

Abstract
Blooms of the dinitrogen-fixing marine cyanobacterium Trichodesmium considerably contribute to new nitrogen inputs into tropical oceans. Intriguingly, only 60% of the Trichodesmium erythraeum IMS101 genome sequence codes for protein, compared with ~85% in other sequenced cyanobacterial genomes. The extensive non-coding genome fraction suggests space for an unusually high number of unidentified, potentially regulatory non-protein-coding RNAs (ncRNAs). To identify the transcribed fraction of the genome, here we present a genome-wide map of transcriptional start sites (TSS) at single nucleotide resolution, revealing the activity of 6,080 promoters. We demonstrate that T. erythraeum has the highest number of actively splicing group II introns and the highest percentage of TSS yielding ncRNAs of any bacterium examined to date. We identified a highly transcribed retroelement that serves as template repeat for the targeted mutation of at least 12 different genes by mutagenic homing. Our findings explain the non-coding portion of the T. erythraeum genome by the transcription of an unusually high number of non-coding transcripts in addition to the known high incidence of transposable elements. We conclude that riboregulation and RNA maturation-dependent processes constitute a major part of the Trichodesmium regulatory apparatus.
The diazotrophic cyanobacterium Trichodesmium contributes significantly to the nitrogen (N) and carbon cycles of tropical and subtropical oceans where it forms large surface accumulations estimated to supply 60–80 Tg N y−1 of 100–200 Tg N y−1 assessed from total marine N2 fixation1.
Extensive research on Trichodesmium demonstrates its unique genetic, cellular, and physiological characteristics related to N2 fixation, nutrient acquisition and assimilation1,2,3. Among the filamentous non-heterocystous diazotrophs of the genus Oscillatoria4, Trichodesmium is unique because it fixes N2 during the day5. It separates this process from photosynthetic oxygen evolution spatially via diazocytes where nitrogenase is localized6,7,8,9,10, and by downregulation of PSII activity when N2 fixation is high7.
Compared with other cyanobacteria, Trichodesmium erythraeum IMS101 (T. erythraeum) possesses a relatively large11 single chromosome of 7,750,108 nt containing 4,451 protein-coding genes (accession number NC_008312). A distinctive feature is its 40% non-protein-coding portion. The cyanobacterial average is only 15%4, raising the question whether T. erythraeum may have an unusually high number of unidentified non-coding RNAs (ncRNAs), which frequently have a regulatory function12.
The genome also harbors a large number of selfish DNA elements such as insertion sequence (IS) elements and transposases, possibly contributing to genome expansion and adaptation. Published numbers differ from 10613 to “more than 150”14 IS elements and approximately 350 entries in the COG transposase categories4. Moreover, there are several genes interrupted by one or multiple introns, primarily group II introns. Group II introns, a purely bacterial intron class, are thought to be evolutionary ancestors of eukaryotic spliceosomal introns15. Only ~25% of eubacterial genomes harbor group II introns, usually only one or a small number15,16. A diversity generating retroelement (DGR) was predicted in T. erythraeum based on sequence similarity to such an element from the Bordetella bacteriophage BPP-117. DGRs introduce sequence diversity into a short, defined section of a protein-coding gene without interrupting it. The mechanism is based on the hypermutation of a variable region (VR) by recombination with a mutated cDNA copy. This copy is generated by the DGR-encoded reverse transcriptase (hereafter referred to as retron-RT) from an RNA template encoded in close vicinity to the retron-RT17,18,19,20. This RNA template is transcribed from a distinct promoter and is called the template repeat (TR). The mutations are induced by the retron-RT during reverse transcription at adenosine residues in the TR-RNA17,18. cDNA synthesis is primed by pairing of homologous sites at the 3′ ends of the target-DNA/TR-RNA sequences19. The mutated cDNA then recombines into the target (VR) sites, replacing the original sequence by a process called mutagenic homing. Consequently, these target sequences are hypermutated at specific positions when the TR and retron-RT are both active. The documented VR sites are located in the 3′ regions of the target genes and usually there are one or two such targets in a DGR-containing bacterial genome20. The presence of a TR transcript is a hallmark of DGR elements, yet no such transcript has been identified in T. erythraeum to date.
Transcriptomic analyses allow the detection of all transcripts including non-coding RNAs (ncRNAs), short unidentified open-reading-frames (ORFs), as well as intron splicing and expression of selfish DNA. In the differential RNA-seq approach (dRNA-seq21), the RNA pool is enriched with primary transcripts, based on the presence of the characteristic 5′ triphosphate that results from the initiation of transcription. dRNA-seq identifies the suite of active transcriptional start sites (TSS), providing a precise genome-wide map of active promoters and insight into the regulatory system.
With this study we address the possible functionality of the extensive non-coding genome fraction of T. erythraeum. We present a genome-wide map of 6,080 TSS combined with extensive verification experiments. We demonstrate that at least 40% of all promoters active under standard lab culture conditions produce non-protein-coding RNAs and that these transcripts accumulate in much larger amounts than mRNA. We identified the DGR-associated TR transcript and demonstrate that this hypermutation-generating system is functional in natural Trichodesmium populations. Based on the determined TR sequence, we identified 12 different genes that are possibly diversified by this DGR. Furthermore, we demonstrate 17 actively splicing group II introns in 10 different host genes in this unusual bacterium.
Results
The primary transcriptome of T. erythraeum
As opposed to the widely used RNA-seq approach where total RNA is randomly sequenced, the applied dRNA-seq approach differentiates between processed or partially degraded RNA and nascent or non-processed (referred to from here as: primary) RNA. The primary RNA is enriched in the 5′ starts of transcripts, allowing the mapping of TSS at single nucleotide resolution. In total, 56,911,484 primary T. erythraeum sequence reads up to 100 nt long were obtained from duplicate laboratory cultures of T. erythraeum under standard growth conditions using Illumina sequencing. These were mapped to the chromosome and the TSS were inferred following the criteria presented in the methods section. According to their location in the genome, TSS were further classified into 4 groups: gTSS drive the expression of annotated genes (including rRNA, tRNA, mRNA), allowing 5′-untranslated-regions (5′UTRs) of up to 200 nt; iTSS and aTSS are located internally or antisense to an annotated gene, driving the expression of internal and antisense RNAs (asRNA, a specific type of ncRNA), respectively; nTSS are all TSS located within intergenic regions and >200 nt from the start of a gene and transcribe ncRNAs. Figure 1 shows the distribution of all four types of TSS along the genome (Fig. 1A) and that nTSS were, at 26.8%, almost as abundant as gTSS at 30% (Fig. 1B). 14% of all TSS were aTSS (Fig. 1B), driving the transcription of asRNAs for almost 15% of all annotated genes in T. erythraeum. The full list of 6,080 identified TSS, together with their expression strength, is presented in Supplementary Table S1, extending the available information on promoters or RNA 5′ ends in T. erythraeum22,23 by three orders of magnitude. We provide a visualization of all mapped reads alongside the genome including all identified TSS, ncRNAs, and asRNAs as Supplementary information (T. erythraeum transcriptome plot), searchable with T. erythraeum locus tags, gene names, or by genome position.
Figure 1. Genome-wide distribution of TSS in T. erythraeum.

(A) Distribution of all TSS ≥ 800 reads along a linear plot of the chromosome (upper part, forward strand; lower part, reverse strand). The classification of each TSS is color-coded: annotated genes (gTSS, gray), gene-internally starting transcripts (iTSS, purple), non-coding transcripts from intergenic spacer regions (nTSS, green), antisense RNAs (aTSS, red). (B) Total counts of different TSS types compared with the portion of coding (CDS) and non-coding nucleotides in the genome (right). aTSS and nTSS both are non-protein-coding, iTSS may or may not code for protein.
An example of the data obtained and how it has been interpreted is shown in Fig. 2 for the sufBCDS gene cassette (Tery_4355–Tery_4358), encoding the Fe-S cluster assembly protein complex. The order of these genes is well conserved in bacteria and the operonic transcriptional organization has been the subject of several studies24,25. Although the gene arrangement in T. erythraeum implies a functional unit, it is interrupted by two long IGRs (intergenic regions) between sufB and sufC, and between sufD and sufS. Our mapping data determined that both of these long IGRs possess active promoters and TSS (Fig. 2, red graph). We conclude that the first gene sufB, the middle two genes sufCD, and the last gene sufS of the sufBCDS gene cassette can, under certain conditions, be transcribed independently from each other. This example illustrates one aspect that can be followed by these data at the genome-wide scale.
Figure 2. Example of the transcriptome data obtained.
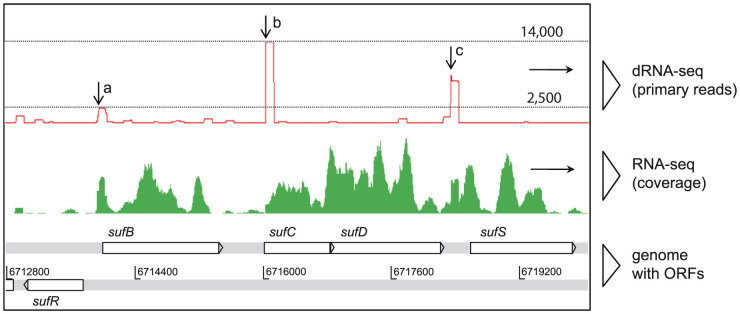
dRNA-seq (top) and RNA-seq (middle) read coverage over the T. erythraeum genomic region 6712800–6720000 containing the FeS-cluster-assembly gene cassette sufBCDS on the forward and repressor sufR on the reverse strand. Numbers on dotted lines are the primary read counts. This example illustrates how TSS can be depicted from the primary reads (black downward arrows) generated by dRNA-seq, whereas the RNA-seq reads offer information on general coverage over the length of a gene or operon. Black horizontal arrows on the right indicate the direction of transcription. In T. erythraeum, separate TSS of different expression strengths for sufB, sufCD, and sufS were found (a, b, c).
The 20 transcripts associated with the highest read counts are presented in Table 1. These 20 transcripts correspond to TSS ranked 1–24 in Supplementary Table S1, because the TSS on ranks 2–6 all belong to one very abundant repeat-derived transcript. Nine of these top 20 transcripts are ncRNAs. These nine ncRNAs alone recruited 55% of all TSS-associated primary reads. Among them is the promoter of one of the two rRNA operons and the RNA component of RNase P, a ribozyme involved in the maturation of tRNAs26. All other ncRNAs in the top 20 have unknown function. Please note that the rRNA operon promoter appears only on rank 13. Whereas mature rRNA is by far the most abundant RNA in the cell, there are ncRNAs whose nascent form is much more abundant (Table 1). The most abundant mRNAs observed among the top 20 transcripts code for two copies of RNA binding proteins, RNP-1a and RNP-1b (Tery_4702 and Tery_4701), two copies of the psaK gene (Tery_3921, Tery_4846, photosystem I reaction center subunit X), an unusually GC-rich mRNA (Tery_3486, 60% GC), a DUF4327 family protein (Tery_2891) widely conserved in cyanobacteria, a transcriptional regulator of the Abr family (Tery_0493), two hypothetical proteins unique to T. erythraeum (Tery_3815 and Tery_2341), and Tery_0600, a protein with remote similarity to a kinesin family-like protein from the brown alga Ectocarpus siliculosus. High read numbers were also associated with genes encoding photosystem I proteins or enzymes involved in tetrapyrrole metabolism such as heme oxygenase (Tery_0335) or coproporphyrinogen oxidase (Tery_1166).
Table 1. The top 20 expressed TSS plus three further highly expressed nTSS (ranking at positions 30, 40, and 56), which were also verified using a northern blot (Fig. 3C). The respective locus tag, the transcript product and type, comments from the annotator, start position and orientation (f, forward; r, reverse strand) and the average read count after normalization is provided. The nTSS read counts are identical for ncf105575–ncf109467 due to their identical sequences, causing each read to be mapped only with 1/5 to each of the 5 identical TSS. Consequently, the true read number for this particular sequence is 5 × 6,110,586 reads. HP, hypothetical protein.
| Locus tag | Transcript/product | Type | Comments | Start | Read # |
|---|---|---|---|---|---|
| ncf104733 | ncRNA (repeat 1) | nTSS | - | 104733f | 6,576,448 |
| ncf105575ncf106548ncf107521ncf108494ncf109467 | ncRNA (repeats 2–6) | nTSS (five consecutive) | reads are split onto identical repeats; together this is the highest read number | 105575–109467f | 6,110,586 × 5 = 30,552,930 |
| ncr1753291 | ncRNA | nTSS | 3′-3′ overlap with DNA photolyase gene; Fig. 3C.1 | 1753291r | 5,935,017 |
| ncf103766 | ncRNA (repeat 0) | nTSS | transposases upstream | 103766f | 2,977,433 |
| Tery_4702 | rnbp1a, RNA binding protein | gTSS | paralogous RNP-1a | 7226151r | 2,406,385 |
| Tery_4701 | rnbp1b,RNA binding protein | gTSS | paralogous RNP-1b | 7225243r | 1,280,025 |
| Tery_3921 | PSI psaK | gTSS | - | 6057068f | 1,137,533 |
| ncf1907319 | ncRNA | nTSS | pterin dehydratase 3′-5′, Fig. 3C.2 | 1907319f | 999,971 |
| Tery_3486 | HP | gTSS | unusually GC rich 60% | 5367312r | 833,051 |
| Tery_0600 | kinesin-like protein | gTSS | in a cassette of integrin genes | 966856f | 730,477 |
| Tery_R0021 | ribozyme RNase P | nTSS | - | 3464228r | 666,840 |
| ncr6410109 | ncRNA | nTSS | 3′-3′ overlap with ABC transporter (phosphonate?), Fig. 3C.3 | 6410109r | 600,168 |
| Tery_R0029 | ribosomal RNA | nTSS | 200 nt upstream of major processing site | 4603568r | 578,931 |
| ncr4997269 | ncRNA | nTSS | as_Tery_3257, Fig. 3C.4 | 4997269r | 576,462 |
| Tery_2891 | conserved HP (DUF4327) | gTSS | - | 4501104r | 516,160 |
| Tery_0493 | AbrB regulator | gTSS | - | 789400f | 489,363 |
| Tery_4846 | PSI psaK | gTSS | - | 7440696r | 477,939 |
| Tery_3815 | unique HP | gTSS | - | 5865700f | 380,696 |
| Tery_2341 | unique HP | gTSS | - | 3616443f | 357,463 |
| ncr1099929 | HP Sta7437_4063 | gTSS | newly identified ORF, HP Sta7437_4063 | 1099929r | 329,410 |
| ncr3647195 | ncRNA | nTSS | in between repeats, Fig. 3C.5 | 3647195r | 272,541 |
| ncr5610167 | misclassified as nTSS due to very long 5′UTR | gTSS | 5′ of apcA, Fig. 3C.6 | 5610167r | 174,396 |
| ncr6950581 | identified here as psbM, encoding PSII protein M | gTSS | 16 nt 3′ of ferredoxin gene Tery4504, Fig. 3C.7 | 6950581r | 126,438 |
To identify possible unknown protein-coding genes we selected the initial set of 1,621 nTSS and 855 aTSS and searched the 300 nt following each of these TSS by blastX against Genbank for matches to protein-coding genes and evaluated the results with the program RNAcode27. We identified 10 additional genes and 11 pseudogenes including 4 remnants of transposases (Supplementary Table S2). Furthermore, this analysis led to the reannotation of Tery_0573 by adding 69 amino acids to the N terminus of this large outer membrane protein and to the annotation of the 5′ exon of an intron-interrupted phage-related lysozyme gene (compare also Table 2). Among the 10 novel genes are petN encoding the cytochrome b6-f complex subunit PetN, and psaM and psbM encoding the photosystem I reaction center subunit XII and photosystem II reaction center protein M, respectively. Another newly identified gene, from which one of the most abundant mRNAs in the cell was transcribed (Table 1), encodes a hypothetical protein similar to Sta7437_4063 of Stanieria cyanosphaera PCC 7437.
Table 2. Verified group II introns in T. erythraeum. The respective locus tag and host gene are provided, followed by information on the presence of a maturase gene in the intron (yes or no), the number of mapped split reads (raw counts, RNA-seq reads), and whether the intron has previously been predicted in the group II intron database29.
| Locus tag | Host gene | Maturase | Mapped split reads | Predicted |
|---|---|---|---|---|
| Tery_4799 | allophycocyanin B alpha subunit, apcD | yes | 337 | yes |
| Tery_4178 | phage related lysozyme (with peptidoglycan-binding domain) | no | 21 | no |
| Tery_2323 | cassette of gas vesicle proteins | no | 37 | no |
| Tery_3633 | phage related lysozyme | no | 18 | no |
| Tery_3633 | phage related lysozyme | yes | 8 | yes |
| Tery_3633 | phage related lysozyme | possibly | 3 | no |
| Tery_3304 | IMP dehydrogenase | yes | 11 | no |
| Tery_0433 | ribonucleotide reductase, nrdJ | yes | 2 | yes |
| Tery_0433 | ribonucleotide reductase, nrdJ | no | 10 | no |
| Tery_0428 | ribonucleotide reductase, nrdJ | yes | 10 | yes |
| Tery_0008 | DNA polymerase III, dnaN | no | 2 | no |
| Tery_0008 | DNA polymerase III, dnaN | no | 1 | no |
| Tery_0008 | DNA polymerase III, dnaN | no | 6 | no |
| Tery_0008 | DNA polymerase III, dnaN | no | 5 | no |
| Tery_2080 | thymidylate synthase | no | 3 | no |
| Tery_1635 | putative metal dependent protease | no | 3 | yes |
| Tery_4732 | domain 1 of group II intron in RNase H | yes | 2 | yes |
Expressed non-coding repeats and other non-coding transcripts in T. erythraeum
The most abundant RNAs (Table 1, ncf104733 and following) originated from a >6,000 bp long tandem repeat array on the forward genome strand consisting of 7 repeats, 736 bp to 973 bp long (Fig. 3A). The ncRNAs produced from this repeat array alone accounted for 44.7% of all TSS-associated primary reads. To verify the high expression of these repeats, northern hybridizations were performed (Fig. 3B). These confirmed their expression: Transcription of repeats 1–6 starts at base 310 of the repeated sequence and the major detectable transcript is ~260 nt. The transcript originating from repeat 0 is somewhat shorter. In view of their high expression, these ncRNAs may be functionally important.
Figure 3. Accumulation of abundant non-coding transcripts.

(A) dRNA-seq read counts over the genomic region 103000–110200 containing 7 tandem repeats, of which repeats 1–6 are >96% identical over their aligned length, repeat 6 is missing the terminal 20 bp and repeat 1 is missing the terminal 130 bp. Repeat 0 is missing the first 228 bp, its aligned sequence is only ~87% identical to repeats 1–6. The TSS at base 310 of the repeated sequence is a T in repeat 1, but a G in repeats 2–6, explaining the different read numbers mapped to these repeats (6,576,448 vs. 6,110,586 reads, on dotted lines), despite 100% sequence identity for the following 216 bp. Orange boxes on the reverse strand show partial duplications of gene Tery_0080. Note that the transcription of the repeats occurs from the forward strand. (B) Northern blot analysis of repeat 0 compared with repeats 1–6. Long precursors were not observed in either of the hybridizations. The sizes of the individual bands were calculated from the original marker on the corresponding membrane with ECapt 15.06 and are prone to an error of approximately 10% (true for all following northern blots). (C) Northern blots for all other unknown ncRNAs from the top 20 (Table 1) including the psbM transcript (panel 7) first classified as nTSS (rank 56 in Supplementary Table S1) and reclassified as gTSS after identification as a new ORF. The apcAB dicistronic mRNA (panel 6) has a very long 5′UTR, initially also misclassified as nTSS, although apcA has a second gTSS closer to the reading frame (black circle). The numbers underneath the images correspond to the panels below, showing the genomic location and size of each of the RNAs (red boxes, to scale). The largest RNA sizes observed in the blots were used, although occasionally they may be read-through due to imperfect termination (C.5 and C.7). Red graph: primary reads. Green graph: RNA-seq. Black double bars: peak rising out of scale. Black horizontal arrows: direction of transcription. For better visualization, only one marker lane is shown here and all images were scaled to fit this marker. Full size blots are available in the Supplementary Fig. S2.
We also examined all other ncRNAs of unknown function within the top 20 expressed transcripts plus 3 additional strongly expressed ncRNAs (Table 1) in northern blot experiments (Fig. 3C) and included one of the newly identified ORFs (psbM, Fig. 3C.7) as an example of a highly expressed nTSS re-classified during the course of this work as gTSS. All these transcripts were readily detected.
A certain class of ncRNAs is associated with the CRISPR (clustered regularly interspaced short palindromic repeats) prokaryotic antiviral defense system that is encoded in the majority of more complex cyanobacterial genomes28. The 3.3 kb intergenic spacer between the genes Tery_1947 and Tery_1949 encompasses four 34 nt direct repeats, which are transcribed and processed and can be folded into an imperfect hairpin (Fig. 4). Therefore, both in length and in secondary structure, these resemble typical CRISPR-associated repeat elements29. These features are hallmarks of an expressed small repeat-spacer array in T. erythraeum, categorized as a CRISPR superclass D by automated repeat-based classification30. However, we could not find any CRISPR-associated (cas) genes in the genome.
Figure 4. Remnant of a CRISPR repeat array.

(A) The genomic location of the four CRSPR repeats (red boxes on the fw-strand) between the genes Tery_1947 and Tery_1949 with primary transcriptome coverage above and below. Due to the size cutoff for the Illumina library preparation (300–500 bp), a TSS was not detected for the CRISPR, but was for two adjacent pseudogenes (vertical arrows). The yellow bars below the CRISPR boxes indicate probe positions for the northern blot (B) that was performed to verify the expression of the repeat spacer array. Only probe 1 detected transcripts; the sizes correspond to a highly transcribed and processed CRISPR array. A precursor was also detected, albeit at very low abundance (420 nt). Full size blots are available in the Supplementary Fig. S2. (C) Secondary structure prediction of the first two repeats with their internal spacer. The repeats form the hairpins characteristic of CRISPR.
Further analysis of non-coding transcripts in the primary transcriptome indicated that 115 (7%) of the nTSS and 4 of the aTSS drive expression of transposase pseudogenes and other IS elements (Supplementary Table S3). Applying ISfinder31 to the non-coding fraction of the genome, we determined that 10.3% of it consists of IS elements. Because 8.5% of all annotated genes encode transposases (Supplementary Table S4), these selfish DNA elements account for 6.1% of the whole genome (i.e., 317,477 nt in IGRs plus 155,034 nt in genes). The fact that selfish DNA is abundant and transcribed in T. erythraeum indicated that we should examine the transcription of other types of mobile elements in the genome such as group II introns.
Transcript maturation in T. erythraeum involves splicing of at least 17 group II introns
By allowing RNA-seq reads to be split during mapping to the genome (Fig. 5A), we obtained direct evidence for the in vivo splicing of 17 group II introns, some of which have been previously described (http://webapps2.ucalgary.ca/~groupii/;16,32,33,34,35). Most were merely predicted, but had not had splicing demonstrated in T. erythraeum. These evidently active introns are located in the genes coding for the allophycocyanin B (AP-B) alpha subunit (apcD), an IMP dehydrogenase, a thymidylate synthase, the gas vesicle protein A, two different phage-related lysozymes36, a putative metal-dependent protease, a ribonucleotide reductase, and the DNA polymerase III (Table 2). Northern blot analysis confirmed the presence of splicing intermediates as well as premature and mature mRNA forms for apcD, as an example (Fig. 5B).
Figure 5. Mapping of split RNA-seq reads for the identification of introns in transcribed genes.

(A) Active group II intron splicing in the gene apcD coding for allophycocyanin-B (AP-B). Transcription and splicing is evident from the transcriptome data. 337 split exon reads (top panel) clearly mark the start and end of the interrupting intron (between the red boxes shown in the genome detail). The dRNA-seq reads (red) mark the TSS of the 5′ exon, as well as two internal TSS of unknown function; the primary read counts are on the dotted lines. White boxes: genes as annotated in the genome. Red boxes: 5′ exon and the missing part of the 3′ exon identified here. Black double bars: peak rising out of scale. The position of the probe used for the northern blot is indicated by an orange bar. (B) Northern blot hybridization of precursor RNA, different splicing intermediates, and the mature, spliced mRNA. Note that signal strength corresponds well to coverage of the probe with the different RNAs. Full size blots are available in the Supplementary Fig. S2.
A Diversity Generating Retroelement and its associated template and variable target repeats (TR and VRs)
An ncRNA belonging to a highly expressed retroelement was detected, originating from the nTSS 2632789r and accumulating as an abundant 265 nt transcript (Fig. 6A). Based on sequence similarity to its likely target genes and due to its location downstream of a specific retron-RT gene (Fig. 6B), we identified this ncRNA as the TR-RNA of a previously predicted DGR17.
Figure 6. A diversity generating retroelement (DGR) targeting 12 different protein-coding genes.

(A) High expression of the DGR template repeat (TR) with a main transcript length of 265 nt and putative precursors. Full size blots are available in the Supplementary Fig. S2. (B) Location of the TR (red box), two neighboring VR genes (with variable regions in light pink), and the retron-type reverse transcriptase (retron-RT) on the genome. The retron-RT is interrupted by a stop codon. The red graphs show the primary reads in this region with the count on the dotted line, for the reverse strand only. (C) The multiple alignment shows the variable regions (VRs) of 12 protein-coding genes of T. erythraeum (including the 3′UTR of these genes) compared against the nucleotide sequence of the TR. The black framed boxes below the alignment indicate the codons corresponding to the reading frames of all 12 VR containing genes; the red X marks the stop codon. The position of hypermutation sites, shown by grey upward arrows, is restricted to the 1st or 2nd codon positions. Each hypervariable nucleotide corresponds to an adenosine (A) in the TR. In the genes Tery_0247 and Tery_2136, a frame shift through an inserted G at position 114 leads to an earlier stop codon (black frames with red X). Filled orange circles show positions where mutations occurred that do not correspond to adenosines in the TR. They accumulate in four genes (orange arrows), indicative of the DGR mechanism being degenerate for these VRs. The red bar below the alignment denotes the putative 3′ homology region (35 bp) necessary for mutagenic homing.
Unprecedented, the DGR of T. erythraeum appears to target at least 12 genes simultaneously (12 VR sites), possibly rewriting 17 codons and thus altering up to 17 carboxy-terminal amino acids in each of the respective proteins. In accordance with the known mutational mechanism17,18, the alignment of all 12 VRs with the TR clearly demonstrates that diversity at a given nucleotide position occurs only where an adenosine occurs in the TR (Fig. 6C, grey arrows). For each VR, the possible sequence diversity is 4∧26 or >10∧15. However, in five of the VRs, random mutations at non-adenosine sites have accumulated (Fig. 6C, orange arrows and circles). 70% (21 of 30) of these mutations do not lead to amino acid changes, as they are at third codon positions or belong to a diversified codon. In the T. erythraeum genome sequence, the necessary retron-RT gene is pseudogenized by a single point mutation generating a stop codon at position 39 (annotated as Tery_1728 and Tery_1729). We resequenced all VR regions, but no differences from the genome sequence were observed, consistent with the pseudogenization of the retron-RT gene in T. erythraeum. The presence of an intact DGR system in natural populations and its inactivation in culture is discussed below.
Discussion
The transcriptome analysis of Trichodesmium revealed several, heretofore, undescribed features. Using the example of the sufBCDS gene cassette, we determined that genes thought to be operonic actually have individual promoters in T. erythraeum. This finding is consistent with observations that SufB can form different complexes with other Suf proteins37 such as SufB(2)C(2) and SufBC(2)D. The different composition of these complexes may rely on the capability to express the corresponding genes in non-stoichiometric amounts. Such situations that allow the separate regulation of individual genes of an apparent operon are most likely more frequent than previously thought, illustrated also by the transcriptional organization of alkane biosynthesis genes in T. erythraeum and other cyanobacteria38. Generally, the long intergenic spacers in the T. erythraeum genome allow for the evolution of promoter sequences that can increase regulatory and metabolic flexibility.
We examined transcription of the coding and non-coding genome fraction of cultured T. erythraeum and determined that, of the 6,080 mapped active TSS, 1,621 were nTSS (26.7%), producing mostly unknown, expressed ncRNAs. This percentage is higher than in other cyanobacteria: 11.8% nTSS in Nodularia spumigena, 12.1% in Synechocystis sp. PCC6803 and 10.3% in Anabaena sp. PCC712039. Notably, our sequencing approach omitted all RNAs < 300 nt, therefore we did not even capture smaller putative ncRNAs. Additionally, using different culture conditions would most likely further increase the number of mapped nTSS. With a very conservative approach, calculating 300 nt as the minimal length of each RNA we sequenced, we determined that at least 18.2% of the non-coding space was transcribed into ncRNAs and 5′UTRs of protein-coding mRNAs. We determined the median 5′UTR length for T. erythraeum to be 52 nt, almost twice as long as the median 5′UTR lengths, only 27 and 29 nt, determined for two strains of Prochlorococcus40, but only slightly longer that the 43 nt in H. pylori21, and in accordance with the 52 nt in Synechocystis PCC680341.
In addition to the high incidence of nTSS in the T. erythraeum genome, we also discovered that ncRNAs have much higher expression levels than mRNAs (Supplementary Figure S1). The top 8 ncRNAs alone, excluding the rRNA-operon and RNase P, accounted for 54% of all TSS-associated primary reads, whereas the top 8 mRNAs accounted for only 9% of these reads (Table 1). Intriguingly, the most highly expressed RNA in the entire dataset originated from a >6 kb array consisting of 7 tandem repeats (Table 1, Fig. 3A,B). Each repeat contains a duplicated 3′ part of Tery_0080, the gene immediately downstream from the array on the reverse strand. Yet, the remaining repeat sequence has no similarity in T. erythraeum or any other genome in Genbank. These long repeats may originate from duplication events involving mobile genetic elements that inserted alien sequence stretches along with duplication events.
We identified several new small proteins that were missing in the T. erythraeum genome annotation. Among them were three genes encoding photosynthetic proteins and one encoding a hypothetical protein, which was among the top 20 transcribed mRNAs in the culture (Supplementary Table S2). Although a few novel ORFs were found, the majority of nTSS and all aTSS give rise to different types of ncRNAs rather than unannotated mRNAs.
The high incidence of actively transcribed repeated sequence elements, transposable elements, and introns is unusual among bacteria studied so far. It has been discussed that transposable elements could contribute to adaptation of a species to its environment42,43,44 through their function as natural genetic engineering systems, thus speeding up genome evolution. Therefore, Trichodesmium's high versatility and ecological success may, in part, be linked to these features.
Since natural Trichodesmium populations can be associated with massive bacteriophage populations45 it is not surprising that we identified a short CRISPR-Cas repeat-spacer array. It exhibits a typical pattern for the processing of crRNAs (CRISPR RNAs) as well as active transcription (Fig. 4). However, T. erythraeum does not have any cas genes. Cyanobacterial CRISPR-cas arrays are prone to genetic rearrangements including their substitution among closely related strains46 or loss28. Therefore, this repeat-spacer array may be a remnant from a previous CRISPR-cas cassette whose main part was lost from this region. Indicative of such a development could be the three pseudogenes present in the same intergenic region (Fig. 4A), as they are all remnants of transposases.
An alternative means to escape phages may be via the diversity generating retroelement (DGR) we identified here. In Bordetella phage BPP-1, the DGR diversifies the gene specifying tropism for host receptor molecules18. In Trichodesmium, the opposite situation, namely a phage resistance mechanism by means of, possibly multiple, surface molecule diversification is easily conceivable. Although mechanistically different, protein diversification by gene mutations is an escape strategy from bacteriophage in the marine cyanobacterium Prochlorococcus47. We show that T. erythraeum has the potential for targeted hypermutation of the 3′ sequence (i.e., the variable repeat DNA sequences, VRs) of 12 protein-coding genes.
To date, the maximum number of VR-containing target genes reported in other bacteria is four20. The targeted sequences in T. erythraeum encode a C-terminal DUF323 (FGE sulfatase) domain, which is considered to recognize specific oligopeptide motifs20. This DUF323 domain is fused to different N terminal regions; mostly protein kinase domains. Moreover, we discovered a cysteine-dependent aspartate-directed protease (Caspase, pfam00656) domain in Tery_2077. Caspases are well known to mediate autocatalytic programmed cell death (PCD), yet are not present in cyanobacteria. Trichodesmium is one of very few bacteria for which PCD-like mechanisms have been described48,49,50 and contains a large number of putative metacaspases48,51 some of which are actively expressed during programmed cell death induction in Trichodesmium49. However, the DGR system appears to have been inactivated during the time between isolation and sequencing of T. erythraeum (T. erythraeum was isolated and brought into culture in 199352 and its genome sequence was available 11 years later). The retron-RT necessary for generating the mutated cDNA from the TR17 is disrupted by a stop codon generated by a point mutation. Although the TR-RNA was highly expressed, resequencing of the VR sequences did not reveal a single mutation compared with the available genome sequence. This is not surprising because selective pressures from the environment are lost in culture. Strikingly, most (7 out of 12) VR sequences have no point mutations at non-adenosine positions, but are identical among each other and the TR, despite the fact that the 3rd positions of most codons can mutate without altering amino acids. Four of the five VRs that differ from the TR at non-adenosine sites (Fig. 6C, orange arrows) also have point mutations in the 35 bp long 3′ conserved sequence stretch (Fig. 6C, red horizontal bar), which is most likely necessary for homing19. These VRs are most likely not targets for the TR any longer and have lost the ability to diversify independent of inactivation of the retron-RT. Most of these non-targeted point mutations are silent, indicating that the proteins harboring these VRs are likely still active, although they are not currently DGR targets. To underline our hypothesis that the T. erythraeum DGR system has been inactivated in culture, but is active in wild populations, we did a search for the TR and associated retron-RT in available metagenomic datasets. Indeed, a dataset from wild Trichodesmium colonies off the coast at Oahu, Hawaii (IMG Submission ID 8735) includes a 9,925 nt-long scaffold (TrichMGDRAFT_c100472) with perfect co-linearity to the corresponding region in the T. erythraeum genome. It harbors an intact retron-RT matching both the Tery_1728 and Tery_1729 genes with 94% nucleotide identity, followed by the TR (95% identity). Furthermore, this scaffold includes two VR-containing genes with a 94% DNA identity to Tery_1727 and 92% to Tery_1731. Therefore, it is very likely that this DGR is active in natural populations. In the future, it will be interesting to investigate the actual activity and regulation of the DGR in wild Trichodesmium populations. Using single cell genomics it should be possible to estimate the diversification rate within a population.
All transcriptome reads can be downloaded from the NCBI Sequence Read Archive (BioProject ID PRJNA237745).
Methods
Culture media and growth conditions
T. erythraeum IMS101 cultures were grown in YBCII medium53 at 25°C, 12:12 light/dark cycle at ~80 μmol photons m−2s−1 white light. For transcriptome analysis, two replicate batch cultures were grown for 9 days in sterile 1 L-Pyrex bottles with gentle air bubbling and were harvested in the middle of the light phase. For Northern blot verification experiments, separate cultures were grown in the same conditions.
Extraction, analysis, library preparation and sequencing of total RNA
RNA from T. erythraeum cultures was prepared as previously described54 with the following modifications. Filters with cells were placed in tubes containing 2 ml RNA resuspension buffer (10 mM sodium acetate, 200 mM D(+)-sucrose, 100 mM NaCl, 5 mM EDTA) and snap-frozen by immersion in liquid nitrogen. Phenolic PGTX buffer (2 ml)55 was added to the frozen samples directly prior to extraction. The quality and quantity of total extracted RNA was analyzed on a Shimadzu MultiNA microchip electrophoresis system.
Total RNA (3–5 μg) from each of the two cultures was used for the preparation of a primary transcriptome cDNA library following the dRNA-seq protocol of Sharma et al.21 with some modifications (vertis Biotechnologie AG, Germany). Total RNA was treated with Terminator-5′P-dependent-exonuclease (TEX) to digest all processed and partly degraded RNA and enrich 5′PPP-containing primary transcripts. Enzymatic treatments are often not 100% effective, which is why the treated RNA was split into two subsamples to allow for a negative control library containing only the processed RNA that was not removed by TEX. The first subsample was prepared as described (+tobacco-acid-5′-pyrophosphatase, 5′ RNA-adapter ligation, randomized first-strand cDNA synthesis, 3′ Illumina adapter ligation) to produce the primary transcriptome library (named +5′PPase from here). From the second subsample tobacco-acid-5′-pyrophosphatase treatment was omitted (named -5′PPase from here). In this negative control, primary transcripts with a triple-phosphate (5′PPP) could not be ligated to the 5′ Illumina adapter, whereas processed transcripts that were not removed by the TEX treatment could be ligated. The first cDNA amplification (18–20 cycles) was followed by cDNA fragmentation and isolation of 300–500 bp long fragments. After ligation of the 3′ Illumina adapter, a final PCR (4 cycles) was performed.
For the preparation of the non-enriched transcriptome (RNA-seq, all transcripts including processed ones), a random primed (N6) cDNA library was prepared from untreated, fragmented total RNA (mixed from both cultures) and Illumina TruSeq adapters ligated to the 5′ and 3′ ends of the cDNA. This library was amplified with 14 PCR cycles. All libraries were sequenced on an Illumina HiSeq 2000 machine using single-read sequencing and 100 bp read length.
The dRNA-seq preparation has an advantage through the enrichment of primary transcripts (non-processed, recently transcribed RNAs) as opposed to the RNA-seq approach, which does not enrich for particular RNA, but gives a general overview of RNA present in the cells, without information on which promoters are active at the time of sampling. To clarify, the TEX treatment removes the bulk of ribosomal RNA (rRNA) because functional rRNA in the cell is processed from a polycistronic transcript and thus contains a 5′P. However, the nascent polycistronic rRNA transcripts are not removed; the resulting sequences represent the activity of the rRNA operon promoter.
Sequence analysis and computational methods
The read quality was verified using FastQC56 and 99% of the sequences had a quality score > = 20 (accuracy > = 99%). Our quality analysis revealed that very few reads had a sequencing quality below 20 at their 3′ ends and all 5′ ends were of very high quality. Accumulation of sequencing errors results in a shorter alignment, but the reads will still be mapped if the alignments reach a certain length. Because we were interested in the 5′ positions of the mapped reads for TSS identification, no trimming was performed. Sequencing reads from all libraries were mapped to the T. erythraeum genome using Segemehl, version 0.1.657, with default parameters. If a read had several best matches on the genome, it was counted only as the corresponding fraction for each of these matches (i.e., for 6 identical repeats, a mapping read was counted 1/6 for each of the repeats). For the identification of splice sites, the RNA-seq reads were remapped using the -S option to obtain higher mapping scores by the splitting of reads58.
The +5′PPase and −5′PPase libraries from dRNA-seq were normalized separately with the TMM normalization method59 implemented in edgeR60 using all genome positions with more than 2 read starts per million and excluding reads mapping to the rRNA operons. The resulting read counts were multiplied by 100. The RNA-seq reads were not normalized.
For the initial TSS mapping, at a specific genome position, two conditions had to be fulfilled: 1) In both of the duplicate +5′PPase libraries, >270 normalized reads had to start at this position, corresponding to 27 and 30 raw reads, respectively. 2) The ratio between these read starts and all reads covering the position had to be >0.541. Beginning with the strongest TSS (most reads) and in decreasing order, all reads from weaker TSS positions within ±10 nt were assigned to this TSS and the weaker TSS were discarded. In a final step, the reads from the negative control library (reads from processed transcripts contaminating the dRNA-seq library, see library preparation) were used to discard false TSS. A TSS was considered to be more likely a processing site than real start site of transcription if the ratio between the number of reads in the dRNA-seq library (+5PPase) and the negative control library (−5PPase) fell below a certain threshold. To define a reliable threshold below which a TSS would be discarded, a randomly selected set of 30 TSS was manually classified as true or false positive resulting in the following exclusion criteria. A TSS was declared “false”, if one of the two ratios was <1 (more reads in the negative control library than in the corresponding dRNA-seq library) and the added +5PPase/−5PPase-ratios of a TSS in both libraries was below 37. The latter criterion ensured that a TSS with a ratio below the threshold in one condition, but with a very good ratio in the other condition, was retained. These criteria for TSS prediction were deliberately conservative (samples of just discarded or just predicted TSS were verified manually) possibly producing false-negative TSS.
To test for statistical significance, the 6,080 predicted TSS were compared with a TSSAR61 TSS annotation. For 5,542 (91.15%) of our predicted TSS, a TSS (p < 0.05, noise threshold = 10) was also annotated by TSSAR within ±10 nt. Furthermore, 249 of the 538 unconfirmed TSS are located in regions that could not be modelled by the zero-inflated Poisson regression applied by TSSAR. The unmodelled regions are small islands with high expression levels and, from manual examination, our predicted TSS appear to be true positives.
For the detection of new short ORFs, up to 300 intergenic nucleotides after each nTSS (less, if a coding sequence (CDS) was encountered before extension by 300 nt) were searched against the NCBI database using blastn and blastx with e < 1e-5. The blastx results were manually analyzed. From the blastn results, multiple alignments were generated using ClustalW62. These alignments were then evaluated for their coding potential with RNAcode27.
For the quantification of IS element remnants in IGRs, a blastx search with a threshold of e < 1e-5 was performed for each IGR against the IS-finder database31 and all nucleotides related to at least one blast hit were counted. The same parameters were used to identify nTSS (TSS in non-coding regions) and aTSS (TSS antisense to an annotated gene) driving pseudogenized transposases (TSS + 300 nt, but only until a CDS was reached). For the identification of IS elements among annotated genes, a blastP search with all CDS features and e < 1e-5 was performed against the IS-finder database.
The Artemis 15.0.0 genome browser63 was used for browsing transcriptome data along the T. erythraeum genome sequence. Secondary structures of RNA molecules were computed on the Mfold64 webserver. The Vienna file format was exported and the final structure visualized using VARNA65.
Northern blot analysis
Blots for the detection of specific highly expressed transcripts were prepared from the separation of 3–5 μg of total RNA on 10% polyacrylamide urea gels or 1.5% agarose gels with 16% formaldehyde, depending on the expected transcript sizes (up to 1,000 nt and above 1,000 nt, respectively). Blotting and hybridization with [α-32P]UTP-labelled transcript probes was performed as previously described54. For the as_repeat1-6 [γ-32P]ATP-labeled oligonucleotide probe, hybridization and washing temperatures were 45°C and 40°C, respectively. Transcript probes were produced from PCR-generated templates using in vitro transcription (MaxiScript kit, Ambion). All DNA oligonucleotides used for PCR and hybridization experiments were purchased from Sigma-Aldrich (Hamburg, Germany) or Life Technologies (Darmstadt, Germany) at the ‘desalted' quality (Supplementary Table S5). Unless stated otherwise, working concentrations of all oligonucleotides were 10 μM.
Author Contributions
U.P., W.R.H. and I.B.F. designed and coordinated the study. N.B. cultivated Trichodesmium, U.P. isolated total RNA and performed all verification experiments. M.K., U.P. and W.R.H. analyzed the data. U.P., W.R.H. and I.B.F. drafted the manuscript and U.P. prepared all figures. All authors reviewed the manuscript.
Supplementary Material
Supplementary Figures S1 and S2
T. erythraeum transcriptome plot
Supplementary Dataset 1
Acknowledgments
This work was supported by the BMBF JOINT GERMAN-ISRAELI RESEARCH PROJECTS, project number GR2378/03F0640A, the GERMAN-ISRAELI RESEARCH FOUNDATION (GIF), project number 1133-13.8/2011 to W.R.H. and I.B.F. and by the EU project MaCuMBA (Marine Microorganisms: Cultivation Methods for Improving their Biotechnological Applications; grant agreement no: 311975 to W.R.H.). We thank Edward F. DeLong for permission to study a metagenomic dataset obtained from Trichodesmium natural colonies off the coast at Oahu, Hawaii (IMG Submission ID 8735).
References
- Carpenter E. J. & Capone D. G. Nitrogen fixation in the marine environment. In: Nitrogen In The Marine Environment, 2nd edition. 141–198 (Elsevier, 2008). [Google Scholar]
- Capone D. G. Trichodesmium, a globally significant marine cyanobacterium. Science 276, 1221–1229 (1997). [Google Scholar]
- Bergman B., Sandh G., Lin S., Larsson J. & Carpenter E. J. Trichodesmium- a widespread marine cyanobacterium with unusual nitrogen fixation properties. FEMS Microbiol. Rev. 37, 286–302 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson J., Nylander J. A. & Bergman B. Genome fluctuations in cyanobacteria reflect evolutionary, developmental and adaptive traits. BMC Evol. Biol. 11, 187 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugdale R. C., Menzel D. W. & Ryther J. H. Nitrogen fixation in the Sargasso Sea. Deep Sea Res. 7, 297–300 (1961). [Google Scholar]
- El-Shehawy R., Lugomela C., Ernst A. & Bergman B. Diurnal expression of hetR and diazocyte development in the filamentous non-heterocystous cyanobacterium Trichodesmium erythraeum. Microbiology 149, 1139 (2003). [DOI] [PubMed] [Google Scholar]
- Berman-Frank I. et al. Segregation of nitrogen fixation and oxygenic photosynthesis in the marine cyanobacterium Trichodesmium. Science 294, 1534–1537 (2001). [DOI] [PubMed] [Google Scholar]
- Sandh G., El-Shehawy R., Diez B. & Bergman B. Temporal separation of cell division and diazotrophy in the marine diazotrophic cyanobacterium Trichodesmium erythraeum IMS101. FEMS Microbiol. Lett. 295, 281–288 (2009). [DOI] [PubMed] [Google Scholar]
- Chen Y.-B., Dominic B., Mellon M. T. & Zehr J. P. Circadian rhythm of nitrogenase gene expression in the diazotrophic filamentous nonheterocystous cyanobacterium Trichodesmium sp. strain IMS 101. J. Bacteriol. 180, 3598–3605 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandh G., Xu L. & Bergman B. Diazocyte development in the marine diazotrophic cyanobacterium Trichodesmium. Microbiology 158, 345–352 (2012). [DOI] [PubMed] [Google Scholar]
- Shih P. M. et al. Improving the coverage of the cyanobacterial phylum using diversity-driven genome sequencing. Proc. Natl. Acad. Sci. 110, 1053–1058 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storz G., Vogel J. & Wassarman K. M. Regulation by small RNAs in bacteria: expanding frontiers. Mol. Cell 43, 880–891 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S. et al. Genome-wide comparison of cyanobacterial transposable elements, potential genetic diversity indicators. Gene 473, 139–149 (2011). [DOI] [PubMed] [Google Scholar]
- Mikheeva L. E., Karbysheva E. A. & Shestakov S. V. The role of mobile genetic elements in the evolution of cyanobacteria. Russ. J. Genet. Appl. Res. 3, 91–101 (2013). [Google Scholar]
- Lambowitz A. M. & Zimmerly S. Group II Introns: Mobile Ribozymes that Invade DNA. Cold Spring Harb. Perspect. Biol. 3, a003616 (2010). 10.1101/cshperspect.a003616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai L. Database for mobile group II introns. Nucleic Acids Res. 31, 424–426 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doulatov S. et al. Tropism switching in Bordetella bacteriophage defines a family of diversity-generating retroelements. Nature 431, 476–481 (2004). [DOI] [PubMed] [Google Scholar]
- Liu M. et al. Reverse transcriptase-mediated tropism switching in Bordetella bacteriophage. Science 295, 2091–2094 (2002). [DOI] [PubMed] [Google Scholar]
- Guo H. et al. Diversity-generating retroelement homing regenerates target sequences for repeated rounds of codon rewriting and protein diversification. Mol. Cell 31, 813–823 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schillinger T. & Zingler N. The low incidence of diversity-generating retroelements in sequenced genomes. Mob. Genet. Elem. 2, 287–291 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma C. M. et al. The primary transcriptome of the major human pathogen Helicobacter pylori. Nature 464, 250–255 (2010). [DOI] [PubMed] [Google Scholar]
- Dominic B., Chen Y. B. & Zehr J. P. Cloning and transcriptional analysis of the nifUHDK genes of Trichodesmium sp. IMS101 reveals stable nifD, nifDK and nifK transcripts. Microbiology 144, 3359–3368 (1998). [DOI] [PubMed] [Google Scholar]
- Post A. F., Rihtman B. & Wang Q. Decoupling of ammonium regulation and ntcA transcription in the diazotrophic marine cyanobacterium Trichodesmium sp. IMS101. ISME J. 6, 629–637 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki A. et al. Light-responsive transcriptional regulation of the suf promoters involved in cyanobacterium Synechocystis sp. PCC 6803 Fe-S cluster biogenesis. FEBS Lett. 580, 5044–5048 (2006). [DOI] [PubMed] [Google Scholar]
- Shen G. et al. SufR coordinates two [4Fe-4S]2+, 1+ clusters and functions as a transcriptional repressor of the sufBCDS operon and an autoregulator of sufR in cyanobacteria. J. Biol. Chem. 282, 31909–31919 (2007). [DOI] [PubMed] [Google Scholar]
- Hess W. R., Fingerhut C. & Schön A. RNase P RNA from Prochlorococcus marinus: contribution of substrate domains to recognition by a cyanobacterial ribozyme. FEBS Lett. 431, 138–142 (1998). [DOI] [PubMed] [Google Scholar]
- Washietl S. et al. RNAcode: robust discrimination of coding and noncoding regions in comparative sequence data. RNA 17, 578–594 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai F., Axen S. D. & Kerfeld C. A. Evidence for the widespread distribution of CRISPR-Cas system in the Phylum Cyanobacteria. RNA Biol. 10, 687–693 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrangou R. CRISPR-Cas systems and RNA-guided interference. Wiley Interdiscip. Rev. RNA 4, 267–278 (2013). [DOI] [PubMed] [Google Scholar]
- Lange S. J., Alkhnbashi O. S., Rose D., Will S. & Backofen R. CRISPRmap: an automated classification of repeat conservation in prokaryotic adaptive immune systems. Nucleic Acids Res. 41, 8034–8044 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siguier P., Perochon J., Lestrade L., Mahillon J. & Chandler M. ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res. 34, D32–36 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candales M. A. et al. Database for bacterial group II introns. Nucleic Acids Res. 40, D187–D190 (2011). 10.1093/nar/gkr1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X.-Q., Yang J. & Meng Q. Four inteins and three group II introns encoded in a bacterial ribonucleotide reductase gene. J. Biol. Chem. 278, 46826–46831 (2003). [DOI] [PubMed] [Google Scholar]
- Meng Q., Wang Y. & Liu X.-Q. An intron-encoded protein assists RNA splicing of multiple similar introns of different bacterial genes. J. Biol. Chem. 280, 35085–35088 (2005). [DOI] [PubMed] [Google Scholar]
- Liu X.-Q. & Yang J. Bacterial thymidylate synthase with intein, group II intron, and distinctive ThyX motifs. J. Bacteriol. 186, 6316–6319 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei J. & Grishin N. V. COG3926 and COG5526: A tale of two new lysozyme-like protein families. Protein Sci. 14, 2574–2581 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chahal H. K. & Outten F. W. Separate FeS scaffold and carrier functions for SufB2C2 and SufA during in vitro maturation of [2Fe2S] Fdx. J. Inorg. Biochem. 116, 126–134 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klähn S. et al. Alkane biosynthesis genes in cyanobacteria and their transcriptional organization. Front. Bioeng. Biotechnol. 2, 00024 (2014). 10.3389/fbioe.2014.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess W. R., Berghoff B. A., Wilde A., Steglich C. & Klug G. Riboregulators and the role of Hfq in photosynthetic bacteria. RNA Biol. 11 (2014). 10.4161/rna.28035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voigt K. et al. Comparative transcriptomics of two environmentally relevant cyanobacteria reveals unexpected transcriptome diversity. ISME J. (2014). 10.1038/ismej.2014.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopf M. et al. Comparative analysis of the primary transcriptome of Synechocystis sp. PCC 6803. DNA Res. (2014). 10.1093/dnares/dsu018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro J. A. Transposable elements as the key to a 21st century view of evolution. Genetica 107, 171–179 (1999). [PubMed] [Google Scholar]
- Touchon M. & Rocha E. P. C. Causes of insertion sequences abundance in prokaryotic genomes. Mol. Biol. Evol. 24, 969–981 (2007). [DOI] [PubMed] [Google Scholar]
- Capy P., Gasperi G., Biémont C. & Bazin C. Stress and transposable elements: co-evolution or useful parasites? Heredity 85, 101–106 (2000). [DOI] [PubMed] [Google Scholar]
- Brown J. M., LaBarre B. A. & Hewson I. Characterization of Trichodesmium-associated viral communities in the eastern Gulf of Mexico. FEMS Microbiol. Ecol. 84, 603–613 (2013). [DOI] [PubMed] [Google Scholar]
- Hein S., Scholz I., Voß B. & Hess W. R. Adaptation and modification of three CRISPR loci in two closely related cyanobacteria. RNA Biol. 10, 852–864 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avrani S., Wurtzel O., Sharon I., Sorek R. & Lindell D. Genomic island variability facilitates Prochlorococcus–virus coexistence. Nature 474, 604–608 (2011). [DOI] [PubMed] [Google Scholar]
- Berman-Frank I., Bidle K. D., Haramaty L. & Falkowski P. G. The demise of the marine cyanobacterium, Trichodesmium spp., via an autocatalyzed cell death pathway. Limnol. Oceanogr. 49, 997–1005 (2004). [Google Scholar]
- Bar-Zeev E., Avishay I., Bidle K. D. & Berman-Frank I. Programmed cell death in the marine cyanobacterium Trichodesmium mediates carbon and nitrogen export. ISME J. 7, 2340–2348 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bidle K. D. & Falkowski P. G. Cell death in planktonic, photosynthetic microorganisms. Nat. Rev. Microbiol. 2, 643–655 (2004). [DOI] [PubMed] [Google Scholar]
- Jiang Q., Qin S. & Wu Q.-Y. Genome-wide comparative analysis of metacaspases in unicellular and filamentous cyanobacteria. BMC Genomics 11, 198 (2010). 10.1186/1471-2164-11-198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prufert-Bebout L., Paerl H. W. & Lassen C. Growth, nitrogen fixation, and spectral attenuation in cultivated Trichodesmium species. Appl. Environ. Microbiol. 59, 1367–1375 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.-B., Zehr J. P. & Mellon M. Growth and nitrogen fixation of the diazotrophic filamentous nonheterocystous cyanobacterium Trichodesmium sp. IMS 101 in defined media: Evidence for a circadian rhythm. J. Phycol. 32, 916–923 (1996). [Google Scholar]
- Pfreundt U., Stal L. J., Voß B. & Hess W. R. Dinitrogen fixation in a unicellular chlorophyll d-containing cyanobacterium. ISME J. 6, 1367–1377 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto F., Thapper A., Sontheim W. & Lindblad P. Analysis of current and alternative phenol based RNA extraction methodologies for cyanobacteria. BMC Mol. Biol. 10, 79 (2009). 10.1186/1471-2199-10-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews S. www.bioinformatics.babraham.ac.uk/projects/fastqc, (2010) (Date of access: 08.07.2014).
- Hoffmann S. et al. Fast mapping of short sequences with mismatches, insertions and deletions using index structures. PLoS Comput. Biol. 5, e1000502 (2009). 10.1371/journal.pcbi.1000502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann S. et al. A multi-split mapping algorithm for circular RNA, splicing, trans-splicing, and fusion detection. Genome Biol. 15, R34 (2014). 10.1186/gb-2014-15-2-r34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M. D. & Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11, R25 (2010). 10.1186/gb-2010-11-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M. D., McCarthy D. J. & Smyth G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amman F. et al. TSSAR: TSS annotation regime for dRNA-seq data. BMC Bioinformatics 15, 89 (2014). 10.1186/1471-2105-15-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin M. A. et al. Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 (2007). [DOI] [PubMed] [Google Scholar]
- Carver T., Harris S. R., Berriman M., Parkhill J. & McQuillan J. A. Artemis: an integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics 28, 464–469 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 31, 3406–3415 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darty K., Denise A. & Ponty Y. VARNA: Interactive drawing and editing of the RNA secondary structure. Bioinformatics 25, 1974–1975 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures S1 and S2
T. erythraeum transcriptome plot
Supplementary Dataset 1