (kb). We tried the models with and without adjusted covariates. A total of 6389 haplotype blocks were constructed by using PLINK and no haplotype was significant in the omnibus test at Bonferroni corrected significance level of 0.05/6389 ~ 8 × 10−6.
Conclusions
Based on the results, we can see that the haplotype containing SNP rs2700464 on ULK4 is strongly associated with our defined hypertension outcome. Daniel et al [17] concluded that ULK4 is associated with high blood pressure and, potentially, hypertension. We also detected that 2 haplotype blocks on LOC64690 had a strong relationship with hypertension. In addition, the interaction effect between age and haplotype was not significant in all models, but power analysis indicated that our sample size was too limited to detect interaction effect, but sufficient for the main effects model.
We focused only on unrelated individuals in our study, ignoring family structures. We may consider including the family structure in further research, and may try to model the complex relationship between family members. In addition, we ran the permutation test for haplotypes in the candidate blocks as well as on the whole chromosome 3. However, the population structure is not preserved for a logistic model when doing permutation tests. Therefore, the permutation p values may not be a good estimate of the asymptotic p values. We may consider using the biased urn method [18] to overcome this problem in further research.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
XS and WX designed the overall study. XS conducted statistical analyses and drafted the manuscript. OEG, XQ, YB, and GL conceived of the study, participated in its design and coordination, and helped to draft the manuscript. All authors read and approved the final manuscript.
Contributor Information
Xiaowei Shen, Email: xshen@uhnresearch.ca.
Osvaldo Espin-Garcia, Email: oesping@uhnresearch.ca.
Xin Qiu, Email: xinqiu@uhnresearch.ca.
Yonathan Brhane, Email: brhane@lunenfeld.ca.
Geoffrey Liu, Email: Geoffrey.Liu@uhn.ca.
Wei Xu, Email: wxu@uhnresearch.ca.
Acknowledgements
The Genetic Analysis Workshop 18 (GAW18) whole genome sequence data were provided by the T2D-GENES Consortium, which is supported by NIH grants U01 DK085524, U01 DK085584, U01 DK085501, U01 DK085526, and U01 DK085545. The other genetic and phenotypic data for GAW18 were provided by the San Antonio Family Heart Study and San Antonio Family Diabetes/Gallbladder Study, which are supported by NIH grants P01 HL045222, R01 DK047482, and R01 DK053889. The Genetic Analysis Workshop is supported by NIH grant R01 GM031575.
This article has been published as part of BMC Proceedings Volume 8 Supplement 1, 2014: Genetic Analysis Workshop 18. The full contents of the supplement are available online at http://www.biomedcentral.com/bmcproc/supplements/8/S1. Publication charges for this supplement were funded by the Texas Biomedical Research Institute.
References
- Kim JJ, Vaziri SA, Elson P, Rini I, Ganapathi MK, Ganapathi R. VEGF single nucleotide polymorphisms and correlation to sunitinib-induced hypertension in metastatic renal cell carcinoma patients [abstract] J Clin Oncol. 2009;27:15s. doi: 10.1200/JCO.2008.21.7695. [DOI] [Google Scholar]
- Davidson S. Research suggests importance of haplotypes over SNPs. Nat Biotechnol. 2000;18:1134–1135. doi: 10.1038/81100. [DOI] [PubMed] [Google Scholar]
- Zhao K, Aranzana MJ, Kim S, Lister C, Shindo C, Tang C, Toomajian C, Zheng H, Dean C, Marjoram P. et al. An Arabidopsis example of association mapping in structured samples. PLoS Genet. 2007;3:e4. doi: 10.1371/journal.pgen.0030004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durrleman S, Simon R. Flexible regression models with cubic splines. Stat Med. 1989;8:551–561. doi: 10.1002/sim.4780080504. [DOI] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Bakker PIW, Daly MJ. et al. PLINK: a toolset for whole-genome association and population-based linkage analysis. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The International HapMap Consortium. The International HapMap Project. Nature. 2003. pp. 789–796. [DOI] [PubMed]
- Patterson NJ, Price AL, Reich D. Population structure and eigenanalysis. PLoS Genet. 2006;2:e190. doi: 10.1371/journal.pgen.0020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- Gabriel SB. The structure of haplotype blocks in the human genome. Science. 2002;296:2225–2229. doi: 10.1126/science.1069424. [DOI] [PubMed] [Google Scholar]
- Wang N. Distribution of recombination crossovers and the origin of haplotype blocks: the interplay of population history, recombination, and mutation. Am J Hum Genet. 2002;71:1227–1234. doi: 10.1086/344398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- Stephens M, Donnelly P. A comparison of Bayesian methods for haplotype reconstruction from population genotype data. Am J Hum Genet. 2003;73:1162–1169. doi: 10.1086/379378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens M, Scheet P. Accounting for decay of linkage disequilibrium in haplotype inference and missing-data imputation. Am J Hum Genet. 2005;76:449–462. doi: 10.1086/428594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens M, Smith N, Donnelly P. A new statistical method for haplotype reconstruction from population data. Am J Hum Genet. 2001;68:978–989. doi: 10.1086/319501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaykin DV, Westfall PH, Young SS, Karnoub MA, Wagner MJ, Ehm MG. Testing association of statistically inferred haplotypes with discrete and continuous traits in samples of unrelated individuals. Hum Hered. 2002;53:79–91. doi: 10.1159/000057986. [DOI] [PubMed] [Google Scholar]
- Gauderman WJ, Morrison JM. QUANTO 1.1: a computer program for power and sample size calculations for genetic-epidemiology studies. 2006. http://hydra.usc.edu/gxe
- Daniel L. Genome-wide association study of blood pressure and hypertension. Nat Genet. 2009;41:677–687. doi: 10.1038/ng.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein MP, Duncan R, Jiang Y, Conneely KN, Allen AS, Satten GA. A permutation procedure to correct for confounders in case-control studies, including tests of rare variation. Am J Hum Genet. 2012;91:215–223. doi: 10.1016/j.ajhg.2012.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]