

Abstract
Ethanol-induced liver injury is a complex process dependent upon the interaction of multiple cell types in the liver, as well as activation of the innate immune response. Increased expression of CYP2E1 in response to high concentrations of ethanol leads to greater production of cytotoxic ethanol metabolites, which in turn contribute to production of reactive oxygen species, oxidative stress, and ultimately, cell death. Necroptotic hepatocyte cell death in response to ethanol is mediated via a CYP2E1-dependent expression of receptor-interacting protein kinase 3 (RIP3), a key component of the necroptosome. In response to alarmins released during ethanol-induced necroptosis, the innate immune response is activated. Macrophage migration inhibitory factor (MIF), a pro-inflammatory multikine involved in many disease processes, is an essential component to this response to injury. MIF expression is increased during ethanol exposure via a CYP2E1-dependent pathway, likely contributing to an exacerbated innate immune response and chronic inflammation after chronic ethanol. This review will discuss the complex interactions between CYP2E1-dependent expression of RIP3 and MIF in the pathophysiology of chronic ethanol-induced liver injury.
Keywords: Alcoholic liver disease, Necroptosis, Chemokines, Cell death, Innate immunity
Graphical abstract
Highlights
-
•
Alcohol induces hepatocellular death via both apoptosis and necroptosis.
-
•
Receptor interacting kinase 3 (RIP3) mediates necroptotic cell death.
-
•
Alcohol-induced injury activates innate immune responses, including MIF.
-
•
Interactions between innate immunity and cell death with ethanol are reviewed.
Overview: alcoholic liver disease
Long-term excessive alcohol consumption can lead to alcoholic liver disease (ALD) [1]. Heavy alcohol consumption is considered 40–80 g/day for males and 20–40 g/day for females; development of ALD can result after such levels of consumption over an extended period of time, usually years [2]. ALD is one of the leading causes of preventable death and accounts for approximately 4% of global mortality [3]. Clinical manifestations of ALD patients include hepatic steatosis, steatohepatitis, liver fibrosis, cirrhosis, and hepatocellular carcinoma. The majority of chronic, heavy drinkers develop steatosis, which is characterized by accumulation of triglyceride droplets in the liver. However, this initial stage of ALD is usually symptomless and reversible if the person reduces alcohol intake [4]. However, further consumption of alcohol may lead to the progression of liver disease. The development of steatohepatitis, characterized by infiltration of leukocytes into the liver, may occur at any stage of ALD and has a high risk of mortality.
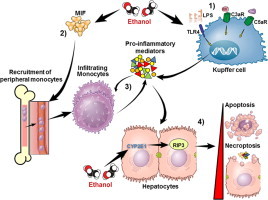
The progression of ALD involves direct effects of ethanol on hepatocytes, resulting in hepatocellular injury and death, as well as indirect effects of ethanol on innate immune pathways active in the liver (Fig. 1). Innate immune signaling is activated in response to bacterial derived signals, termed pathogen-associated molecular patterns (PAMPs), such as lipopolysaccharide (LPS), and danger-associated molecular patterns (DAMPs), which serve as alarmins that indicate the presence of cellular injury and debris. Un-resolved activation of innate immune pathways then contributes to the acceleration of hepatocellular injury, with an eventual loss of hepatic function. Understanding the molecular and cellular mechanisms linking ethanol metabolism to hepatocellular injury and death to dysregulated innate immune activity is of critical importance for developing rationally-designed therapeutic interventions to prevent and treat ALD. Here we will provide an overview of one such pathway linking ethanol metabolism to a specific pathway of cytochrome P4502E1 (CYP2E1)-dependent, RIP3-mediated necroptotic cell death and subsequent activation of innate immune signaling via the chemokine/cytokine macrophage inhibitory factor (MIF) that contributes to the progression of ALD.
Fig. 1.
Dynamic interactions between parenchymal and non-parenchymal cells in the liver during the progression of ALD. Parenchymal cells/hepatocytes carry out the primary metabolic functions of the liver. One particularly important function of hepatocytes is to metabolize ethanol. In the context of chronic ethanol exposure, expression of CYP2E1 is increased in hepatocytes and Kupffer cells. Ethanol metabolism via this pathway increases redox stress within the liver, resulting in a condition of “organelle stress” whereby the hepatocytes exhibit impaired metabolic function. Although hepatocytes are the most abundant cell type in the liver, normal physiologic function depends on interaction between hepatocytes and non-parenchymal cells (NPCs). NPCs include liver sinusoidal endothelial cells, natural killer cells, natural killer T cells, and Kupffer cells, the liver resident macrophage. Kupffer cells are an important site for the production of inflammatory mediators, such as TNFα, during ethanol exposure. Kupffer cells and hepatocytes are also important sources of chemokines, such as MCP-1 and MIF, which serve to recruit peripheral leukocytes to the liver in response to hepatocellular injury and inflammation.
Ethanol metabolism
Ethanol is metabolized in the liver parenchymal cells, hepatocytes, primarily in the area near the central vein [5]. Alcohol dehydrogenase metabolizes ethanol to acetaldehyde, and subsequently, acetaldehyde is converted to acetate via acetaldehyde dehydrogenase. Ethanol is also metabolized by cytochrome P450 2E1 (CYP2E1) to acetaldehyde; CYP2E1 expression is induced in response to high concentrations of ethanol. Hepatocytes are the primary site of CYP2E1 expression; however, CYP2E1 is induced in Kupffer cells, the resident macrophage in the liver, in response to chronic, heavy ethanol consumption (Fig. 1) [6]. Acetaldehyde is a highly reactive aldehyde; therefore, production of this metabolite can lead to oxidation of lipids and nucleic acids, as well as the formation of protein adducts. Additionally, ethanol metabolism via CYP2E1 leads to the generation of reactive oxygen species (ROS) [2,7]. The resultant oxidative stress has multiple deleterious effects on hepatocytes, including dysregulation of fatty acid synthesis and oxidation, changes in the turnover of macromolecules, as well as mitochondrial dysfunction and cellular stress, which can lead to hepatocyte death [8,9].
Mechanisms of ethanol-induced hepatocyte death
Multiple mechanisms of cell injury are associated with hepatocyte death during progression of ALD (Fig. 2). Apoptosis, or programmed cell death, is implicated as one of the major modes of cell injury in the liver during ALD. Apoptosis is a caspase-dependent cell death mechanism, mediated by two distinct pathways called the extrinsic (death-receptor-mediated) or the intrinsic (mitochondria-mediated) pathway [10]. In addition to apoptosis, non-apoptotic cell death pathways, including necrosis, are implicated in ethanol-induced liver injury; however, little is known about the molecular mechanisms of alcohol-induced necrosis [11].
Fig. 2.
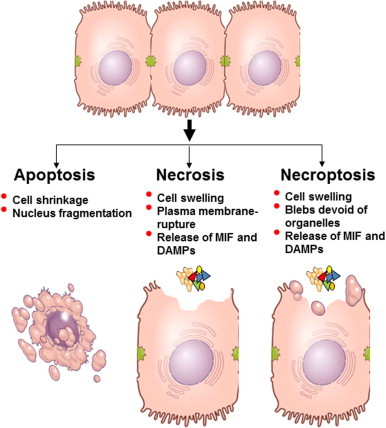
Mechanisms of cell death. Three potential pathways of hepatocellular cell death are illustrated: apoptosis, necrosis and necroptosis. Apoptosis is characterized by cell shrinking and blebbing, as well as fragmentation of the nucleus and DNA. Apoptosis proceeds under regulation from mitochondria and/or receptor signaling, particularly the receptors for TNFα and Fas. Apoptosis is considered to be the least inflammatory mode of cell death, as there is little release of cellular contents (DAMPs) that would activate inflammatory and chemotactic responses. Necrotic cell death is due to acute injury; cells swell and burst, leading to a release of cytoplasmic contents into the extracellular space. These DAMPs trigger pathways termed sterile inflammation that can lead to further hepatocellular injury [41]. MIF, which is present in pre-formed pools in cells, can also be released by necrotic cells, likely contributing to the sterile inflammatory response evoked by necrotic cells. Necroptosis, or programmed necrosis, morphologically resembles necrosis; however, it is RIP3-mediated process and has been indicated as a mediator of ethanol-induced liver injury in mice [13].
Recently, another mode of non-apoptotic cell death, necroptosis, has been characterized in a variety of cell types [12] and implicated in the development of chronic ethanol-induced liver injury in mice [13]. Necroptosis, like apoptosis, is a highly regulated pathway of cell death; however, morphologically, the process resembles necrosis. Exposure to death ligands, including TNFα or CD95, initiates necroptotic signaling cascades that are initially similar to pathways initiating apoptosis; however, the metabolic status of the cell determines the subsequent steps of cell death that ultimately lead to apoptosis or necroptosis [12]. If apoptosis is inhibited, exposure to the Fas ligand or TNFα can even exacerbate cell death via necroptosis. Receptor-interacting protein (RIP) kinases, in particular RIP3, are key activators of necroptosis (Fig. 3). RIP3, an inducible protein, is implicated in TNFα-induced non-apoptotic cell death in a variety of cell types [14,15]. RIP3 has a receptor homotypic interacting motif (RHIM) domain at its C-terminus that mediates interaction with RIP1, while the N-terminal domain contains the kinase activity [12]. Interaction of RIP1 with the RHIM-containing C-terminal domain of RIP3 initiates signals necessary for execution of necroptosis [15]. Under certain conditions, RIP3 can also execute TNFα-induced cell death in a RIP1-independent manner [16].
Fig. 3.
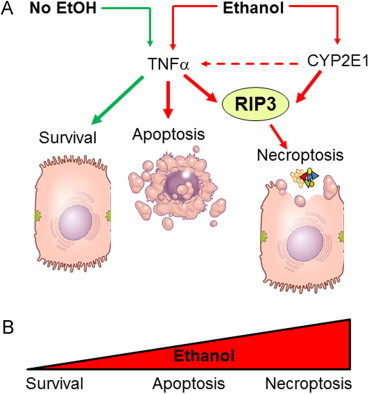
Ethanol-induced necroptosis is RIP3-dependent. (A) In a healthy hepatocyte, under physiologic conditions, TNFα-activated signaling is cytoprotective. TNF receptor activation results in the activation NFκB, which promotes cell survival. Cell survival is also promoted by TNFα-mediated inhibition of p53-dependent apoptosis and induction of anti-apoptotic Bcl-2 family of proteins. In contrast, under conditions of cellular stress, such as during chronic ethanol exposure, there is a shift in the cellular response to TNFα from cytoprotection to cell death. Initially, this response shift to apoptosis, the less inflammatory mode of cell death. However, when expression of CYP2E1 is increased, there is an induction of RIP3, as well as loss of mitochondrial integrity. This combination likely shifts the mode of cell death to the more inflammatory necroptotic pathway. This shift in hepatocyte response to TNFα, coupled with an increase in TNFα expression in the liver, leads to an increase in RIP3-mediated cell death [13]. This shift in the continuum of hepatocellular response to TNFα results in a switch from TNFα being cell protective to cell killing, first to the less inflammatory mode of apoptosis, but ultimately to the more injurious necroptotic mode of cell death (B).
Chronic exposure to ethanol in mice increases the expression of RIP3 in hepatocytes and RIP3 expression is increased in the livers of patients with alcoholic liver disease [13]. While the mechanisms for the induction of RIP3 in response to chronic, heavy ethanol exposure are not well understood, importantly, this induction is dependent on the expression of CYP2E1. Taken together, these data link ethanol metabolism in the liver to RIP3-dependent cell death (Fig. 3). This shift to the more injurious/pro-inflammatory pathway of RIP3-mediated necroptosis contributes to the exacerbation of chronic inflammation in the liver during ethanol exposure.
Innate immune responses in alcoholic liver disease
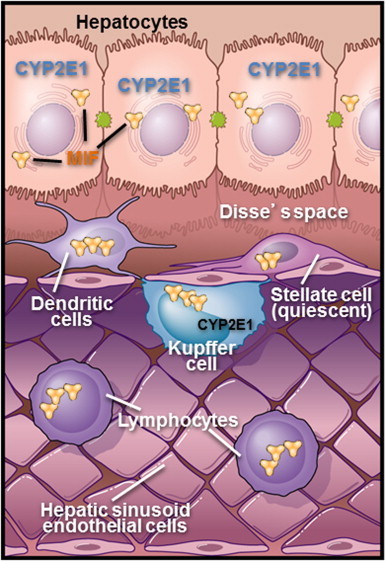
The liver acts an important innate and adaptive immune organ with a broad range of actions, involving both cellular and soluble factors. This complex, dynamic organ houses a large population of natural killer, natural killer T cells and Kupffer cells (Fig. 1). The liver is also the principal site of production of complement proteins, as well as other molecules important for orchestrating an immune response [17]. Many cells types contribute to ALD pathogenesis and the essential role of Kupffer cell activation and sensitization in ALD has been well characterized [4] (Fig. 1).
In addition to contribution of the cellular components of the innate immune system to ALD, soluble innate immune factors have been identified as key factors in the progression of ALD. The role of soluble mediators, particularly pro-inflammatory cytokines [18], in early progression of ALD has been well documented. However, additional innate immune mediators, e.g. complement anaphylatoxins and chemokines, also contribute to promotion of the pathogenesis of ALD, but are much less understood.
Cytokines and alcoholic liver disease
A variety of cytokines are upregulated in the liver in response to ethanol exposure; in particular, TNFα is a critical mediator of ALD. TNFα is produced by many cells types; however, in the liver it is primarily produced by Kupffer cells [19]. TNFα production is an important mediator for initiating injury in response to ethanol. Furthermore, LPS–TLR-4 signaling, which leads to production of TNFα, is important for development of ALD [20]. Ethanol exhibits a sensitizing effect on monocytes, as monocytes from patients with ALD, as well as rodents after heavy, chronic ethanol exposure, produce increased amounts of TNFα in response to LPS challenge compared to monocytes from healthy individuals [18,21]. IL-1β is an another potent pro-inflammatory mediator and is elevated in patients with ALD, as well as in animal models after heavy, chronic ethanol exposure [22]. Clinical trials are currently underway to test the efficacy of IL1 receptor antagonists (Anakinra) in the treatment of alcoholic hepatitis [23].
Complement and alcoholic liver disease
Complement is an important mediator of innate and adaptive immune responses. Complement proteins exhibit opsonizing, chemotactic and cell lytic properties. In the context of ALD, complement components C1q, C3, and C5 are known to contribute to the progression of ethanol-induced liver injury in mice [24,25].
Chemokines and alcoholic liver disease
While the role of cytokines in ALD, particularly TNFα, has been extensively studied, the role of chemokines is not well understood. Chemokines are a subset of cytokines that specialize in recruitment of cells during homeostasis and inflammation through chemotaxis, movement of cells toward a chemical signal or gradient [26]. Mandrekar et al. explored the role of MCP-1 (CCL2), a potent chemoattractant for monocytes, in ethanol-induced liver injury. Ethanol feeding to mice increased MCP-1 in both Kupffer cells and hepatocytes. Increased expression of MCP-1 was associated with increased liver injury, as well as, increased expression of pro-inflammatory mediators. Additionally, MCP-1 promotes hepatic triglyceride accumulation by inhibiting action of peroxisome proliferator-activated receptor-α (PPAR-α), a protein to protein that promotes oxidation of fatty acids [27]. This was the first study to establish a role for chemokines in promoting liver injury, and surprisingly, promoting hepatic steatosis.
Macrophage migration inhibitory factor (MIF)
Macrophage migration inhibitory factor (MIF) is a soluble factor, initially described as a T cell-derived mediator that inhibited movement of macrophages, which is involved in many disease processes [28–30]. Since its original discovery, MIF has been shown to be a multipotent innate immune mediator that is involved in many diseases, including multiple liver diseases [31]. Although MIF is known to contribute to ALD [32], its precise role(s) in the progression of ALD is still under continued investigation.
MIF expression and function
Determining the exact cellular source of MIF under specific pathophysiological conditions has remained elusive because there are many sources of MIF. MIF is released by many cell types including macrophages, which are considered a major source of MIF, other leukocytes, endothelial cells, epithelial cells, as well as the pituitary gland. It is likely that multiple cells types contribute to increased MIF in the systemic circulation [31,33].
One of the first described functions of MIF was its ability to inhibit glucocorticoid (GC) action. GCs exhibit potent anti-inflammatory properties, such suppression of inflammation and leukocyte recruitment. Because MIF is induced by glucocorticoids and acts as a negative regulator to glucocorticoid mediated responses, it helps maintain normal immune function despite increases in anti-inflammatory signals. One method GCs utilize to reduce inflammation involves activating MAP kinase phosphatase (MKP). MKPs dephosphorylate MAP kinases, which are essential signaling proteins during immune responses, thus inactivating them and suppressing pro-inflammatory immune functions [34]. MIF counteracts GCs by activating MAP kinase signaling and promoting leukocyte recruitment [35,36].
MIF and alcoholic liver disease
Expression of MIF in liver is increased in patients with alcoholic hepatitis or cirrhosis, with expression increased in both hepatocytes and monocytes. Further, circulating concentrations of MIF are correlated with plasma aspartate aminotransferase in patients with alcoholic hepatitis or cirrhosis [32].
The underlying mechanisms by which ethanol promotes expression and release of MIF are likely complex, but it is clear that this response is at least partially dependent on CYP2E1 [37]. While regulation of MIF expression and secretion is not well understood, recent studies have provided insights into possible mechanisms of control. One mechanism involves microRNA-451 directly interacting with the 3′ UTR of MIF and regulates MIF expression. Golgi-Associated Protein p115 was shown to be essential for releasing MIF from cytoplasmic pools (Fig. 4). When p115 was depleted in human monocytes, cells exhibited reduced ability to release MIF into the extracellular environment [38]. Cells stimulated with LPS in vitro secreted both MIF and p115 into their supernatant, further demonstrating the association between MIF and p115 in the control of MIF secretion [39].
Fig. 4.
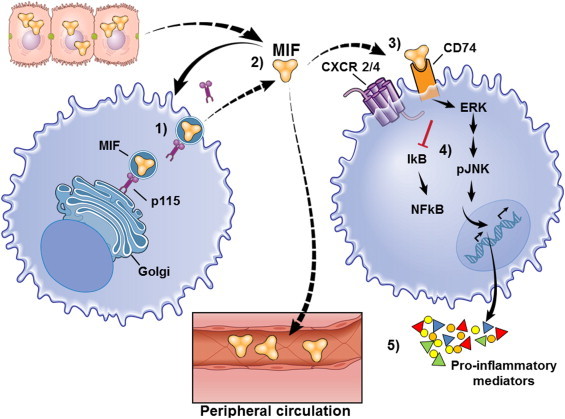
MIF signaling. (1) MIF is stored in intracellular pools in unstimulated cells, associated with Golgi-associated protein p115. (2) Upon stimulation of a cell, a macrophage in this illustration, MIF is released into the extracellular environment, where is dissociates from p115. MIF can act in a paracrine, endocrine or autocrine fashion. (3) Once it reaches its target cell, MIF engages its cognate receptor complex, CD74–CXCR2/4. To date, only CD74, also known as major histocompatibility class II (MHC II) invariant chain, has been identified as a cognate receptor for MIF. As a part of the MHC II antigen presenting complex, the invariant chain prevents MHC II from binding proteins in the endoplasmic reticulum (ER) and acts as a protein chaperone in the ER. Though most CD74 is associated with MHC II, a small proportion of protein is located at the cell surface, where it is expressed as a transmembrane protein. (4) Interaction with the receptor complex initiates intracellular signaling events, including MAP kinase signal transduction. CD74’s short cytoplasmic tail is not believed to possess direct signaling capacity; instead CD74 forms signaling complexes with CD44 and chemokine receptors CXCR2/4 [42]. CD44 is a multifunctional surface protein that acts as a receptor for ECM proteins and is involved with leukocyte activation. CD74 alone was able to bind MIF; however, activation of MAP kinase signaling required association and phosphorylation of CD44 to the signaling complex. Additionally, MIF signaling activates MAP kinases via interaction with CXCR4 [43]. CD74–CXCR4 complex is also important for endocytosis of MIF and subsequent intracellular signaling [44]. (5) Once MIF-dependent MAP kinase signaling is activated numerous cellular responses are possible, including production of cytokines and other pro-inflammatory mediators, such as TNFα and TLR4, and adhesion molecules, as well as inhibition of p53-dependent apoptosis. MIF can also lead to sustained MAP kinases signaling, which is usually transient [45,46]. Much less is known about the direct chemotactic properties of MIF, but they are likely linked to its ability to bind CXCR2/4. Interaction between MIF and CXCR2/4 mediates monocyte chemotaxis by promoting expression of integrins that assist in cell migration, arrest and extravasation [47].
Summary
Hepatocellular injury in response to chronic ethanol exposure is dependent on expression and activity of CYP2E1. Metabolism of ethanol via this pathway is associated with a shift in the mode of hepatocyte cell death in response to inflammatory mediators, from the relatively homeostatic pathway of apoptosis to the more injurious pathway of necroptosis (Fig. 3). Multiple DAMPs are released from necroptotic hepatocytes, and as a result, there are multiple innate immune pathways activated to respond to this injury.
One key pathway activated in response to ethanol-induced injury is MIF. MIF has been implicated in the pathogenesis of many inflammatory diseases including sepsis, colitis, metabolic disorders, and multiple types of arthritis [31]. MIF is an important for modulation of expression of pro-inflammatory mediators, including TNFα, IFNγ, IL-1β, TLR4, and is produced by and exerts its effects upon many different cells types, including directly promoting chemotaxis (Fig. 5). MIF contributes to the progression of ethanol-induced liver injury at multiple stages in the pathophysiology of ALD and ASH. The primary role of MIF is associated with regulation of expression of pro-inflammatory and chemotactic mediators, and recruitment of peripheral monocytes. MIF contributes to the pathophysiology of ethanol-induced liver injury in mice and suggest that inhibition of MIF activity with small molecule inhibitors of MIF, such as ISO-1 [40], may be an important therapeutic approach to the treatment of ALD.
Fig. 5.
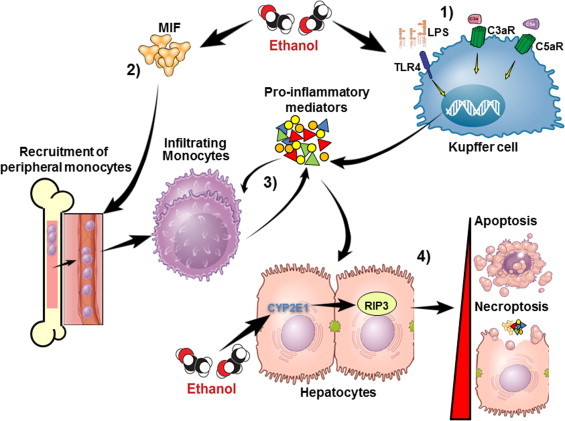
The role of MIF in ethanol-induced liver injury. (1) Ethanol exposure leads to activation of Kupffer cells. (2) Ethanol exposure also leads to increased expression of MIF. Studies modeling chronic ethanol feeding to mice have revealed that ethanol exposure increased the expression of MIF mRNA in the liver, as well as the concentration of MIF in the circulation. MIF production appears to be dependent on CYP2E1-mediated ethanol metabolism. CYP2E1−/− mice do not induce the expression of MIF mRNA after chronic ethanol feeding compared to wild-type mice, suggesting that induction of MIF is CYP2E1 dependent. Another contributor to induction of MIF during ethanol exposure may be related to localized hypoxia in the liver during ethanol metabolism [48,49]. Ischemia–reperfusion, which similar to ethanol exposure, results in hypoxia and is associated with increased serum MIF, as well as increased expression of MIF mRNA and protein [50]. MIF also enhances its own expression under hypoxic conditions [48]. Taken together, it is likely that ethanol metabolism via CYP2E1, in conjunction with localized ethanol-induced hypoxia in the liver, contributes to increased MIF expression during chronic ethanol feeding. (3) Activated Kupffer cells and MIF contribute to increased production of pro-inflammatory mediators. MIF additionally acts as a chemokine and recruits peripheral monocytes to the liver. MIF is involved in both the early and chronic innate immune responses in the liver after ethanol feeding and is critical for recruitment of infiltrating monocytes after chronic ethanol feeding [37]. During ethanol-induced liver injury, expression of MCP-1 mRNA was increased in a MIF-dependent fashion. These observations indicate that MIF directly, and/or indirectly via MCP-1, recruits monocytes from peripheral immune reservoirs. (4) It is not yet clear which happens first, but a complex feedback loop is formed between infiltrating monocytes and pro-inflammatory mediators. Ethanol-mediated infiltration of monocytes and increased pro-inflammatory mediators in the liver leads to death of hepatocytes.
Acknowledgements
This work was supported in part by grants from the NIH: P20 AA017837, 5U01 AA020821 and R37 AA011876 to LEN and R21AA020941 to SR. Additionally, this work was also supported in part by the ABMRF/The Foundation for Alcohol Research (SR) and the Case Western Reserve University/Cleveland Clinic CTSA: UL1RR024989.
References
- 1.Tome S., Lucey M.R. Review article: current management of alcoholic liver disease. Alimentary Pharmacology & Therapeutics. 2004;19:707–714. doi: 10.1111/j.1365-2036.2004.01881.x. 15043511 [DOI] [PubMed] [Google Scholar]
- 2.Gramenzi A., Caputo F., Biselli M., Kuria F., Loggi E., Andreone P., Bernardi M. Review article: alcoholic liver disease—pathophysiological aspects and risk factors. Alimentary Pharmacology & Therapeutics. 2006;24:1151–1161. doi: 10.1111/j.1365-2036.2006.03110.x. 17014574 [DOI] [PubMed] [Google Scholar]
- 3.Paula H., Asrani S.K., Boetticher N.C., Pedersen R., Shah V.H., Kim W.R. Alcoholic liver disease-related mortality in the United States: 1980–2003. American Journal of Gastroenterology. 2010;105:1782–1787. doi: 10.1038/ajg.2010.46. 20179691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Altamirano J., Bataller R. Alcoholic liver disease: pathogenesis and new targets for therapy. Nature Reviews. Gastroenterology & Hepatology. 2011;8:491–501. doi: 10.1038/nrgastro.2011.134. 21826088 [DOI] [PubMed] [Google Scholar]
- 5.Maher J.J. Exploring alcohol’s effects on liver function. Alcohol Health and Research World. 1997;21:5–12. 15706758 [PMC free article] [PubMed] [Google Scholar]
- 6.Thakur V., McMullen M.R., Pritchard M.T., Nagy L.E. Regulation of macrophage activation in alcoholic liver disease. Journal of Gastroenterology and Hepatology. 2007;22(Suppl. 1):S53–S56. doi: 10.1111/j.1440-1746.2006.04650.x. 17567466 [DOI] [PubMed] [Google Scholar]
- 7.Lieber C.S. Microsomal ethanol-oxidizing system (MEOS): the first 30 years (1968–1998)—a review. Alcoholism, Clinical and Experimental Research. 1999;23:991–1007. 10397283 [PubMed] [Google Scholar]
- 8.Lieber C.S. ALCOHOL: its metabolism and interaction with nutrients. Annual Review of Nutrition. 2000;20:395–430. doi: 10.1146/annurev.nutr.20.1.395. 10940340 [DOI] [PubMed] [Google Scholar]
- 9.Wu D., Cederbaum A.I. Alcohol, oxidative stress, and free radical damage. Alcohol Research & Health: The Journal of the National Institute on Alcohol Abuse and Alcoholism. 2003;27:277–284. 15540798 [PMC free article] [PubMed] [Google Scholar]
- 10.Elmore S. Apoptosis: a review of programmed cell death. Toxicologic Pathology. 2007;35:495–516. doi: 10.1080/01926230701320337. 17562483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Malhi H., Guicciardi M.E., Gores G.J. Hepatocyte death: a clear and present danger. Physiological Reviews. 2010;90:1165–1194. doi: 10.1152/physrev.00061.2009. 20664081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vandenabeele P., Galluzzi L., Vanden Berghe T., Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nature Reviews. Molecular Cell Biology. 2010;11:700–714. doi: 10.1038/nrm2970. 20823910 [DOI] [PubMed] [Google Scholar]
- 13.Roychowdhury S., McMullen M.R., Pisano S.G., Liu X., Nagy L.E. Absence of receptor interacting protein kinase 3 prevents ethanol-induced liver injury. Hepatology. 2013;57:1773–1783. doi: 10.1002/hep.26200. 23319235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.He S., Wang L., Miao L., Wang T., Du F., Zhao L., Wang X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137:1100–1111. doi: 10.1016/j.cell.2009.05.021. 19524512 [DOI] [PubMed] [Google Scholar]
- 15.Zhang D.W., Shao J., Lin J., Zhang N., Lu B.J., Lin S.C., Dong M.Q. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325:332–336. doi: 10.1126/science.1172308. 19498109 [DOI] [PubMed] [Google Scholar]
- 16.Upton J.W., Kaiser W.J., Mocarski E.S. Virus inhibition of RIP3-dependent necrosis. Cell Host & Microbe. 2010;7:302–313. doi: 10.1016/j.chom.2010.03.006. 20413098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gao B., Jeong W.-I., Tian Z. Liver: an organ with predominant innate immunity. Hepatology. 2008;47:729–736. doi: 10.1002/hep.22034. 18167066 [DOI] [PubMed] [Google Scholar]
- 18.Wang H.J., Gao B., Zakhari S., Nagy L.E. Inflammation in alcoholic Liver disease. Annual Review of Nutrition. 2012;32:343–368. doi: 10.1146/annurev-nutr-072610-145138. 22524187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.An L., Wang X., Cederbaum A.I. Cytokines in alcoholic liver disease. Archives of Toxicology. 2012;86:1337–1348. doi: 10.1007/s00204-012-0814-6. 22367091 [DOI] [PubMed] [Google Scholar]
- 20.Yin M., Wheeler M.D., Kono H., Bradford B.U., Gallucci R.M., Luster M.I., Thurman R.G. Essential role of tumor necrosis factor alpha in alcohol-induced liver injury in mice. Gastroenterology. 1999;117:942–952. doi: 10.1016/s0016-5085(99)70354-9. 10500078 [DOI] [PubMed] [Google Scholar]
- 21.McClain C.J., Cohen D.A. Increased tumor necrosis factor production by monocytes in alcoholic hepatitis. Hepatology. 1989;9:349–351. doi: 10.1002/hep.1840090302. 2920991 [DOI] [PubMed] [Google Scholar]
- 22.Tilg H., Wilmer A., Vogel W., Herold M., Nölchen B., Judmaier G., Huber C. Serum levels of cytokines in chronic liver diseases. Gastroenterology. 1992;103:264–274. doi: 10.1016/0016-5085(92)91122-k. 1612333 [DOI] [PubMed] [Google Scholar]
- 23.National Institute on Alcohol Abuse and Alcoholism, The Cleveland Clinic, University of Massachusettes WUoL, Efficacy Study of Anakinra, pentoxifylline, and zinc compared to Methylprenisolone in severe acute alcoholic hepatitis, Clinical Trials.gov [Internet], National Institutes of Health (NIAAA), 2013 [cited 2014 Jun 24].
- 24.Cohen J.I., Roychowdhury S., McMullen M.R., Stavitsky A.B., Nagy L.E. Complement and alcoholic liver disease: role of C1q in the pathogenesis of ethanol-induced liver injury in mice. Gastroenterology. 2010;139:664–674. doi: 10.1053/j.gastro.2010.04.041. 20416309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pritchard M.T., McMullen M.R., Stavitsky A.B., Cohen J.I., Lin F., Medof M.E., Nagy L.E. Differential contributions of C3, C5, and decay-accelerating factor to ethanol-induced fatty liver in mice. Gastroenterology. 2007;132:1117–1126. doi: 10.1053/j.gastro.2007.01.053. 17383432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Le Y., Zhou Y., Iribarren P., Wang J. Chemokines and chemokine receptors: their manifold roles in homeostasis and disease. Cellular & Molecular Immunology. 2004;1:95–104. 16212895 [PubMed] [Google Scholar]
- 27.Mandrekar P., Ambade A., Lim A., Szabo G., Catalano D. An essential role for monocyte chemoattractant protein-1 in alcoholic liver injury: regulation of proinflammatory cytokines and hepatic steatosis in mice. Hepatology. 2011;54:2185–2197. doi: 10.1002/hep.24599. 21826694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bloom B.R., Bennett B. Mechanism of a reaction in vitro associated with delayed-type hypersensitivity. Science. 1966;153:80–82. doi: 10.1126/science.153.3731.80. 5938421 [DOI] [PubMed] [Google Scholar]
- 29.David J.R. Delayed hypersensitivity in vitro: its mediation by cell-free substances formed by lymphoid cell–antigen interaction. Proceedings of the National Academy of Sciences of the United States of America. 1966;56:72–77. doi: 10.1073/pnas.56.1.72. 5229858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Isaacs A., Lindenmann J. Virus interference. I. The interferon. Proceedings of the Royal Society B: Biological Sciences. 1957;147:258–267. doi: 10.1098/rspb.1957.0048. [DOI] [PubMed] [Google Scholar]
- 31.Calandra T., Roger T. Macrophage migration inhibitory factor: a regulator of innate immunity. Nature Reviews. Immunology. 2003;3:791–800. doi: 10.1038/nri1200. 14502271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kumagi T., Akbar F., Horiike N., Onji M. Increased serum levels of macrophage migration inhibitory factor in alcoholic liver diseases and their expression in liver tissues. Clinical Biochemistry. 2001;34:189–193. doi: 10.1016/s0009-9120(01)00214-4. 11408016 [DOI] [PubMed] [Google Scholar]
- 33.Bernhagen J., Calandra T., Mitchell R.A., Martin S.B., Tracey K.J., Voelter W., Manogue K.R. MIF is a pituitary-derived cytokine that potentiates lethal endotoxaemia. Nature. 1993;365:756–759. doi: 10.1038/365756a0. 8413654 [DOI] [PubMed] [Google Scholar]
- 34.Korhonen R., Moilanen E. Mitogen-activated protein kinase phosphatase 1 as an inflammatory factor and drug target. Basic & Clinical Pharmacology & Toxicology. 2014;114:24–36. doi: 10.1111/bcpt.12141. 24112275 [DOI] [PubMed] [Google Scholar]
- 35.Onodera S., Nishihira J., Iwabuchi K., Koyama Y., Yoshida K., Tanaka S., Minami A. Macrophage migration inhibitory factor up-regulates matrix metalloproteinase-9 and -13 in rat osteoblasts: relevance to intracellular signaling pathways. Journal of Biological Chemistry. 2002;277:7865–7874. doi: 10.1074/jbc.M106020200. 11751895 [DOI] [PubMed] [Google Scholar]
- 36.Gregory J.L., Hall P., Leech M., Morand E.F., Hickey M.J. Independent roles of macrophage migration inhibitory factor and endogenous, but not exogenous glucocorticoids in regulating leukocyte trafficking. Microcirculation. 2009;16:735–748. doi: 10.3109/10739680903210421. 19905972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barnes M.A., McMullen M.R., Roychowdhury S., Pisano S.G., Liu X., Stavitsky A.B., Bucala R. Macrophage migration inhibitory factor contributes to ethanol-induced liver injury by mediating cell injury, steatohepatitis, and steatosis. Hepatology. 2013;57:1980–1991. doi: 10.1002/hep.26169. 23174952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bandres E., Bitarte N., Arias F., Agorreta J., Fortes P., Agirre X., Zarate R. microRNA-451 regulates macrophage migration inhibitory factor production and proliferation of gastrointestinal cancer cells. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research. 2009;15:2281–2290. doi: 10.1158/1078-0432.CCR-08-1818. 19318487 [DOI] [PubMed] [Google Scholar]
- 39.Merk M., Baugh J., Zierow S., Leng L., Pal U., Lee S.J., Ebert A.D. The Golgi-associated protein p115 mediates the secretion of macrophage migration inhibitory factor. Journal of Immunology. 2009;182:6896–6906. doi: 10.4049/jimmunol.0803710. 19454686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Al-Abed Y., Dabideen D., Aljabari B., Valster A., Messmer D., Ochani M., Tanovic M. ISO-1 Binding to the tautomerase active site of MIF inhibits its pro-inflammatory activity and increases survival in severe sepsis. Journal of Biological Chemistry. 2005;280:36541–36544. doi: 10.1074/jbc.C500243200. 16115897 [DOI] [PubMed] [Google Scholar]
- 41.Kubes P., Mehal W.Z. Sterile inflammation in the liver. Gastroenterology. 2012;143:1158–1172. doi: 10.1053/j.gastro.2012.09.008. 22982943 [DOI] [PubMed] [Google Scholar]
- 42.Borghese F., Clanchy F.I. CD74: an emerging opportunity as a therapeutic target in cancer and autoimmune disease. Expert Opinion on Therapeutic Targets. 2011;15:237–251. doi: 10.1517/14728222.2011.550879. 21208136 [DOI] [PubMed] [Google Scholar]
- 43.Lue H., Dewor M., Leng L., Bucala R., Bernhagen J. Activation of the JNK signalling pathway by macrophage migration inhibitory factor (MIF) and dependence on CXCR4 and CD74. Cellular Signalling. 2011;23:135–144. doi: 10.1016/j.cellsig.2010.08.013. 20807568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schwartz V., Krüttgen A., Weis J., Weber C., Ostendorf T., Lue H., Bernhagen J. Role for CD74 and CXCR4 in clathrin-dependent endocytosis of the cytokine MIF. European Journal of Cell Biology. 2012;91:435–449. doi: 10.1016/j.ejcb.2011.08.006. 22014447 [DOI] [PubMed] [Google Scholar]
- 45.Kleemann R., Hausser A., Geiger G., Mischke R., Burger-Kentischer A., Flieger O., Johannes F.J. Intracellular action of the cytokine MIF to modulate AP-1 activity and the cell cycle through Jab1. Nature. 2000;408:211–216. doi: 10.1038/35041591. 11089976 [DOI] [PubMed] [Google Scholar]
- 46.Grieb G., Merk M., Bernhagen J., Bucala R. Macrophage migration inhibitory factor (MIF): a promising biomarker. Drug News & Perspectives. 2010;23:257–264. doi: 10.1358/dnp.2010.23.4.1453629. 20520854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bernhagen J., Krohn R., Lue H., Gregory J.L., Zernecke A., Koenen R.R., Dewor M. MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nature Medicine. 2007;13:587–596. doi: 10.1038/nm1567. 17435771 [DOI] [PubMed] [Google Scholar]
- 48.Zhang Y., Talwar A., Tsang D., Bruchfeld A., Sadoughi A., Hu M., Omonuwa K. Macrophage migration inhibitory factor mediates hypoxia-induced pulmonary hypertension. Molecular Medicine. 2012;18:215–223. doi: 10.2119/molmed.2011.00094. 22113497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Arteel G.E., Iimuro Y., Yin M., Raleigh J.A., Thurman R.G. Chronic enteral ethanol treatment causes hypoxia in rat liver tissue in vivo. Hepatology. 1997;25:920–926. doi: 10.1002/hep.510250422. 9096598 [DOI] [PubMed] [Google Scholar]
- 50.Liu A., Fang H., Dirsch O., Jin H., Dahmen U. Early release of macrophage migration inhibitory factor after liver ischemia and reperfusion injury in rats. Cytokine. 2012;57:150–157. doi: 10.1016/j.cyto.2011.11.009. 22136975 [DOI] [PubMed] [Google Scholar]