

Abstract
Mitochondria are principal regulators of cellular function and metabolism through production of ATP for energy homeostasis, maintenance of calcium homeostasis, regulation of apoptosis and fatty acid oxidation to provide acetyl CoA for fueling the electron transport chain. In addition, mitochondria play a key role in cell signaling through production of reactive oxygen species that modulate redox signaling. Recent findings support an additional mechanism for control of cellular and tissue function by mitochondria through complex mitochondrial–nuclear communication mechanisms and potentially through extracellular release of mitochondrial components that can act as signaling molecules. The activation of stress responses including mitophagy, mitochondrial number, fission and fusion events, and the mitochondrial unfolded protein response (UPRMT) requires mitochondrial–nuclear communication for the transcriptional activation of nuclear genes involved in mitochondrial quality control and metabolism. The induction of these signaling pathways is a shared feature in long-lived organisms spanning from yeast to mice. As a result, the role of mitochondrial stress signaling in longevity has been expansively studied. Current and exciting studies provide evidence that mitochondria can also signal among tissues to up-regulate cytoprotective activities to promote healthy aging. Alternatively, mitochondria release signals to modulate innate immunity and systemic inflammatory responses and could consequently promote inflammation during aging. In this review, established and emerging models of mitochondrial stress response pathways and their potential role in modulating longevity are discussed.
Keywords: Mitochondria, Longevity, Retrograde response, Mitochondrial unfolded protein response, Mitochondrial signaling
Graphical abstract
Highlights
-
•
Activation of mitochondrial stress responses is associated with longevity.
-
•
Mitochondria release signals to induce expression of quality control genes.
-
•
Mitochondria can mediate stress signaling between tissues.
Introduction
The concept that mitochondrial function declines during aging has been a basic tenet of the biology of aging for many years. Paradoxically, it has been shown, first in yeast and later in mice, that perturbations of some mitochondrial electron transport chain (ETC) complexes increase lifespan [1], [2], [3]. These results are counterintuitive, yet they have the potential to significantly shift the way we think about the role of mitochondrial function in general and especially in aging. Recent studies have provided evidence to support the hypothesis that up-regulation of mitochondrial stress responses contributes to enhanced longevity in the long-lived mitochondrial mutants [3], [4], [5]. These stress responses include mitochondrial turnover, fission and fusion events, regulation of mitochondrial number (induction of mitochondrial biogenesis), retrograde signaling, and the mitochondrial unfolded protein response UPRMT. Compromised mitochondrial stress responses could contribute to age-related accumulation of damaged proteins, reduced oxidative phosphorylation, increased reactive oxygen species production, and induction of cellular apoptosis. As a result, maintenance of mitochondrial stress responses has gained recognition as a potential pro-longevity mechanism in the aging field. Understanding the molecular mechanisms by which mitochondrial stress responses might lead to longevity is key for the development of interventions to prevent age-associated diseases and improve healthspan. Here, we review recent studies that have shed light on the relationship between mitochondrial stress signaling and longevity.
Yeast retrograde response
Retrograde signaling in yeast is a mitochondrial-to-nuclear signal transduction pathway resulting in the induction of nuclear-encoded mitochondrial genes in response to mitochondrial stress (see Fig. 1). Ron Butow discovered the yeast retrograde response in 1987 [6]. Butow showed that yeast lacking mitochondrial DNA (rho° cells) modulate the transcript levels of nuclear-encoded mitochondrial genes [6]. A genome-wide analysis of rho° cells and yeast treated with mitochondrial electron transport chain (ETC) inhibitors demonstrated alterations in a wide-range of nuclear-encoded genes [7]. The majority of the genes up-regulated encode for proteins that facilitate a metabolic switch from aerobic to anaerobic respiration [8].
Fig. 1.
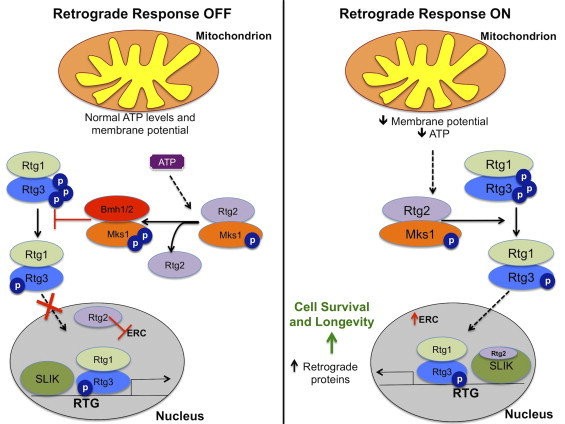
Yeast retrograde response. Under normal conditions, ATP competitively binds to Mks1, a negative regulator of the retrograde response (RTG), releasing Rtg2 and allowing Mks-1 to bind to the 14-3-3 protein Bmh1/2, another negative regulator of the RTG. Consequently, this inhibits the nuclear translocation of the Rtg1/3 complex. Rtg2 has also been shown to suppress the formation of extrachromosomal rDNA circles (ERCs). Under conditions of reduced mitochondrial membrane potential or mitochondrial stress, Rtg2 is stabilized by Mks1. Stabilized Rtg2 promotes the dephosphorylation of Rtg3. Subsequently, Rtg1/3 complex translocates to the nucleus where is turns on the transcription of retrograde genes. Rtg2 can also modulate the retrograde response by interacting with the transcriptional co-activator SAGA-like (SLIK) complex. As a result, cell survival is promoted.
A number of studies have defined the molecular pathways involved in the retrograde response [9], [10], [11]. Defective mitochondria release a retrograde signal to activate the nuclear-localization of the Rtg1/3 complex that consists of two basic helix–loop–helix leucine zipper transcription factors [11]. This complex regulates the expression of genes encoding for enzymes in mitochondrial metabolism, peroxisomal biogenesis and stress response pathways [8], [12]. The activation of the Rtg1/3 complex is dependent on the phosphorylation of Rtg3 [11]. A cytoplasmic protein, Rtg2 promotes the partial dephosphorylation of Rtg3 through an interaction with Mks-1, a negative regulator of the retrograde response [11], [13]. ATP competitively binds to Mks-1 releasing Rtg2 and allowing Mks-1 to bind to the 14-3-3 protein Bmh1/2, which inhibits the translocation of the Rtg1/3 complex [10], [14]. Therefore, low levels of ATP promote the interaction between Rtg2 and Mks-1 and activation of the retrograde response. The exact mechanism of Rtg3 dephosphorylation is currently not known.
Regulation of the retrograde response
More recent work has investigated the mitochondrial signal that triggers the retrograde response. One potential signal is mitochondrial membrane potential. Loss of membrane potential was shown to trigger the retrograde response in rho° and cox4 null yeast [15]. Conversely, restoring membrane potential blocks the induction of the retrograde response [15]. To date, it is not known how Rtg2 senses the loss of membrane potential to modulate the translocation of the Rtg1/3 complex. The production of reactive oxygen species (ROS) is an unlikely candidate for the retrograde signal as supplementing rho° cells with a free radical quencher does not reverse the retrograde response [15]. Rho° strains effectively use glycolysis to compensate for reduced oxidative phosphorylation, and ATP concentrations are relatively unchanged compared to wild-type yeast. Therefore, it is unlikely that reduced ATP levels serve as a retrograde signal. However, the impact of ATP levels cannot be completely ruled out as a recent study revealed that the interaction between Mks-1 and Rtg2 are dependent on physiological ATP levels [14].
Rtg2 has also been shown to modulate the retrograde response by interacting with the transcriptional co-activator SAGA-like (SLIK) complex, a conserved histone acetyltransferase-coactivator of gene expression [16]. Rtg2 is a component of SLIK and is present at the promoter of retrograde genes [16]. Deletion of a critical component of the SLIK complex, GCN5, suppresses the retrograde response in rho° cells [17]. Recently, the retrograde response has also been linked to TOR signaling. Lst8p, a component of the target of rapamycin complex I (TORC1), negatively regulates the retrograde response both upstream and downstream of Rtg2 [10], [18]. In addition, inhibition of TORC1, via rapamycin, induces the expression of retrograde genes [18]. However, induction of the retrograde response by mitochondrial dysfunction does not require TOR. Rather, it is speculated that TOR regulates the retrograde response through sensing nutrient availability [18].
Retrograde response and longevity
The relationship between the retrograde response and longevity was first implicated in a long-lived rho° yeast strain. Specifically, deletion of Rtg2 was shown to reverse replicative lifespan extension in rho° cells [5]. Conversely, replacing mtDNA in rho° cells with intact mtDNA through cytoduction abrogates the retrograde response and reverses lifespan extension [5]. Reduced respiratory capacity is not responsible for lifespan extension since treatment with mitochondrial ETC, inhibitors does not alter lifespan in wild-type yeast, and other respiratory deficient yeast that exhibit basal retrograde signaling do not have increased lifespan [5]. This was confirmed by another study showing that rho° longevity is not due to reduced oxidative capacity since nuclear petite mutants of the same yeast background did not extend lifespan compared to wild-type control [19]. Furthermore, increased reactive oxygen species production does not appear to be responsible for lifespan extension associated with retrograde signaling because overexpression of antioxidant enzymes does not suppress lifespan extension in rho° cells [5]. Retrograde response-mediated lifespan extension is not restricted to replicative lifespan, since it was also shown to mediate chronological lifespan in yeast [20]. Finally, the retrograde response has also been linked to stress resistance [21]. For example, yeast cells grown on a non-fermentable carbon source, raffinose, are resistant to acetic acid program cell death (AA-PCD) and show induction of the retrograde response [21].
The metabolic state is critical for initiating the retrograde response and regulating lifespan in rho° cells. In yeast lacking RAS2, a critical factor in sensing nutrient availability, the retrograde response and longevity is repressed in rho° cells [5]. Caloric restriction, initiated in yeast by limiting the availability of glucose or non-essential amino acids, induces citric acid cycle and retrograde response genes [22], [23]. However, inhibiting the retrograde response by deletion in the RTG2 or RTG3 genes, does not suppress lifespan extension-mediated by caloric restriction [22]. Thus, caloric restriction mediates longevity independent of the retrograde response pathway.
Retrograde response and ERCs
A hallmark phenotype of yeast aging is the accumulation of extrachromosomal rDNA circles (ERCs), which have detrimental effects on cellular homeostasis [24]. Surprisingly, activation of the retrograde response is associated with the accumulation of ERCs. This is consistent with the fact that activation of the retrograde response increases with age. Rho° cells have a greater accumulation of ERCs compared to wild-type cells partly due to activation of retrograde response [25]. Eliminating ERCs by deletion of FOB1, a gene that encodes for a protein required for ERC formation, further extends rho° cell lifespan by 50% [25]. Rtg2 suppresses ERC production and disruption of the SLIK complex by GCN5 deletion lessens the ERC load in rho° cells due to the release of Rtg2 from the SLIK complex [17]. However, disruption of SLIK suppresses the retrograde response and lifespan extension of rho° cells [17]. Together, this suggests that lifespan extension in rho° cells is mediated by retrograde signaling, not by ERC accumulation.
Alternative mitochondrial–nuclear signaling pathways in yeast
A recently discovered mitochondrial–nuclear transduction pathway, coined mitochondrial back-signaling, was also shown to mediate longevity [26]. This pathway is activated in response to a deletion in the AFO1/MRPL25 gene, which encodes for a protein found in the large subunit of the mitochondrial ribosome. Deletion of this gene results in a 60% increase in replicative lifespan and enhanced resistance to oxidative stress [26]. Lifespan extension and resistance to oxidative stress through this pathway requires an active target of rapamycin complex 1 (TORC1) and transcription factor Sfp1, a regulator of cytoplasmic translation. Another mitochondrial–nuclear transduction pathway independent of retrograde signaling mediates nuclear genomic silencing in a Sir2 histone deacetylase dependent manner [27]. Disruption in the mitochondrial translation complex activates this pathway and promotes longevity. These two pathways warrant further investigation, as they will help elucidate mechanisms of pro-longevity.
Retrograde response in mammals
A number of mammalian cell types lacking mitochondrial DNA were shown to have higher expression levels of genes encoding for enzymes in glycolysis, mitochondrial and peroxisomal metabolism, lipid transport, inflammation and oxidative stress [28], [29]. Structural modeling and alignment identified Myc and Max as putative mammalian homologs for Rtg1 and Rtg3, respectively [30]. In mammalian rho° cells, Myc transcript and protein levels are significantly elevated [29]. RNAi knockdown of Myc suppressed the induction of a number of retrograde-related genes in rho° cells, strongly suggesting that the retrograde response is evolutionarily conserved [29]. Furthermore, induction of the retrograde response via mild uncoupling delays senescence as indicated by increased population doubling [31]. Together, these results hint that the retrograde response may positively influence mammalian aging as was demonstrated in yeast.
Mitochondrial unfolded protein response
The mitochondrial unfolded protein response (UPRMT) is a mitochondrial–to-nuclear signal transduction pathway or retrograde response; initiated by the accumulation of unfolded proteins in the mitochondria and resulting in the induction of specific stress response proteins [32], [33]. A model for the UPRMT was first proposed in Caenorhabditis elegans in which mitochondrial stress involving unfolded proteins was shown to up-regulate the ATP-dependent mitochondrial matrix ClpP protease, a protease that cleaves mis-folded proteins (see Fig. 2) [32], [34]. The cleaved proteins (peptides) are exported out from the mitochondria to the cytosol through the mitochondrial matrix peptide exporter HAF-1 [35]. The exported peptides serve as a signal for the activation of the bZip transcription factor ATFS-1. ATFS-1 has a mitochondrial targeting sequence and nuclear localization sequence. Under basal conditions, ATFS-1 is imported into the mitochondria and degraded by the Lon protease in the mitochondrial matrix; however, when mitochondria are stressed, ATFS-1 accumulates in the cytosol [35]. This accumulation leads to the trafficking of ATFS-1 to the nucleus. ATFS-1 activates UBL-5 to form a complex with the transcription factor DVE-1 to transcriptionally activate mitochondrial chaperones such as heat shock protein 6 (Hsp6) and Hsp 10 [34], [35], [36]. Furthermore, microarray analysis in C. elegans revealed that the UPRMT also induces the transcription of coenzyme Q biosynthesis, mitochondrial fission, glycolysis, and biogenesis genes [36], [37], [38]. These findings suggest that the UPRMT alters mitochondrial metabolism to promote mitochondrial function and cell survival during stress. Recent studies have shown that the UPRMT can be activated in response to mitochondrial stress, including depletion of the mitochondrial genome [39], deletions in mitochondrial ETC genes [3], inhibition of mitochondrial protease activity [36], [37], and reagents that induce reactive oxygen species production [39].
Fig. 2.
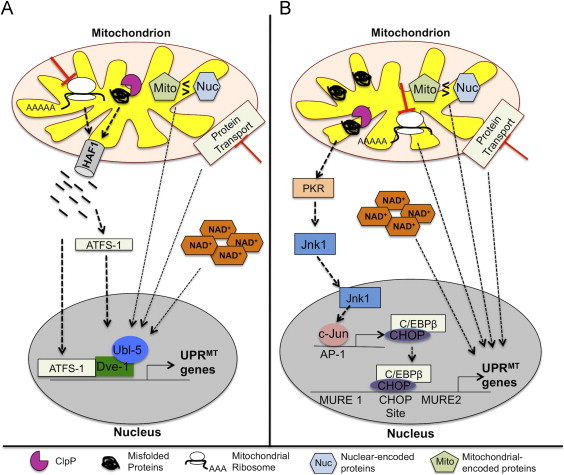
Mitochondrial unfolded protein response (UPRMT) in C. elegans and mammals. (A) In C. elegans, mis-folded proteins are degraded by the ClpP protease and the cleaved products are exported to the cytosol through the HAF-1 peptide exporter. These peptides serve as a signal for activation of transcription factor, ATFS-1. Subsequently, ATFS-1 activates Ubl-5 to form a complex with transcription factor Dve-1 to transcriptionally activate UPRMT genes. (B) Currently, the signaling pathways involved in the UPRMT in response to mitochondrial protein stress are not completely defined in mammals. The accumulation of mis-folded proteins has been shown to activate the UPRMT through the Jnk/c-Jun pathway. In response to mis-folded proteins, protein kinase R (PKR) is activated and activates the nuclear translocation of Jnk via phosphorylation, which activates the transcription factor, c-Jun. Activated c-Jun in turn binds to the activation protein-1 (AP-1) element to initiate the transcription of CHOP. Subsequently, CHOP and co-transcriptional activator, C/EBPβ, induce the expression of UPRMT genes. Interestingly, activation of the Jnk/c-Jun pathway requires the ClpP protease suggesting that cleaved peptides could potentially serve as an initial signal for the induction of the UPRMT.
The UPRMT has not been studied extensively in mammals, but UPRMT components are elevated in mitochondrial DNA depleted mammalian cells, suggesting a conserved signaling mechanism in response to mitochondrial stress [33], [40]. Two potential mammalian orthologues of transcription factors involved in the response in C. elegans include SATB1, an orthologue for DVE-1, which is a CREB-binding protein (CBP) partner [41] and UBL-5 [42]. However, to date no studies have shown the involvement of these transcription factors in the transcriptional control of mitochondrial stress proteins in mammals. The conserved regulatory element in promoters of the UPRMT related genes (e.g., Hsp60, Hsp10, mtDnaJ, ClpP, YME1L1, PMPCB, TIMM17A, NDUFB2, Trx2 and endoG) implicate the transcription factors CHOP and C/EBPβ as putative transcription factors for this pathway [37]. Transcriptional activation of the CHOP gene is regulated by the UPRMT through AP-1 (activation protein-1) element [43]. Transcriptional activation of CHOP is under the control of the Jnk/c-Jun pathway. There is evidence that transcriptional factor, c-Jun binds to the AP-1 element of CHOP suggesting that the transcriptional activation of CHOP is under the control of the Jnk/c-Jun pathway [44], [45]. Consistent with this, Cos-7 cells with increased UPRMT activity induced by a mutation in mitochondrial ornithine transcarbamlyase (OTC) have elevated levels of phosphorylated-Jnk [46]. The induction of the UPRMT triggered by an OTC mutation, is dependent on protein kinase R (PKR) and PKR activity is required for the activation of c-Jun and transcriptional activation of CHOP [46]. Furthermore, PKR activation requires ClpP protease, suggesting a potential involvement of mitochondrial peptide release for UPRMT activation, analogous to the one observed in C. elegans [46]. Together these data predict one putative signaling pathway involved in the mammalian UPRMT, but leave many unanswered questions.
The UPRMT and longevity
A link between UPRMT and longevity was first revealed in two long-lived C. elegans mitochondrial mutants (isp-1 and clk-1) [3]. RNAi knockdown of either UBL-5 or DVE-1 reversed lifespan extension in both isp-1 and clk-1 mutants. Since that first study, there have been a number of other reports implicating the UPRMT in longevity. For example, a recent study showed that mitochondrial biogenesis and components of the UPRMT were up-regulated in the previously reported long-lived Complex IV deficiency mouse, Surf1−/− [1], [4]. Another recent study showed that longer-lived mice have decreased expression of mitochondrial ribosomal protein 5 (mrps-5), indicating that reduced mitochondrial translation correlates with longevity [47]. In fact, RNAi knockdown of mrps-5 extends lifespan and strongly induces the UPRMT in C. elegans. Supplementation with the free radical scavenger, N-acetylcysteine, had no effect on lifespan suggesting that longevity is independent of ROS production [47]. However, lifespan extension was suppressed with RNAi knockdown of the UPRMT component, HAF-1.
Studies in C. elegans showed that the inhibition of Complex IV using RNAi against cco-1 and restricted to neuronal tissue increased lifespan and induced the UPRMT not only in neurons but also in a heterologous tissue, the intestine [3]. These findings suggest that there is not only intracellular signaling resulting in UPRMT activation, but that there is also signaling between tissues resulting in UPRMT activation. It is possible that cells in one tissue may sense stress and induce a nuclear response that would lead to the extracellular release of a signal, coined mitokine [3], to generate UPRMT response in a distal tissue. It is not clear whether cell-nonautonomous induction of the UPRMT is required for enhanced longevity in the cco-1 worms.
Mitochondrial systemic signaling also exists in Drosophila. In flies, muscle-specific disruption of Complex I was shown to promote healthy longevity and preserve mitochondrial and muscle function with age [48]. These findings were dependent on the induction of the UPRMT since RNAi knockdown of either ClpX and Hsp60C suppressed climbing performance and lifespan. Not surprisingly, reducing ClpX and Hsp60C expression levels also suppressed climbing performance and lifespan in wild-type flies suggesting the requirement of the UPRMT for muscle homeostasis [48]. In contrast to C. elegans, UPRMT induction in Drosophila appears to require mitochondrial dependent ROS production. Overexpression of the mitochondrial antioxidant enzymes catalase and glutathione peroxidase suppressed UPRMT in Complex-1 defective flies [48]. To eliminate other potential mechanisms by which Complex-1 could mediate longevity, Hsp60 and Hsp60c were overexpressed in muscle of Drosophila. Muscle-specific overexpression of UPRMT chaperones had no effect on lifespan however, mitochondrial and muscle function were preserved with age [48]. It is possible that limiting UPRMT in one tissue in Drosophila is not sufficient to enhance whole organismal lifespan and may require whole body overexpression. However, despite the lack of effect on lifespan, muscle-specific overexpression of UPRMT chaperones did result in improved healthspan.
A number of other pro-longevity models, independent of perturbations of mitochondrial ETC, complexes result in the induction of the UPRMT. For example, there is a long-standing association between the sirtuin-1 signaling pathway and longevity. Supplementation of NAD+ precursors, nicotinamide (NAM) and NAM ribose (NR), significantly induces the UPRMT and antioxidant response in both C. elegans and mammals [49]. In C. elegans, UPRMT induction is associated with enhanced longevity and partial protection against age-related decline in mitochondrial function [49]. Lifespan extension via supplementation of NAD+ precursors or Sir2.1 overexpression is suppressed by RNAi knockdown of UBL-5. However, it is not clear whether lifespan extension mediated by NAD+/Sirt1 pathway is singly dependent on UPRMT induction or also requires an antioxidant response, as UBL-5 was also shown to regulate the antioxidant response in C. elegans [49]. In addition, the life-extending intervention, rapamycin, is also a potent inducer of the UPRMT in both C. elegans and mammals [47]. Lifespan extension is suppressed with RNAi knockdown of UBL-5 in C. elegans suggesting that rapamycin-mediated longevity requires the UPRMT. Similar observations were made with resveratrol. In these models, it is proposed that the UPRMT activation is due to an imbalance of mitochondrial to nuclear-encoded proteins (i.e., mitonuclear imbalance) [47].
The causal relationship between UPRMT and longevity has been called into question by a recent study showing that the UPRMT is not necessary for longevity. In previous studies, RNAi used against HAF-1 and UBL-5 did not fully attenuate the UPRMT in cco-1 and mrps-5 RNAi treated worms [3], [47], [49]. In fact, UPRMT induction was still significantly higher when compared to untreated worms. Furthermore, employing RNAi knockdown of ATFS-1 to inhibit the UPRMT had no effect on lifespan in the isp-1 mutant or C. elegans with RNAi knockdown of cco-1, letm-1 and Y24D9A.8 despite a reduction in UPRMT in response to reduced ATFS-1 [50]. In addition, some C. elegans mitochondrial mutants with increased UPRMT were reported having no change in lifespan or they were short-lived compared to wild-type control [50]. These data suggest that UPRMT is not causally linked to longevity nor does it promote longevity. In support of this, deletion of ClpP, a key modulator of the UPRMT was shown to result in increased lifespan in fungi [51]. Thus, these studies highlight the complex relationship between UPRMT in longevity.
Mitochondrial-derived peptides, newly discovered modulators of cytoprotective activities
Mitochondrial-derived peptides (MDPs) are a newly discovered set of mitochondrial stress signals that are generated within the mitochondrial genome. Humanin, the first discovered MDP, is released during mitochondrial dysfunction, resulting in an increase of cytoprotective activities and metabolic adaptation [52]. In fact, humanin was discovered using a functional expression screening for peptides and proteins that could suppress neuronal cell death caused by multiple types of familial Alzheimer’s disease [52], [53]. There is controversy over whether humanin is of mitochondrial or nuclear origin. However, evidence points toward mitochondrial origin; humanin has 99% sequence similarity to mitochondrial-encoded 16s RNA, humanin is absent in cells depleted of the mitochondrial genome, and humanin is formylated, a hallmark post-translation modification of mitochondrial-encoded proteins [54], [55]. To date, it is not clear whether humanin is translated in the mitochondria and then exported to the cytoplasm or vice versa. Nevertheless, humanin has gained significant recognition for its versatile cytoprotective activities.
Humanin has several modes of action. Intracellular humanin binds to IGFBP-3, Bax, and tBid [53], [56], [57]. As a result, humanin suppresses apoptosis and promotes cell survival during stress. Intracellular levels of humanin are regulated by its interaction with Trim11, which promote ubiquitin-mediated degradation [58], [59]. Humanin is also present in human plasma, suggesting that cells secrete humanin. Cells sense circulating humanin through two plasma membrane receptors, the trimeric CNTFR/gp130/WSX-1 receptor and the FPRL1/2 receptor [60], [61]. A number of cytoprotective activities are initiated upon binding to these receptors. Studies have shown that humanin is neuroprotective, ameliorates atherosclerotic plaque formation, and is protective in rodent models of diabetes [62], [63], [64], [65], [66], [67]. Humanin was also shown to restore physiological ATP levels in lymphocytes isolated from MELAS patients [68]. Together these studies implicate humanin as a potential therapeutic target for a number of diseases.
Recent studies suggest that MDPs, like humanin, may be a key pro-longevity factor due to its versatile cytoprotective activities. Offspring of centenarians have three-fold higher levels of humanin. Rats have a significant loss of humanin in brain and skeletal muscle with age [69]. Conversely, circulating humanin levels significantly decline with age in mice and humans [62], [64], [69]. Humanin may be one promising intervention for age-related diseases and to improve healthspan.
Mitochondrial-derived damage associated-molecular patterns
For many years, studies have shown that mitochondrial dysfunction is tightly associated to the immune response and inflammation [70], [71]. Recent work has identified a number of mitochondrial-derived signals that modulate the immune response and inflammation. These signals, also known as mitochondrial damage associated-molecular patterns (DAMPs), have been shown to be in the form of mitochondrial DNA (mtDNA), N-formyl peptides and mitochondrial proteins. Recently mitochondrial dysfunction related to impaired autophagy was shown to lead to the export of mtDNA to the cytoplasm activating caspase-1 and promoting the secretion of IL-1β and IL-18 in macrophages [72]. Degraded mtDNA has also been shown to strongly induce production of pro-inflammatory cytokines in mouse astrocytes [73]. MtDNA is a ligand for the toll-like receptor 9 (TLR9), which is critical for the synthesis of pro-inflammatory cytokines. Activation of TLR9 can have significant downstream effects on vascular function. In fact, mtDNA plasma levels are significantly elevated in hypertensive rats [74]. Together these data suggest that mtDNA can serve as an endogenous and exogenous danger signal to activate an inflammatory response. Another group of mitDAMPs is N-formyl peptides with mitochondrial origin. These peptides have high affinity toward Formyl Peptide Receptors (FPR), a class of G-coupled receptors involved in chemotaxis, a critical action in the inflammatory response [75]. N-formyl peptides stimulate the expression of pro-inflammatory cytokines, activate innate immune response and cause systemic inflammation and lung injury in rats [76], [77], [78].
There are a number of mitochondrial proteins that have been implicated to serve as mitDAMPs. For example, a recent study showed that neurons treated with whole mitochondrial protein extracts had an increased expression of pro-inflammatory cytokines and induced cell death [79]. Serum cytochrome c levels are severely elevated in patients with systemic inflammatory response syndrome and in patients on hemodialysis, a procedure known to cause inflammation [80], [81]. Mitochondrial transcription factor A (TFAM), an implicated mitDAMP, can activate monocytes and plasmacytoid dendritic cell responses [82], [83]. TFAM was shown to induce the expression of pro-inflammatory cytokines for microglial activation, a predecessor of neuronal cell death [79]. In fact, neurons incubated in conditioned media from TFAM-treated microglia induced cell death [79].
The discovery of mitDAMPs provides evidence that mitochondria can directly regulate immune response and systemic inflammation. These findings beg the question: do mitDAMPs contribute to inflammaging? It was recently shown that circulating mtDNA is significantly elevated in the elderly population [84]. The fact that mitDAMPs have potent effects on the vascular system suggests that mitDAMPs could contribute to age-related diseases such as cardiovascular disease and atherosclerosis. It is also tempting to speculate that mitDAMPs are one potential contributor to age-related neurodegeneration since extracellular TFAM can activate microglia and induce neuronal cell death, common features of the aging brain [79]. Overall, mitDAMPs have significant clinical relevance and may be a promising target to ameliorate inflammaging.
Concluding remarks/summary
In just the last few years, mitochondrial research has significantly evolved, especially in the field of aging. Communication between mitochondria and nucleus is a well-established phenomenon that occurs as a result of mitochondrial dysfunction. This phenomenon is observed in longer-lived species with perturbations in the mitochondrial electron transport chain. Until recently the signaling pathways that elicit mitochondrial–nuclear communication were unknown. The discovery of these pathways has allowed the field to closely assess the relationship between mitochondrial–nuclear signaling and longevity. In the majority of the studies reviewed here, this relationship has been evaluated by genetic manipulation of critical components of either the retrograde response or UPRMT in long-lived species. In most cases, knockdown of these components reversed lifespan extension in long-lived mitochondrial mutants suggesting that these signaling pathways are essential for longevity. However, recent work published by Kaeberlein’s group [50] does not support a causal link between the UPRMT and longevity. In agreement with these findings, overexpression of the UPRMT components Hsp60 and ClpP does not extend longevity in Drosophila, but does extend healthy aging. Thus, the UPRMT may not be a viable target to increase lifespan, but should not be ruled out as a target for the improvement of age-related diseases. These studies highlight the complex relationship between longevity and the UPRMT.
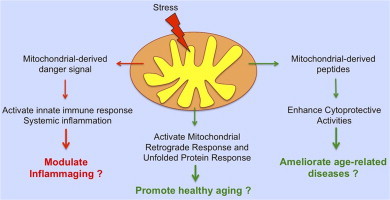
An emerging concept in the aging field is the idea that mitochondria can signal between tissues to upregulate cytoprotective activities to promote healthy aging and organismal fitness (see Fig. 3). The discovery of the mitochondrial-derived peptide, humanin, has unraveled other modes by which mitochondria can regulate signal transduction pathways. Even more interestingly, humanin can serve as a cell-nonautonomous signal. In support of this concept, Dillin’s laboratory found that the UPRMT is activated in a cell-nonautonomous fashion in C. elegans [3]. Also, there are a number of reports showing that mitochondria can release a danger signal, mitDAMP to activate immune response and inflammation in other tissues and organs. Despite the controversy over the origin of humanin (nuclear or mitochondrial), it is a novel peptide that has significant implications in the development of therapeutics to ameliorate metabolic and age-related diseases. Therefore, future studies should aim to uncover additional peptides that have cytoprotective activities similar to humanin.
Fig. 3.
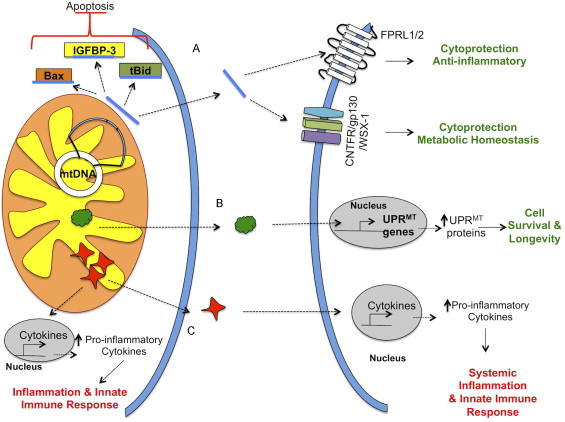
Signaling factors that mediate mitochondrial crosstalk between cells and tissues. Mitochondrial dysfunction can result in the release of signaling factors that can act both intracellularly and extracellularly. (A) For example, humanin, a mitochondrial-derived peptide that is generated within the mitochondrial genome, is released by the mitochondria to suppress apoptosis through binding to IGFBP-3 and pro-apoptotic factors, Bax and tBid. Humanin can also exert its cytoprotective, anti-inflammatory and metabolic-protective properties through its binding to the FPRL1/2 and CNFTR/gp130/WSX-1 receptors. (B) Mitokines are recently hypothesized signaling factors that are released by cells to induce the UPRMT in distal cells. (C) Mitochondria can release danger signals, also known as mitochondrial damage associated-molecular patterns (mitDAMPs), to promote inflammation and activate the innate immune response both intracellularly and extracellularly. MitDAMPs induce the expression of pro-inflammatory cytokines endogenously and in distal cells.
Acknowledgements
A Senior Scholar Award by the Ellison Medical Foundation to H.V.R., the Barrett Scholarship Fund by the Oklahoma Medical Research Foundation to S.H., and a Grant-in-Aid of Research from the National Academy of Sciences, administered by Sigma Xi, The Scientific Research Society (G20140315432081) to S.H. supported this work.
References
- 1.Dell’agnello C., Leo S., Agostino A., Szabadkai G., Tiveron C., Zulian A., Prelle A., Roubertoux P., Rizzuto R., Zeviani M. Increased longevity and refractoriness to Ca(2+)-dependent neurodegeneration in Surf1 knockout mice. Human Molecular Genetics. 2007;16:431–444. doi: 10.1093/hmg/ddl477. 17210671 [DOI] [PubMed] [Google Scholar]
- 2.Lapointe J., Hekimi S. Early mitochondrial dysfunction in long-lived Mclk1+/− mice. Journal of Biological Chemistry. 2008;283:26217–26227. doi: 10.1074/jbc.M803287200. 18635541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Durieux J., Wolff S., Dillin A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell. 2011;144:79–91. doi: 10.1016/j.cell.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pulliam D.A., Deepa S.S., Liu Y., Hill S., Lin A.L., Bhattacharya A., Shi Y., Sloane L., Viscomi C., Zeviani M. Complex IV deficient Surf1−/− mice initiate mitochondrial stress responses. Biochemical Journal. 2014 doi: 10.1042/BJ20140291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kirchman P.A., Kim S., Lai C.Y., Jazwinski S.M. Interorganelle signaling is a determinant of longevity in Saccharomyces cerevisiae. Genetics. 1999;152:179–190. doi: 10.1093/genetics/152.1.179. 10224252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Parikh V.S., Morgan M.M., Scott R., Clements L.S., Butow R.A. The mitochondrial genotype can influence nuclear gene expression in yeast. Science. 1987;235:576–580. doi: 10.1126/science.3027892. 3027892 [DOI] [PubMed] [Google Scholar]
- 7.Epstein C.B., Waddle J.A., Hale W.t., Davé V., Thornton J., Macatee T.L., Garner H.R., Butow R.A. Genome-wide responses to mitochondrial dysfunction. Molecular Biology of the Cell. 2001;12:297–308. doi: 10.1091/mbc.12.2.297. 11179416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu Z., Butow R.A. A transcriptional switch in the expression of yeast tricarboxylic acid cycle genes in response to a reduction or loss of respiratory function. Molecular and Cellular Biology. 1999;19:6720–6728. doi: 10.1128/mcb.19.10.6720. 10490611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liao X., Butow R.A. RTG1 and RTG2: two yeast genes required for a novel path of communication from mitochondria to the nucleus. Cell. 1993;72:61–71. doi: 10.1016/0092-8674(93)90050-z. 8422683 [DOI] [PubMed] [Google Scholar]
- 10.Ferreira Junior J.R., Spírek M., Liu Z., Butow R.A. Interaction between Rtg2p and Mks1p in the regulation of the RTG pathway of Saccharomyces cerevisiae. Gene. 2005;354:2–8. doi: 10.1016/j.gene.2005.03.048. 15967597 [DOI] [PubMed] [Google Scholar]
- 11.Sekito T., Thornton J., Butow R.A. Mitochondria-to-nuclear signaling is regulated by the subcellular localization of the transcription factors Rtg1p and Rtg3p. Molecular Biology of the Cell. 2000;11:2103–2115. doi: 10.1091/mbc.11.6.2103. 10848632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chelstowska A., Butow R.A. RTG genes in yeast that function in communication between mitochondria and the nucleus are also required for expression of genes encoding peroxisomal proteins. Journal of Biological Chemistry. 1995;270:18141–18146. doi: 10.1074/jbc.270.30.18141. 7629125 [DOI] [PubMed] [Google Scholar]
- 13.Dilova I., Chen C.Y., Powers T. Mks1 in concert with TOR signaling negatively regulates RTG target gene expression in S. cerevisiae. Current Biology. 2002;12:389–395. doi: 10.1016/s0960-9822(02)00677-2. 11882290 [DOI] [PubMed] [Google Scholar]
- 14.Zhang F., Pracheil T., Thornton J., Liu Z. Adenosine triphosphate (ATP) is a candidate signaling molecule in the mitochondria-to-nucleus retrograde response pathway. Genes. 2013;4:86–100. doi: 10.3390/genes4010086. 24605246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miceli M.V., Jiang J.C., Tiwari A., Rodriguez-Quiñones J.F., Jazwinski S.M. Loss of mitochondrial membrane potential triggers the retrograde response extending yeast replicative lifespan. Frontiers in Genetics. 2011;2:102. doi: 10.3389/fgene.2011.00102. 22303396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pray-Grant M.G., Schieltz D., McMahon S.J., Wood J.M., Kennedy E.L., Cook R.G., Workman J.L., Yates J.R. 3rd, Grant P.A. The novel SLIK histone acetyltransferase complex functions in the yeast retrograde response pathway. Molecular and Cellular Biology. 2002;22:8774–8786. doi: 10.1128/MCB.22.24.8774-8786.2002. 12446794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim S., Ohkuni K., Couplan E., Jazwinski S.M. The histone acetyltransferase GCN5 modulates the retrograde response and genome stability determining yeast longevity. Biogerontology. 2004;5:305–316. doi: 10.1007/s10522-004-2568-x. 15547318 [DOI] [PubMed] [Google Scholar]
- 18.Giannattasio S., Liu Z., Thornton J., Butow R.A. Retrograde response to mitochondrial dysfunction is separable from TOR1/2 regulation of retrograde gene expression. Journal of Biological Chemistry. 2005;280:42528–42535. doi: 10.1074/jbc.M509187200. 16253991 [DOI] [PubMed] [Google Scholar]
- 19.Woo D.K., Poyton R.O. The absence of a mitochondrial genome in rho0 yeast cells extends lifespan independently of retrograde regulation. Experimental Gerontology. 2009;44:390–397. doi: 10.1016/j.exger.2009.03.001. 19285548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barros M.H., Bandy B., Tahara E.B., Kowaltowski A.J. Higher respiratory activity decreases mitochondrial reactive oxygen release and increases life span in Saccharomyces cerevisiae. Journal of Biological Chemistry. 2004;279:49883–49888. doi: 10.1074/jbc.M408918200. 15383542 [DOI] [PubMed] [Google Scholar]
- 21.Guaragnella N., Zdralević M., Lattanzio P., Marzulli D., Pracheil T., Liu Z., Passarella S., Marra E., Giannattasio S. Yeast growth in raffinose results in resistance to acetic-acid induced programmed cell death mostly due to the activation of the mitochondrial retrograde pathway. Biochimica et Biophysica Acta. 2013;1833:2765–2774. doi: 10.1016/j.bbamcr.2013.07.017. 23906793 [DOI] [PubMed] [Google Scholar]
- 22.Jiang J.C., Jaruga E., Repnevskaya M.V., Jazwinski S.M. An intervention resembling caloric restriction prolongs life span and retards aging in yeast. FASEB Journal: Official Publication of the Federation of American Societies for Experimental Biology. 2000;14:2135–2137. doi: 10.1096/fj.00-0242fje. [DOI] [PubMed] [Google Scholar]
- 23.Wang J., Jiang J.C., Jazwinski S.M. Gene regulatory changes in yeast during life extension by nutrient limitation. Experimental Gerontology. 2010;45:621–631. doi: 10.1016/j.exger.2010.02.008. 20178842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sinclair D.A., Guarente L. Extrachromosomal rDNA circles—a cause of aging in yeast. Cell. 1997;91:1033–1042. doi: 10.1016/s0092-8674(00)80493-6. 9428525 [DOI] [PubMed] [Google Scholar]
- 25.Borghouts C., Benguria A., Wawryn J., Jazwinski S.M. Rtg2 protein links metabolism and genome stability in yeast longevity. Genetics. 2004;166:765–777. doi: 10.1534/genetics.166.2.765. 15020466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heeren G., Rinnerthaler M., Laun P., von Seyerl P., Kössler S., Klinger H., Hager M., Bogengruber E., Jarolim S., Simon-Nobbe B. The mitochondrial ribosomal protein of the large subunit, Afo1p, determines cellular longevity through mitochondrial back-signaling via TOR1. Aging. 2009;1:622–636. doi: 10.18632/aging.100065. 20157544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Caballero A., Ugidos A., Liu B., Öling D., Kvint K., Hao X., Mignat C., Nachin L., Molin M., Nyström T. Absence of mitochondrial translation control proteins extends life span by activating sirtuin-dependent silencing. Molecular Cell. 2011;42:390–400. doi: 10.1016/j.molcel.2011.03.021. 21549315 [DOI] [PubMed] [Google Scholar]
- 28.Miceli M.V., Jazwinski S.M. Nuclear gene expression changes due to mitochondrial dysfunction in ARPE-19 cells: implications for age-related macular degeneration. Investigative Ophthalmology & Visual Science. 2005;46:1765–1773. doi: 10.1167/iovs.04-1327. 15851580 [DOI] [PubMed] [Google Scholar]
- 29.Miceli M.V., Jazwinski S.M. Common and cell type-specific responses of human cells to mitochondrial dysfunction. Experimental Cell Research. 2005;302:270–280. doi: 10.1016/j.yexcr.2004.09.006. 15561107 [DOI] [PubMed] [Google Scholar]
- 30.Srinivasan V., Kriete A., Sacan A., Jazwinski S.M. Comparing the yeast retrograde response and NF-kappaB stress responses: implications for aging. Aging Cell. 2010;9:933–941. doi: 10.1111/j.1474-9726.2010.00622.x. 20961379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Passos J.F., Saretzki G., Ahmed S., Nelson G., Richter T., Peters H., Wappler I., Birket M.J., Harold G., Schaeuble K. Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLoS Biology. 2007;5:e110. doi: 10.1371/journal.pbio.0050110. 17472436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Benedetti C., Haynes C.M., Yang Y., Harding H.P., Ron D. Ubiquitin-like protein 5 positively regulates chaperone gene expression in the mitochondrial unfolded protein response. Genetics. 2006;174:229–239. doi: 10.1534/genetics.106.061580. 16816413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martinus R.D., Garth G.P., Webster T.L., Cartwright P., Naylor D.J., Hoj P.B., Hoogenraad N.J. Selective induction of mitochondrial chaperones in response to loss of the mitochondrial genome. European Journal of Biochemistry/FEBS. 1996;240:98–103. doi: 10.1111/j.1432-1033.1996.0098h.x. [DOI] [PubMed] [Google Scholar]
- 34.Haynes C.M., Petrova K., Benedetti C., Yang Y., Ron D. ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Developmental Cell. 2007;13:467–480. doi: 10.1016/j.devcel.2007.07.016. 17925224 [DOI] [PubMed] [Google Scholar]
- 35.Haynes C.M., Yang Y., Blais S.P., Neubert T.A., Ron D. The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans. Molecular Cell. 2010;37:529–540. doi: 10.1016/j.molcel.2010.01.015. 20188671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nargund A.M., Pellegrino M.W., Fiorese C.J., Baker B.M., Haynes C.M. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science. 2012;337:587–590. doi: 10.1126/science.1223560. 22700657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aldridge J.E., Horibe T., Hoogenraad N.J. Discovery of genes activated by the mitochondrial unfolded protein response (mtUPR) and cognate promoter elements. PloS One. 2007;2:e874. doi: 10.1371/journal.pone.0000874. 17849004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cristina D., Cary M., Lunceford A., Clarke C., Kenyon C. A regulated response to impaired respiration slows behavioral rates and increases lifespan in Caenorhabditis elegans. PLoS Genetics. 2009;5:e1000450. doi: 10.1371/journal.pgen.1000450. 19360127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yoneda T., Benedetti C., Urano F., Clark S.G., Harding H.P., Ron D. Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperones. Journal of Cell Science. 2004;117:4055–4066. doi: 10.1242/jcs.01275. 15280428 [DOI] [PubMed] [Google Scholar]
- 40.Zhao Q., Wang J., Levichkin I.V., Stasinopoulos S., Ryan M.T., Hoogenraad N.J. A mitochondrial specific stress response in mammalian cells. EMBO Journal. 2002;21:4411–4419. doi: 10.1093/emboj/cdf445. 12198143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang M., Poplawski M., Yen K., Cheng H., Bloss E., Zhu X., Patel H., Mobbs C.V. Role of CBP and SATB-1 in aging, dietary restriction, and insulin-like signaling. PLoS Biology. 2009;7:e1000245. doi: 10.1371/journal.pbio.1000245. 19924292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bozaoglu K., Curran J.E., Elliott K.S., Walder K.R., Dyer T.D., Rainwater D.L., VandeBerg J.L., Comuzzie A.G., Collier G.R., Zimmet P. Association of genetic variation within UBL5 with phenotypes of metabolic syndrome. Human Biology. 2006;78:147–159. doi: 10.1353/hub.2006.0033. 17036923 [DOI] [PubMed] [Google Scholar]
- 43.Horibe T., Hoogenraad N.J. The chop gene contains an element for the positive regulation of the mitochondrial unfolded protein response. PloS One. 2007;2:e835. doi: 10.1371/journal.pone.0000835. 17848986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jaeschke A., Karasarides M., Ventura J.J., Ehrhardt A., Zhang C., Flavell R.A., Shokat K.M., Davis R.J. JNK2 is a positive regulator of the cJun transcription factor. Molecular Cell. 2006;23:899–911. doi: 10.1016/j.molcel.2006.07.028. 16973441 [DOI] [PubMed] [Google Scholar]
- 45.Weiss C., Schneider S., Wagner E.F., Zhang X., Seto E., Bohmann D. JNK phosphorylation relieves HDAC3-dependent suppression of the transcriptional activity of c-Jun. EMBO Journal. 2003;22:3686–3695. doi: 10.1093/emboj/cdg364. 12853483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rath E., Berger E., Messlik A., Nunes T., Liu B., Kim S.C., Hoogenraad N., Sans M., Sartor R.B., Haller D. Induction of dsRNA-activated protein kinase links mitochondrial unfolded protein response to the pathogenesis of intestinal inflammation. Gut. 2012;61:1269–1278. doi: 10.1136/gutjnl-2011-300767. 21997551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Houtkooper R.H., Mouchiroud L., Ryu D., Moullan N., Katsyuba E., Knott G., Williams R.W., Auwerx J. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature. 2013;497:451–457. doi: 10.1038/nature12188. 23698443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Owusu-Ansah E., Song W., Perrimon N. Muscle mitohormesis promotes longevity via systemic repression of insulin signaling. Cell. 2013;155:699–712. doi: 10.1016/j.cell.2013.09.021. 24243023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mouchiroud L., Houtkooper R.H., Moullan N., Katsyuba E., Ryu D., Cantó C., Mottis A., Jo Y.S., Viswanathan M., Schoonjans K. The NAD(+)/Sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell. 2013;154:430–441. doi: 10.1016/j.cell.2013.06.016. 23870130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bennett C.F., Vander Wende H., Simko M., Klum S., Barfield S., Choi H., Pineda V.V., Kaeberlein M. Activation of the mitochondrial unfolded protein response does not predict longevity in Caenorhabditis elegans. Nature Communications. 2014;5:3483. doi: 10.1038/ncomms4483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fischer F., Weil A., Hamann A., Osiewacz H.D. Human CLPP reverts the longevity phenotype of a fungal ClpP deletion strain. Nature Communications. 2013;4:1397. doi: 10.1038/ncomms2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hashimoto Y., Niikura T., Tajima H., Yasukawa T., Sudo H., Ito Y., Kita Y., Kawasumi M., Kouyama K., Doyu M. A rescue factor abolishing neuronal cell death by a wide spectrum of familial Alzheimer’s disease genes and Abeta. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:6336–6341. doi: 10.1073/pnas.101133498. 11371646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guo B., Zhai D., Cabezas E., Welsh K., Nouraini S., Satterthwait A.C., Reed J.C. Humanin peptide suppresses apoptosis by interfering with Bax activation. Nature. 2003;423:456–461. doi: 10.1038/nature01627. 12732850 [DOI] [PubMed] [Google Scholar]
- 54.Harada M., Habata Y., Hosoya M., Nishi K., Fujii R., Kobayashi M., Hinuma S. N-formylated humanin activates both formyl peptide receptor-like 1 and 2. Biochemical and Biophysical Research Communications. 2004;324:255–261. doi: 10.1016/j.bbrc.2004.09.046. 15465011 [DOI] [PubMed] [Google Scholar]
- 55.Tajima H., Niikura T., Hashimoto Y., Ito Y., Kita Y., Terashita K., Yamazaki K., Koto A., Aiso S., Nishimoto I. Evidence for in vivo production of Humanin peptide, a neuroprotective factor against Alzheimer’s disease-related insults. Neuroscience Letters. 2002;324:227–231. doi: 10.1016/s0304-3940(02)00199-4. 12009529 [DOI] [PubMed] [Google Scholar]
- 56.Ikonen M., Liu B., Hashimoto Y., Ma L., Lee K.W., Niikura T., Nishimoto I., Cohen P. Interaction between the Alzheimer’s survival peptide humanin and insulin-like growth factor-binding protein 3 regulates cell survival and apoptosis. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:13042–13047. doi: 10.1073/pnas.2135111100. 14561895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhai D., Luciano F., Zhu X., Guo B., Satterthwait A.C., Reed J.C. Humanin binds and nullifies bid activity by blocking its activation of Bax and Bak. Journal of Biological Chemistry. 2005;280:15815–15824. doi: 10.1074/jbc.M411902200. 15661737 [DOI] [PubMed] [Google Scholar]
- 58.Niikura T., Hashimoto Y., Tajima H., Ishizaka M., Yamagishi Y., Kawasumi M., Nawa M., Terashita K., Aiso S., Nishimoto I. A tripartite motif protein TRIM11 binds and destabilizes Humanin, a neuroprotective peptide against Alzheimer’s disease-relevant insults. European Journal of Neuroscience. 2003;17:1150–1158. doi: 10.1046/j.1460-9568.2003.02553.x. 12670303 [DOI] [PubMed] [Google Scholar]
- 59.Ishikawa H., Tachikawa H., Miura Y., Takahashi N. TRIM11 binds to and destabilizes a key component of the activator-mediated cofactor complex (ARC105) through the ubiquitin–proteasome system. FEBS Letters. 2006;580:4784–4792. doi: 10.1016/j.febslet.2006.07.066. 16904669 [DOI] [PubMed] [Google Scholar]
- 60.Ying G., Iribarren P., Zhou Y., Gong W., Zhang N., Yu Z.X., Le Y., Cui Y., Wang J.M. Humanin, a newly identified neuroprotective factor, uses the G protein-coupled formylpeptide receptor-like-1 as a functional receptor. Journal of Immunology. 2004;172:7078–7085. doi: 10.4049/jimmunol.172.11.7078. [DOI] [PubMed] [Google Scholar]
- 61.Hashimoto Y., Kurita M., Aiso S., Nishimoto I., Matsuoka M. Humanin inhibits neuronal cell death by interacting with a cytokine receptor complex or complexes involving CNTF receptor alpha/WSX-1/gp130. Molecular Biology of the Cell. 2009;20:2864–2873. doi: 10.1091/mbc.E09-02-0168. 19386761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hoang P.T., Park P., Cobb L.J., Paharkova-Vatchkova V., Hakimi M., Cohen P., Lee K.W. The neurosurvival factor Humanin inhibits beta-cell apoptosis via signal transducer and activator of transcription 3 activation and delays and ameliorates diabetes in nonobese diabetic mice. Metabolism: Clinical and Experimental. 2010;59:343–349. doi: 10.1016/j.metabol.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Oh Y.K., Bachar A.R., Zacharias D.G., Kim S.G., Wan J., Cobb L.J., Lerman L.O., Cohen P., Lerman A. Humanin preserves endothelial function and prevents atherosclerotic plaque progression in hypercholesterolemic ApoE deficient mice. Atherosclerosis. 2011;219:65–73. doi: 10.1016/j.atherosclerosis.2011.06.038. 21763658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bachar A.R., Scheffer L., Schroeder A.S., Nakamura H.K., Cobb L.J., Oh Y.K., Lerman L.O., Pagano R.E., Cohen P., Lerman A. Humanin is expressed in human vascular walls and has a cytoprotective effect against oxidized LDL-induced oxidative stress. Cardiovascular Research. 2010;88:360–366. doi: 10.1093/cvr/cvq191. 20562421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xu X., Chua C.C., Gao J., Hamdy R.C., Chua B.H. Humanin is a novel neuroprotective agent against stroke. Stroke: A Journal of Cerebral Circulation. 2006;37:2613–2619. doi: 10.1161/01.STR.0000242772.94277.1f. [DOI] [PubMed] [Google Scholar]
- 66.Xu X., Chua C.C., Gao J., Chua K.W., Wang H., Hamdy R.C., Chua B.H. Neuroprotective effect of humanin on cerebral ischemia/reperfusion injury is mediated by a PI3K/Akt pathway. Brain Research. 2008;1227:12–18. doi: 10.1016/j.brainres.2008.06.018. 18590709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Niikura T., Sidahmed E., Hirata-Fukae C., Aisen P.S., Matsuoka Y. A humanin derivative reduces amyloid beta accumulation and ameliorates memory deficit in triple transgenic mice. PloS One. 2011;6:e16259. doi: 10.1371/journal.pone.0016259. 21264226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kariya S., Hirano M., Furiya Y., Ueno S. Effect of humanin on decreased ATP levels of human lymphocytes harboring A3243G mutant mitochondrial DNA. Neuropeptides. 2005;39:97–101. doi: 10.1016/j.npep.2004.11.004. 15752543 [DOI] [PubMed] [Google Scholar]
- 69.Muzumdar R.H., Huffman D.M., Atzmon G., Buettner C., Cobb L.J., Fishman S., Budagov T., Cui L., Einstein F.H., Poduval A. Humanin: A novel central regulator of peripheral insulin action. PloS One. 2009;4:e6334. doi: 10.1371/journal.pone.0006334. 19623253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Franceschi C., Bonafè M., Valensin S., Olivieri F., De Luca M., Ottaviani E., De Benedictis G. Inflamm-aging. An evolutionary perspective on immunosenescence. Annals of the New York Academy of Sciences. 2000;908:244–254. doi: 10.1111/j.1749-6632.2000.tb06651.x. 10911963 [DOI] [PubMed] [Google Scholar]
- 71.Tschopp J. Mitochondria: sovereign of inflammation? European Journal of Immunology. 2011;41:1196–1202. doi: 10.1002/eji.201141436. 21469137 [DOI] [PubMed] [Google Scholar]
- 72.Nakahira K., Haspel J.A., Rathinam V.A., Lee S.J., Dolinay T., Lam H.C., Englert J.A., Rabinovitch M., Cernadas M., Kim H.P. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nature Immunology. 2011;12:222–230. doi: 10.1038/ni.1980. 21151103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mathew A., Lindsley T.A., Sheridan A., Bhoiwala D.L., Hushmendy S.F., Yager E.J., Ruggiero E.A., Crawford D.R. Degraded mitochondrial DNA is a newly identified subtype of the damage associated molecular pattern (DAMP) family and possible trigger of neurodegeneration. Journal of Alzheimer's Disease. 2012;30:617–627. doi: 10.3233/JAD-2012-120145. 22460333 [DOI] [PubMed] [Google Scholar]
- 74.Veiko N.N., Konorova I.L., Neverova M.E., Fidelina O.V., Mkrtumova N.A., Ershova E.S., Kon’kova M.S., Postnov A. Delayed appearance of hypertension in spontaneously hypertensive rat (SHR) injected with CpG-rich DNA early in ontogenesis. Biomeditsinskaia khimiia. 2010;56:686–699. doi: 10.18097/pbmc20105606686. [DOI] [PubMed] [Google Scholar]
- 75.Rabiet M.J., Huet E., Boulay F. Human mitochondria-derived N-formylated peptides are novel agonists equally active on FPR and FPRL1, while Listeria monocytogenes-derived peptides preferentially activate FPR. European Journal of Immunology. 2005;35:2486–2495. doi: 10.1002/eji.200526338. 16025565 [DOI] [PubMed] [Google Scholar]
- 76.Zhang Q., Raoof M., Chen Y., Sumi Y., Sursal T., Junger W., Brohi K., Itagaki K., Hauser C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464:104–107. doi: 10.1038/nature08780. 20203610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hauser C.J., Sursal T., Rodriguez E.K., Appleton P.T., Zhang Q., Itagaki K. Mitochondrial damage associated molecular patterns from femoral reamings activate neutrophils through formyl peptide receptors and P44/42 MAP kinase. Journal of Orthopaedic Trauma. 2010;24:534–538. doi: 10.1097/BOT.0b013e3181ec4991. 20736789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Raoof M., Zhang Q., Itagaki K., Hauser C.J. Mitochondrial peptides are potent immune activators that activate human neutrophils via FPR-1. Journal of Trauma. 2010;68:1328–1332. doi: 10.1097/TA.0b013e3181dcd28d. (Discussion 1332–1324) [DOI] [PubMed] [Google Scholar]
- 79.Little J.P., Simtchouk S., Schindler S.M., Villanueva E.B., Gill N.E., Walker D.G., Wolthers K.R., Klegeris A. Mitochondrial transcription factor A (Tfam) is a pro-inflammatory extracellular signaling molecule recognized by brain microglia. Molecular and Cellular Neurosciences. 2014;60:88–96. doi: 10.1016/j.mcn.2014.04.003. [DOI] [PubMed] [Google Scholar]
- 80.Eleftheriadis T., Pissas G., Antoniadi G., Liakopoulos V., Stefanidis I. Damage-associated molecular patterns derived from mitochondria may contribute to the hemodialysis-associated inflammation. International Urology and Nephrology. 2014;46:107–112. doi: 10.1007/s11255-013-0417-z. 23515931 [DOI] [PubMed] [Google Scholar]
- 81.Adachi N., Hirota M., Hamaguchi M., Okamoto K., Watanabe K., Endo F. Serum cytochrome c level as a prognostic indicator in patients with systemic inflammatory response syndrome. Clinica Chimica Acta: International Journal of Clinical Chemistry. 2004;342:127–136. doi: 10.1016/j.cccn.2003.12.011. 15026273 [DOI] [PubMed] [Google Scholar]
- 82.Crouser E.D., Shao G., Julian M.W., Macre J.E., Shadel G.S., Tridandapani S., Huang Q., Wewers M.D. Monocyte activation by necrotic cells is promoted by mitochondrial proteins and formyl peptide receptors. Critical Care Medicine. 2009;37:2000–2009. doi: 10.1097/CCM.0b013e3181a001ae. 19384205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Julian M.W., Shao G., Bao S., Knoell D.L., Papenfuss T.L., VanGundy Z.C., Crouser E.D. Mitochondrial transcription factor A serves as a danger signal by augmenting plasmacytoid dendritic cell responses to DNA. Journal of Immunology (Baltimore, MD: 1950) 2012;189:433–443. doi: 10.4049/jimmunol.1101375. 22675199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pinti M., Cevenini E., Nasi M., De Biasi S., Salvioli S., Monti D., Benatti S., Gibellini L., Cotichini R., Stazi M.A. Circulating mitochondrial DNA increases with age and is a familiar trait: implications for “inflamm-aging”. European Journal of Immunology. 2014;44:1552–1562. doi: 10.1002/eji.201343921. 24470107 [DOI] [PubMed] [Google Scholar]