

Abstract
De novo mutations enrich the sequence diversity and carry the clue of evolutional selection. Recent studies suggest the de novo mutations could be one of the risk factors for complex diseases. We conducted a survey of de novo mutations using the whole genome sequence data but only available on the odd autosomes of Mexican American families provided by Genetic Analysis Workshop 18. We extracted 8 three-generation families who have sequencing data available from 20 large pedigrees. By comparing the known single nucleotide variants (SNVs) in dbSNP129 and the de novo variants transmitted in the Mexican American families, we were able to estimate a de novo mutation rate of 1.64(±0.42) × 10−8 per position per haploid genome. This result is consistent with the estimates in literature that required many extensive validation efforts, such as genotyping and further resequencing. Our analysis suggests the importance of using family samples for studying rare variants.
Background
De novo mutations enrich the sequence diversity and carry the clue of evolutional selection [1]. Because of the technological advances in whole genome sequencing, genome-wide de novo mutation survey becomes possible. Recent studies show that de novo mutations, including de novo copy number variations, are strongly associated with multiple diseases, such as autism and schizophrenia [2]. Currently de novo mutations are often studied in family trios by comparing the parents' and child's whole genome sequence data, as well as the publicly available dbSNP database [3]. Variants observed in offspring, but not in their parents, are often considered as potential de novo mutations. However, even highly accurate sequencing data will have inevitable errors that lead to false variant callings and possible mendelian errors. Therefore, the de novo mutation candidates observed by comparing offspring's and their parents' sequencing data can be false positive [4]. Thus, researchers often resequence or genotype the candidates to confirm the true de novo mutations [1-4]. This procedure could be time and money consuming. Here we propose an approach using 3-generation families to detect de novo mutations (a) using the parents and grandparents to search for de novo mutation candidates, and (b) using offspring sequence data to confirm true de novo mutations. We applied this approach to the Genetic Analysis Workshop 18 (GAW18) data and found our results consistent with previous genotyping and further resequencing validation efforts. This result suggested our approach is reliable. With the continuously decreasing cost of whole genome sequencing, this approach should be efficient to detect de novo mutations.
Methods
GAW18 data include 20 large Mexican American pedigrees as part of the Type 2 Diabetes Genetic Exploration by Next-generation sequencing in Ethnic Samples (T2D-GENES) project. Whole genome sequence data on the odd autosomes are provided to the GAW18 participants. Our analysis focused on the 464 individuals who were whole genome sequenced, resulting in 12 million SNVs. Among those, more than 6.1 million SNVs are novel and not present in dbSNP129. Among the novel SNVs, 5,086,136 SNVs have minor allele frequencies less than 0.5% (Figure 1). As our goal is to detect de novo mutations, our analysis is restricted to these novel and rare SNVs in order to reduce the false-positive rate. When a real de novo mutation is observed in an individual, there is a 50% probability of it being transmitted to each of the individual's children. Thus, the transmission of variants from an individual to the individual's offspring can be used as a validation procedure in detecting the de novo mutations. Therefore, we selected families with sequenced data available for at least 3 generations. A total of 8 three-generation families were selected (Figure 2). For each of the families in Figure 2, we examined every rare and novel variant and considered it as a de novo mutation candidate if it is present in a parent (the child in the triangle) but absent in both grandparents. We next examined whether a de novo mutation candidate is transmitted from a parent to the parent's offspring. Only a de novo mutation candidate who transmitted to his/her offspring is declared as a true de novo mutation. Among the 8 families in Figure 2, 4 families (including 1 a family and 3 e families) were used to identify de novo mutations in males, and 4 families (including 2 b families, 1 c, and 1 d families) were used to identify de novo mutations in females, depending on whether the parent is male or female. We further categorized the 8 families into 2 family types according to the number of offspring: type I included families a, b, d, and e, and type II included family c. Let No be the number of de novo mutations observed in a family and L be the sequence length of all odd autosomes in human. For a type I family, the total number of de novo mutations is then estimated as 2No because only half of them are expected to be transmitted. Because humans have a pair chromosomes, the mutation rate µ is estimated as N0/L. For a type II family, mutation rate µ is estimated as 2No/3L because 75% of de novo mutations are expected to transmit to 1 of the 2 children. As families d and e have both parents with sequencing data available, it is possible to further exclude any of variants present in both parents, further reducing the false discovery rate.
Figure 1.
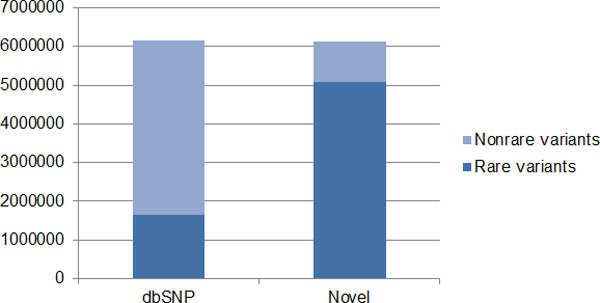
Comparison between the distribution of SNVs in dbSNP129 and novel SNVs.
Figure 2.
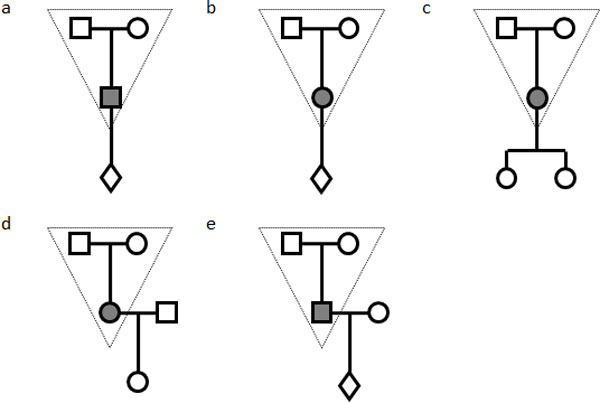
A summary of selected family types. We identified 1 a family, 2 b families, 1 c family, 1 d family, and 3 e families. The upper trios (in the dashed triangles) are used to identify de novo mutation candidates, and the third generations are used to confirm a true de novo mutation. Family a and e measure the de novo mutations in males. Family b, c, and d measure the de novo mutations in females.
Results
We analyzed the sequencing data after quality controls provided by GAW18. By investigating the first 2 generations in the 8 families, we were able to identify a total of 13,584 de novo mutation candidates. Among these candidates, 186 were successfully transmitted to the grandchildren. On average, 23.25 (±5.62) de novo mutations on the odd autosomes per family were discovered (Table 1). Considering there is an average of 1.35 billion base pairs on the odd chromosomes, we estimated an average mutation rate (µ) of 1.64(±0.42) × 10−8 per position per haploid genome, which falls in the range between 1.1 × 10−8 and 3.8 × 10−8 reported in the literature [4-6]. We did not observe a significant difference between the de novo mutations in males (1.61 × 10−8 ) and females (1.67 × 10−8).
Table 1.
Summary of de novo mutation numbers in each family.
| Family ID | Family type | Paternal age | Maternal age | Observed de novo mutations No | De novo mutation rate µ |
|---|---|---|---|---|---|
| Fam2_1 | e | 35 | 31 | 27 | 2.00 × 10−8 |
| Fam2_2 | a | 26 | 24 | 25 | 1.85 × 10−8 |
| Fam2_3 | c | 25 | 23 | 27 | 1.33 × 10−8 |
| Fam10_1 | d | 29 | 23 | 33 | 2.44 × 10−8 |
| Fam10_2 | b | 26 | 29 | 20 | 1.48 × 10−8 |
| Fam10_3 | b | 21 | 25 | 19 | 1.41 × 10−8 |
| Fam16_1 | e | 31 | 27 | 18 | 1.33 × 10−8 |
| Fam27_1 | e | 26 | 21 | 17 | 1.26 × 10−8 |
| Average | 1.64(±0.42) × 10−8 | ||||
We used the UCSC genome browser (http://genome.ucsc.edu/) [7,8] and SIFT (http://sift.jcvi.org/) [9] to map and predict the protein functions of the 186 de novo mutations. Seven of them are in exon regions and 2 are nonsynonymous SNVs. One of the nonsynonymous SNVs is in the gene PDZ domain containing 2 (PDZD2) on chromosome 5; the other is in gene spastic ataxia of Charlevoix-Saguenay (sacsin) (SACS) on chromosome 13. PDZ domains are protein-protein recognition modules that play a central role in organizing diverse cell signaling assemblies, most often in the cytoplasmic tails of transmembrane receptors and channels. PDZD2 and its secreted form (sPDZD2) are possibly involved in functional maturation of human fetal PPC-derived ICCs and the early stages of prostate tumorigenesis [10,11]. SACS encodes the sacsin protein, which is highly expressed in the central nervous system. Mutations in this gene will cause autosomal recessive spastic ataxia of Charlevoix-Saguenay, but the detail of its function is still unknown [12,13].
CpG sites are known as the mutation hotspots in mammals [14]. In the great apes, the de novo mutation rate on the CpG sites is estimated to be 11 times higher than that on the non-CpG sites [4,15]. We extracted the CpG islands from UCSC genome browser and examined the locations of the identified de novo mutations. Of our confirmed 186 de novo mutations, only 1 is located on the CpG islands. Considering the coverage of CpG islands on the odd autosomes, we expect we underestimated the CpG mutations. In the remaining 185 non-CpG mutations, we observed 127 transition mutations and 58 transversion mutations. The transition-to-transversion ratio is 2.2, similar to previous estimates [4,6].
Furthermore, we examined the relationships between the age of parents and the de novo mutation rate in the child using the first 2 generations in the 8 families by constructing linear models. In general, the de novo mutation rate in the child increases with the child's parents' ages, especially with the father's age. This is consistent with the previous report that the de novo mutation rate in offspring is positively correlated with the paternal age [1]. Nevertheless, no significant association effect was observed because of the small sample size in this study.
Discussion
We conducted an analysis of the whole genome sequences on odd autosomes of 8 three-generation families to identify de novo mutations. We found this 3-generation approach is efficient, although no further resequencing of the candidate variants was performed. In the 8 selected Mexican American families, we estimated a mutation rate of 1.64(±0.42) × 10−8 per position per haploid human genome, which is consistent with the previous estimates [4-6].
Among the 13,584 de novo mutation candidates observed in 8 three-generation families, only 186 are observed in grandchildren. This is remarkably less than the expected number of transmissions, suggesting that most de novo mutation candidates can be attributed to SNV calling errors. Because the goals in a whole genome sequencing project are to detect rare and possible de novo variants and test for association of these to a complex disease, how to account for the false-positive calls of SNVs is extremely important in an association study. Our analysis suggests sequencing family members is an efficient way to detect these SNV calling errors. For example, our analysis suggests that a variant observed in offspring but not in their parents in a simple trio can usually be treated as an SNV calling error, and should be excluded in downstream analyses. Previous studies suggest family data has many statistical advantages in detecting rare disease variants [16,17]. Thus, our results suggest whole-genome sequencing family members is worthwhile when most current whole genome sequencing projects only focus on unrelated subjects. It should be pointed out that the recruitment of multigeneration pedigrees is more difficult than family trios. However, many multigeneration pedigrees have already been collected in traditional linkage studies, such as the pedigrees used here. We expect the proposed method can be useful in detecting de novo mutations.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
XZ designed the overall study, HW conducted statistical analyses, HW and XZ drafted the manuscript. All authors read and approved the final manuscript.
Contributor Information
Heming Wang, Email: heming.wang@case.edu.
Xiaofeng Zhu, Email: xiaofeng.zhu@case.edu.
Acknowledgements
The work was supported by the National Institutes of Health, grants HL086718 and HL053353 from National Heart, Lung, Blood Institute, and HG003054 and HG005854 from the National Human Genome Research Institute. The Genetic Analysis Workshop is supported by the National Institutes of Health, grant R01 GM031575.
The GAW18 whole genome sequence data were provided by the T2D-GENES Consortium, which is supported by NIH grants U01 DK085524, U01 DK085584, U01 DK085501, U01 DK085526, and U01 DK085545. The other genetic and phenotypic data for GAW18 were provided by the San Antonio Family Heart Study and San Antonio Family Diabetes/Gallbladder Study, which are supported by NIH grants P01 HL045222, R01 DK047482, and R01 DK053889. The Genetic Analysis Workshop is supported by NIH grant R01 GM031575.
This article has been published as part of BMC Proceedings Volume 8 Supplement 1, 2014: Genetic Analysis Workshop 18. The full contents of the supplement are available online at http://www.biomedcentral.com/bmcproc/supplements/8/S1. Publication charges for this supplement were funded by the Texas Biomedical Research Institute.
References
- Kong A, Frigge ML, Masson G, Besenbacher S, Sulem P, Magnusson G, Gudjonsson SA, Sigurdsson A, Jonasdottir A, Wong WS. et al. Rate of de novo mutations and the importance of father's age to disease risk. Nature. 2012;488:471–475. doi: 10.1038/nature11396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, Yamrom B, Yoon S, Krasnitz A, Kendall J. et al. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1000 Genomes Project Consortium; Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA, Hurles ME, McVean GA. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roach JC, Glusman G, Smit AF, Huff CD, Hubley R, Shannon PT, Rowen L, Pant KP, Goodman N, Bamshad M. et al. Analysis of genetic inheritance in a family quartet by whole-genome sequencing. Science. 2010;328:636–639. doi: 10.1126/science.1186802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondrashov AS. Direct estimates of human per nucleotide mutation rates at 20 loci causing mendelian diseases. Hum Mutat. 2003;21:12–27. doi: 10.1002/humu.10147. [DOI] [PubMed] [Google Scholar]
- Nachman MW, Crowell SL. Estimate of the mutation rate per nucleotide in humans. Genetics. 2000;156:297–304. doi: 10.1093/genetics/156.1.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita PA, Rhead B, Zweig AS, Hinrichs AS, Karolchik D, Cline MS, Goldman M, Barber GP, Clawson H, Coelho A. et al. The UCSC Genome Browser database: update 2011. Nucleic Acids Res. 2011;39:D876–D882. doi: 10.1093/nar/gkq963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karolchik D, Hinrichs AS, Furey TS, Roskin KM, Sugnet CW, Haussler D, Kent WJ. The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 2004;32:D493–D496. doi: 10.1093/nar/gkh103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- Leung KK, Suen PM, Lau TK, Ko WH, Yao KM, Leung PS. PDZ-domain containing-2 (PDZD2) drives the maturity of human fetal pancreatic progenitor-derived islet-like cell clusters with functional responsiveness against membrane depolarization. Stem Cells Dev. 2009;18:979–990. doi: 10.1089/scd.2008.0325. [DOI] [PubMed] [Google Scholar]
- Harris BZ, Lim WA. Mechanism and role of PDZ domains in signaling complex assembly. J Cell Sci. 2001;114:3219–3231. doi: 10.1242/jcs.114.18.3219. [DOI] [PubMed] [Google Scholar]
- Grieco GS, Malandrini A, Comanducci G, Leuzzi V, Valoppi M, Tessa A, Palmeri S, Benedetti L, Pierallini A, Gambelli S. et al. Novel SACS mutations in autosomal recessive spastic ataxia of Charlevoix-Saguenay type. Neurology. 2004;62:103–106. doi: 10.1212/01.WNL.0000104491.66816.77. [DOI] [PubMed] [Google Scholar]
- Engert JC, Berube P, Mercier J, Dore C, Lepage P, Ge B, Bouchard JP, Mathieu J, Melancon SB, Schalling M. et al. ARSACS, a spastic ataxia common in northeastern Quebec, is caused by mutations in a new gene encoding an 11.5-kb ORF. Nat Genet. 2000;24:120–125. doi: 10.1038/72769. [DOI] [PubMed] [Google Scholar]
- Coulondre C, Miller JH, Farabaugh PJ, Gilbert W. Molecular basis of base substitution hotspots in Escherichia coli. Nature. 1978;274:775–780. doi: 10.1038/274775a0. [DOI] [PubMed] [Google Scholar]
- Chimpanzee Sequencing and Analysis Consortium. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature. 2005;437:69–87. doi: 10.1038/nature04072. [DOI] [PubMed] [Google Scholar]
- Zhu X, Feng T, Li Y, Lu Q, Elston RC. Detecting rare variants for complex traits using family and unrelated data. Genet Epidemiol. 2010;34:171–187. doi: 10.1002/gepi.20449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng T, Elston RC, Zhu X. Detecting rare and common variants for complex traits: sibpair and odds ratio weighted sum statistics (SPWSS, ORWSS) Genet Epidemiol. 2011;35:398–409. doi: 10.1002/gepi.20588. [DOI] [PMC free article] [PubMed] [Google Scholar]