

Abstract
Aberrant production of nitric oxide (NO) by inducible NO synthase (iNOS) has been implicated in the pathogenesis of endothelial dysfunction and vascular disease. Mechanisms responsible for the fine-tuning of iNOS activity in inflammation are still not fully understood. Zinc is an important structural element of NOS enzymes and is known to inhibit its catalytical activity. In this study we aimed to investigate the effects of zinc on iNOS activity and expression in endothelial cells. We found that zinc down-regulated the expression of iNOS (mRNA+protein) and decreased cytokine-mediated activation of the iNOS promoter. Zinc-mediated regulation of iNOS expression was due to inhibition of NF-κB transactivation activity, as determined by a decrease in both NF-κB-driven luciferase reporter activity and expression of NF-κB target genes, including cyclooxygenase 2 and IL-1β. However, zinc did not affect NF-κB translocation into the nucleus, as assessed by Western blot analysis of nuclear and cytoplasmic fractions. Taken together our results demonstrate that zinc limits iNOS-derived high output NO production in endothelial cells by inhibiting NF-κB-dependent iNOS expression, pointing to a role of zinc as a regulator of iNOS activity in inflammation.
Keywords: Endothelial cells, Inducible nitric oxide synthase, Zinc, NF-κB, Gene expression
Graphical abstract
Highlights
-
•
Zinc inhibits iNOS-dependent nitrite accumulation in endothelial cells.
-
•
Zinc decreases cytokine-induced iNOS expression in endothelial cells.
-
•
Zinc inhibits iNOS promoter activity.
-
•
NF-kB silencing abolishes cytokine-induced iNOS expression.
-
•
Zinc inhibits the transactivation activity of NF-κB.
Introduction
Endothelial dysfunction is characterized by a loss of endothelial control over vascular tone, thrombosis, and vessel wall remodeling [1]. Hallmarks of endothelial dysfunction is a decreased bioavailability of nitric oxide (NO) and increased inflammation [1]. In endothelial cells NO can be produced from l-arginine in a reaction catalyzed by the constitutive type 3 isoform of NO synthase or endothelial NOS (eNOS or NOS3; EC 1.14.13.39) [2]. Under pro-inflammatory conditions endothelial cells express also the inducible isoform of NOS (iNOS or NOS2) [2,3]. Although both NOS isoforms catalyze the same biochemical reaction, eNOS and iNOS are very different enzymes. The expression of eNOS is constitutive in endothelial cells, and produces low nanomolar levels of NO. The biochemical activity of eNOS is tightly regulated by its intracellular localization and by post-translational mechanisms, including Ca2+ signaling, and phosphorylation. Instead, iNOS is not expressed under resting non-inflammatory conditions in endothelial cells, but after its induction by pro-inflammatory stimuli iNOS produces low micromolar amounts of NO independently on Ca2+ fluctuation until it is degraded by proteolytic enzymes [4]. iNOS is known to exert important regulatory and protective effects on the vasculature [2–4]. However, aberrant iNOS-derived NO production has been shown to be involved in pathological conditions, e.g. the blood pressure fall in septic shock [5], as well as in the pathogenesis of chronic inflammatory diseases, including atherosclerosis [6,7]. Although in recent years both post-translational and post-transcriptional mechanisms regulating iNOS activity have been discovered [4,8], the regulation of iNOS expression remains the main regulatory step to control iNOS activity [4,5].
Accumulating evidence indicates that iNOS-derived NO is also involved in zinc homeostasis [9–11]. It has been hypothesized that zinc released by high output NO synthesis may induce endothelial resistance against oxidative stimuli and disease [12–14], as recently reviewed [15]. It was shown that both NO donors and iNOS-derived NO induce intracellular zinc release from zinc sulfur clusters, as found in proteins such as metallothionein [9,11–14,16–21]. We and others have demonstrated that iNOS-derived NO controls zinc trafficking in endothelial cells [14,16,18,20,22] and protects endothelial cells against H2O2-mediated damage via zinc-mediated increase of cellular glutathione de novo synthesis [12]. Interestingly, the same pathway was activated also by exogenously added zinc [13].
Some evidence of a possible regulatory role of zinc on iNOS activity exists. Zinc is an important structural element of iNOS, holding together the two subunits via tetrahedral coordination of four cysteines, two belonging to one monomer and the other two to the second protein monomer [23]. Thus, zinc displacement form the zinc–sulfur cluster of iNOS by NO can contribute to the control of iNOS activity [24]. Moreover, it is well known that transition metals, including copper and zinc, profoundly affect NOS enzyme activity at different levels. Stuehr and Griffith have reported in a review that in vitro iNOS activity in activated macrophages is more than 90% inhibited by 100 µM zinc [26]. In keratinocytes zinc supplementation was shown to decrease iNOS protein levels [27], but the molecular mechanisms responsible of zinc-mediated iNOS regulation were not further investigated.
In this study we analyzed the effects of zinc on iNOS activity in aortic endothelial cells. We could demonstrate that zinc limits high output NO production in endothelial cells by inhibiting NF-κB-dependent expression of iNOS. Therefore, zinc might participate to control iNOS signaling and contribute to the stabilization of the endothelium in inflammation by limiting NF-κB-mediated responses.
Materials and methods
Chemicals
Non-fat dry milk was purchased from BioRad (Munich, Germany), materials for western blot (NuPAGE LDS sample buffer, NuPAGE reducing agent, NuPAGE 10% Bis-Tris pre-cast gels) from Invitrogen GmbH (Karlsruhe, Germany), Ponceaus S from SERVA Electrophoresis GmbH (Heidelberg, Germany), Hybond P transfer membrane from Amersham Biosciences (Munich, Germany), mouse monoclonal anti-human β-actin, horseradish peroxidase (HRP)-conjugated goat anti-mouse antiserum and HRP-conjugated goat anti-rabbit antiserum from BD biosciences (Erembodegem, Belgium), pro-inflammatory cytokines (PeproTech INC, Rocky Hill, USA), and L-N5-(1-iminoethyl)-ornithine-dihydrochloride (L-NIO) from Alexis Biochemicals (Lörrach, Germany). Unless otherwise specified, chemicals were purchased from Sigma (Deisenhofen, Germany), cell culture plastics from Greiner (Frickenhausen, Germany) and cell culture material from PAA (Pashing, Austria).
Isolation and culture of rat aorta endothelial cells
Rat aorta endothelial cells were isolated by outgrowth from aortic rings, expressed the phenotype vWFhigh Ox43high eNOShigh and were cultured as described [13,28].
Culture of A549/8 iNOS cells
The human alveolar epithelium-like A549/8 cells stably transfected with a 16 kB fragment of the human iNOS promoter in front of a luciferase reporter gene and a neomycin resistance gene was a kind gift of Prof. Dr. H. Kleinert (Institute of Pharmacology, Johannes Gutenberg-University of Mainz) [29]. Cells were grown in RPMI 1640/10% FCS supplemented with 1 mg/ml Geneticin G418. Experiments were performed in the absence of G418.
Zinc determination
The zinc concentrations of cell culture media, sera and buffers were determined by flame atomic adsorption spectrometry as described previously [13]. We found that FBS contained 24.98±2.00 µM zinc and the complete medium 2.28±0.32 µM zinc. Increase in intracellular “free” zinc concentration after addition of zinc to culture media was measured by loading with Zinquin as described [13].
Measurement of iNOS derived NO production
Activity of recombinant bovine iNOS (155.5 U/ml) was determined at room temperature in a NOS reaction buffer at pH 7.4 containing 50 mM HEPES, 0.15 mM NADPH, 1 mM arginine, 1 mM magnesium acetate, 18 µM tetrahydrobiopterin, and 180 µM DTT in the presence or absence of zinc. The nitrite concentration was measured in aliquots of the reaction mixture collected at different time points by chemiluminescence by using triiodide reduction in CLD 60 (Echophyics GmbH, München, Germany), as described [30]. The results corresponding to 10 min incubations are shown. Nitrite accumulation in culture supernatants during 24 h of incubation with pro-inflammatory cytokines (as indicated in the figure legend) in the absence or presence of the NOS inhibitor L-NIO was determined using the diazotization reaction (Griess assay), as described previously [28]. The nitrite concentrations in the cell culture supernatant were normalized against the number of live cells as assessed by neutral red staining [13].
Treatments with pro-inflammatory cytokines and zinc
Endothelial cells (5×104 cells/well) were cultured in 12-well plates for 48 h and then incubated for 12 h with a mix of pro-inflammatory cytokines (1000 U/ml TNFα+400 U/ml IL-1β+200 U/ml INFγ) to induce the expression of the inducible nitric oxide synthase (iNOS) and high output NO synthesis. NO synthesis was inhibited by addition of the specific NOS-inhibitor L-NIO (500 µM). Zinc (50 mM stock in 0.9% NaCl) was diluted in culture medium to the indicated concentrations [12,13].
The effects of iNOS-derived NO±zinc on cell viability was assessed with three different methods. Cell integrity was assessed by neutral red staining, as described [12,13]. The amount of cells accumulating neutral red into their lysosomes (viable cells) is expressed as percentage of live cells vs. untreated cells. The effects of the treatments on mitochondrial dehydrogenase activities was measured by analyzing the reduction of a tetrazolium salt to formazan [31]. Sodium 3′-[1-(phenylaminocarbonyl)-3,4-tetrazolium]-bis(4-methoxy-6-nitro) benzene sulfonic acid hydrate (XTT; Roche Diagnostic GmbH, Mannheim, Germany) was used following the manufacturer’s instructions. Briefly, 300 µl of a solution containing XTT was added to the culture medium and incubated for 4 h at 37 °C, 5% CO2. Absorption was measured in triplicates at 450 nm in a FLUOstar OPTIMA (BMG Labtech, Offenburg, Germany). Cell proliferation was assessed as DNA synthesis by analyzing the incorporation of 5-bromo-2′-deoxyuridine with a commercial kit (BrdU Cell Proliferation ELISA, ZytoMed). Briefly, endothelial cells (104 cells/well) were cultured in 96-well plates for 24 h, and then treated in quadruplicates as indicated. Six hours after the beginning of the treatments 20 µl of a solution containing BrdU (provided in the kit) was added to the culture medium and incubated for 18 h. Cells were then fixed and stained with an anti-BrdU mouse monoclonal antibody (1:200) for 1 h at RT. The signal was detected using a horseradish peroxidase-conjugated goat anti-mouse antibody (1:2000) and tretra-methylbenzidine as a substrate. XTT was added to the culture medium and incubated for 4 h at 37 °C, 5% CO2. Absorption was measured at 450 nm in a FLUOstar OPTIMA.
RNA interference, RNA extraction and real time RT-PCR
Endothelial cells (1×105 cells/well) were grown in a 6-well plate for 48 h, treated for 24 h as indicated and then lysed with 350 µl RLT-lysis buffer (Qiagen, Hilden, Germany). For gene silencing experiments 105 cells were transfected with 3 nM silencing RNA (siRNA) and 4.5 µl of HiPerfect transfection reagent (Qiagen) following the manufacturer’s instruction (see also [29]). After 48 h, the culture medium was changed and the cells were treated for 24 h with pro-inflammatory cytokines at the concentrations indicated or left untreated. The siRNA sequence targeting RelA (p65) was purchased from Qiagen (Rn-RelA-2-HPsiRNA). Silencing was considered successful when RelA (p65) mRNA was 2-fold decreased as compared to cells treated with the transfection reagent only (mock control). The silencing sequence with the maximal efficiency has been selected by comparing silencing efficiency, i.e. a decrease in mRNA expression of Rel A after transfection as compared to the mock control (cells treated with transfection reagent only). Other sequences tested and not selected were Rn-RelA-1-HPsiRNA, Rn-RG2:727889_2_HPsiRNA, and Rn-RG2:727889_3_HPsiRNA. As a control for unspecific effects, a non-silencing (NS) siRNA sequence was used (Qiagen). The positive control was an siRNA sequence directed against rat Gapdh mRNA (Dharmacon), which has been used also for determining the best conditions for transfection. The silencing efficiency was verified in each experiment by measuring the expression of Gapdh and RelA (p65) mRNA. RNA was isolated by using RNeasy mini kit (Qiagen) following the manufacturer’s instruction. The RNA (0.5 µg) was then reversely transcribed in MyCycler Personal Thermal Cycler (Bio-Rad Laboratories GmbH, Munich, Germany) using a QuantiTect Reverse Transcription Kit (Qiagen) following the manufacturer’s instructions. The cDNA (5 ng) or control RNA was used as a template for real time PCR performed in triplicate, using SYBR GreenER™ qPCR SuperMix for ABI PRISM® (Invitrogen, Karlsruhe, Germany), or TaqMan Universal PCR Master Mix in ABI PRISM 7900 (Applied Biosystems) and 18 sr RNA as a housekeeping gene. Primers were purchased from Applied Biosystems: transporter 1 (Znt1) (NM_022853, assay Rn00575737-m1), and 18S rRNA (Eukaryotic 18S rRNA, endogenous control VIC/TAMRA primer limited); or from Qiagen: RelA (p65) (QT01580012), inducible NO synthase, nitric oxide type 2 (Nos2) (QT00068740), interleukin 1beta (il1b) QT00181657 cyclooxygenase 2 (Cox2, official name: prostaglandin-endoperoxide synthase 2, Ptgs2, QT00192934), and 18SrRNA (Hs-RRN18S-1-SG). Primer specificity was verified by melting curve analysis. Results were analyzed as described [13].
Preparation of total and nuclear protein extracts, and western blot analysis
Total protein lysate was prepared as previously described [12,13] from endothelial cells grown in a 6-well plate and treated for 24 h with pro-inflammatory cytokines±zinc, as indicated in the figure legends. Nuclear protein extracts were obtained from endothelial cells grown in 100 mm dishes (107 cells/plate) for 48 h and then treated for 30 min with pro-inflammatory cytokines±zinc as indicated. Nuclei were extracted with Nuclear extract kit (Active Motif, Carlsbad, USA) following the manufacturer’s protocol. Protein concentration was determined by the Lowry assay (DC Protein Assay, Bio-Rad). Total cell lysates (30 µg) and nuclear cells extracts (10 µg) were denatured and were loaded together with protein markers (Magic Mark, Invitrogen) on a 10% NuPAGE Bis-Tris pre-cast gel following the manufacture’s protocols. The proteins were transferred on PVDF membrane Hybond P (Amersham Biosciences, GE Healthcare, Freiburg, Germany). Staining was performed as described [13] with mouse anti-NOS2 or mouse anti RelA (p65) antibodies (BD Biosciences, Heidelberg, Germany) diluted 1:1000 or anti-human-actin (1:5000) and then incubated with HRP-conjugated goat anti-mouse antibody diluted 1:5000 in blocking buffer. The bands were visualized by autoradiography on Hyperfilm ECL (Amersham Biosciences) using SuperSignal west Pico Chemiluminescent Substrate (Pierce, Thermo Lifescience, Bonn, Germany).
Determination NF-κB-dependent gene expression activation and iNOS promoter activity
To measure the effects of zinc on cytokine-dependent κB responsive element activation in endothelial cells, endothelial cells were transfected with a reporter vector pGL4.32[luc2P/NF-κB-RE/Hygro] with 5 NF-κB responsive elements controlling the expression of a luciferase (Promega, Mannheim, Germany). Endothelial cells (105 cells/well) were seeded on to a 6-well plate and grown for 48 h, and then transfected with 2 µg plasmid, 0.2 µg Renilla luciferase expression vector (pRL3-SV40), and 8 µl lipofectin (Invitrogen). 24 h after transfection cells were treated with pro-inflammatory cytokines±zinc for 6 h and then lysed in 150 µl of a lysis buffer (reporter gene assay lysis buffer, Roche Diagnistic GmbH), centrifuged at 13,000g for 2 min at 4 °C in Universal 30RF (Hettich Zentrifugen), quick frozen in liquid N2, and stored at −80 °C until luciferase assays was performed.
To specifically determine the effects of zinc on cytokines-dependent transactivation of the iNOS promoter, A549/8 iNOS cells were treated for 6 h as indicated and then lysed as described above.
Luciferase assay was carried out in a luciferase assay buffer (30 mM tricine, 0.1 mM EDTA, 15 mM MgSO4, 10 mM DTT) supplemented with 0.27 mM coenzyme-A (Applichem, Darmstadt, Germany), 0.53 mM ATP and with 0.5 mM of the substrate d-luciferine (Applichem). Cell lysate (20 µl) was assayed in double for luminescence using a microtiter plate reader with automatic injection (FLUOstar OPTIMA, BMG). The light units (LU) of the luciferase assay were normalized by protein content and were calculated as relative luciferase activity given in percent as compared to the controls (=100%).
Statistical analysis
Real-time PCR data were processed as described [13]. Data are reported as mean±SEM. For statistical analysis, we used ANOVA followed by an appropriate post hoc multiple comparison test (Tukey or Student’s T test).
Results
Zinc reverses iNOS activity-dependent inhibition of endothelial cell proliferation
We aimed to study whether zinc might affect iNOS activity in endothelial cells. The expression of iNOS was induced by treating primary rat aorta endothelial cells with a mixture of pro-inflammatory cytokines (IL-1β+INFγ+TNFα), which leads to a significant accumulation of nitrite in the supernatant found after 6–8 h of incubation (Supplementary Fig. S1). Accumulation of nitrite was fully inhibited by treatment with 500 µM of the specific NOS inhibitor L-NIO (Supplementary Fig. S1).
Fig. 1.
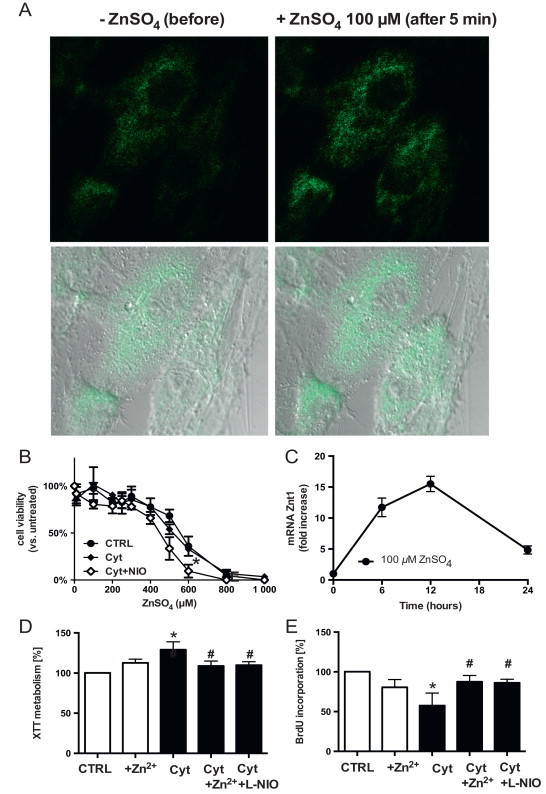
Non-toxic zinc concentrations increases intracellular free zinc, induces the expression of Znt1 and rescues endothelial cells from cytokines-dependent inhibition of cell proliferation. (A) Increases in intracellular “free” zinc concentration 5 min after addition of 100 µm zinc (right panels) as compared to the untreated control (left panel) as assessed by loading cells with zinquin. (B) Toxicity of zinc treatment in the presence or absence of pro-inflammatory cytokines (Cyt=IL-1β, TNF-alpha, IFN-gamma), or of the NOS inhibitor L-NIO, as assessed by neutral red staining. (C) Kinetics of zinc-mediated increases in Znt1 mRNA levels. (D) Cytokines increase metabolic activity of the cells as measured by conversion of XTT in the supernatant of the cells after 4 h of incubation. Both zinc and NOS inhibition abolish this effect. (E) Treatment with pro-inflammatory cytokines decreases cell proliferation, as assessed by a decrease in BrdU accumulation within 24 h. Addition of zinc or inhibition of NOS activity by L-NIO abolish the effect of the cytokines.
To increase intracellular “free” zinc concentrations, a zinc solution obtained by dissolving ZnSO4 in NaCl 0.9% was added directly to the culture medium (containing 2.28±0.32 µM zinc). This treatment induced a concentration-dependent increase in the intracellular fluorescence intensity of the zinc-specific fluorophore Zinquin, which could be observed immediately (<30 s) after addition of zinc to the medium [13]. Fig. 1A shows the increases of Zinquin fluorescence before and 5 min after adding 100 µM zinc. After the addition of 100 µM ZnSO4, intracellular Zinquin fluorescence increased approximately 3 folds. As a result of an increase in “free” bioavailable zinc, the expression of zinc transporter 1 (ZnT-1) (Fig. 1C) and metallothionein (MT1a) [13] is induced in the endothelial cells.
The toxicity of the treatment was tested by treating endothelial cells with different concentrations of ZnSO4 in the absence or in the presence of cytokines, or the NOS-inhibitor L-NIO (Fig. 1B). Under the experimental conditions chosen the toxicity of 24 h incubation with zinc in cytokine-treated cells was not different from untreated cells (half maximal lethal dose (LD50)=547±85 µM with zinc vs. LD50=579±91 µM without zinc). To test the effects of zinc supplementation on metabolic activity and proliferation of cytokine-treated cells, we measured the mitochondrial dehydrogenase catalyzed reduction [31] of XTT into formazan (Fig. 1D) and the incorporation of BrdU into the DNA (Fig. 1E), respectively. We found that cytokines increased metabolic activity of the cells by about 20% (Fig. 1D, Cyt), but decreased cell proliferation by about 40% (Fig. 1E, Cyt) as compared to untreated cells (Fig. 1D, E, CTRL). These effects were inhibited by adding the specific NOS inhibitor L-NIO (500 µM; Fig. 1D, E, Cyt+L-NIO), indicating that NOS-derived NO production is responsible for the cytokine-induced effects. Addition of non-toxic zinc concentrations to cytokine treated cells exerted the same effects as NOS inhibition on the effects induced by cytokines on cell proliferation and mitochondrial dehydrogenase activity, respectively (Fig. 1D, E, 100 µM ZnSO4).
Zinc inhibits iNOS activity and decreases cytokine-induced iNOS expression in endothelial cells
Next we aimed to analyze whether zinc might affect iNOS-dependent NO production in endothelial cells. We found that concentrations >50 µM zinc affect the catalytic activity of recombinant iNOS in vitro, as tested by measuring the accumulation of nitrite in the reaction buffer by chemiluminescence analysis (Fig. 2A). In endothelial cells, addition of zinc decreased nitrite accumulation in the supernatant of cytokine-treated endothelial cells as measured after 24 h of incubation (Fig. 2B). We found that zinc added together with cytokines shown in Fig. 2B, or 6 h after the treatment with cytokines (not shown) strongly decreased iNOS expression as shown by Western blotting (Fig. 2C).
Fig. 2.
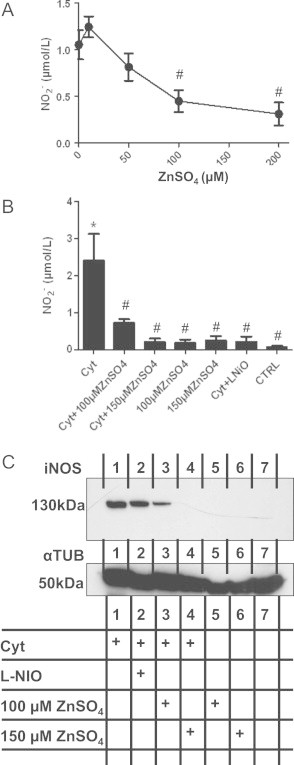
Zinc inhibits iNOS expression and activity in endothelial cells. (A) Inhibition of recombinant iNOS activity as assessed by measuring the accumulation of nitrite into the reaction buffer after 10 min of incubation with the indicated concentrations of zinc by reductive chemiluminescence. #T-test p<0.05 vs. control without zinc. (B) Inhibition of cytokine-induced accumulation of nitrite into the supernatant of endothelial cells by zinc as assessed after 24 h of incubation. ANOVA p<0.001; ⁎p<0.05 vs. CTRL (untreated); #p<0.05 vs. Cyt-treated cells. (C) Inhibition of cytokine-induced iNOS protein expression by zinc as assessed by western blot analysis. Staining of α-tubulin was used as a loading control.
Zinc inhibits iNOS expression by affecting NF-κB-dependent gene transcription
To verify whether these effects were mainly dependent on zinc-mediated inhibition of iNOS transcription, iNos (Nos2) mRNA levels were measured by real-time RT-PCR. Cytokines increased the levels of Nos2 mRNA already after 2–4 h of incubation (not shown), reaching the maximum after 6–8 h (Fig. 3A shows the results after 6 hours of incubation). In the presence of zinc, Nos2 mRNA levels strongly decreased. Similarly, the expression of two NF-κB-dependent inflammatory genes Cox2 (Fig. 3B) and IL-1β (Fig. 3C) was strongly affected by zinc, as assessed after 6 h of incubation. Interestingly, inhibition of iNOS during cytokines exposure results in about the same effect on mRNA for iNOS, COX2, IL-1β as zinc exposure.
Fig 3.
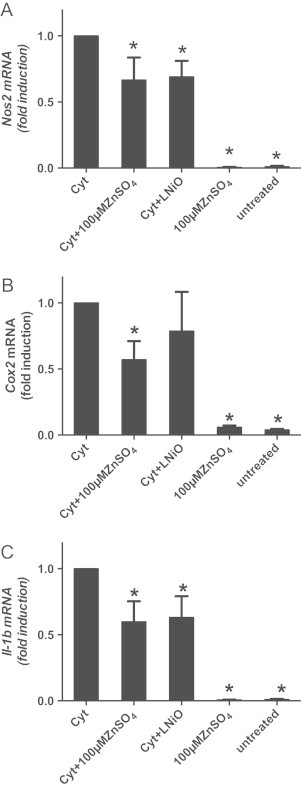
Zinc inhibits cytokine-induced expression of pro-inflammatory genes. Zinc supplementation of cytokine-treated cells for 24 h decreased the mRNA levels of (A) Nos2, (B) IL-1β, (C) Cox2 mRNA as assessed by real time RT-PCR. ⁎p<0.05 vs. Cyt-treated cells.
Next, we studied whether NF-κB was involved in zinc-mediated inhibition of iNOS expression. We knocked down the expression of the NF-κB (p50/p65) by transfecting endothelial cells with an siRNA directed against p65, which decreased mRNA levels of p65 to a minimum of 90%. Cytokine-induced iNOS expression in the absence of NF-κB was decreased by almost 99% (Fig. 4A), confirming that NF-κB is the dominant transcription factor involved in cytokine-induced iNOS expression in our cells. To analyze whether zinc affects NF-κB translocation into the nucleus, the presence of p65 in the cytoplasmic as well as the in the nuclear fraction of endothelial cells was assessed by western blot analysis (Fig. 4B). Treatment with zinc did not affect the cytokine-induced translocation of p65 into the nucleus. In fact, the amount of p65 found in the nucleus of cytokine-treated cells after a 1 h-incubation with cytokines+zinc is not different as found in cells treated with cytokines only (Fig. 4B).
Fig 4.
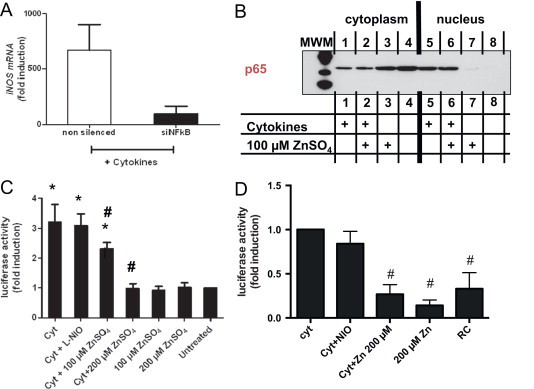
Zinc affects NF-κB-dependent activation of the iNOS promoter without affecting NF-κB translocation into the nucleus. (A) NF-κB is the dominant transcription factor regulating cytokine-induced expression of iNOS under the experimental conditions used as assessed by knocking down the expression of NF-κB via RNA interference. T-test p<0.05 (B) zinc does not affect cytokine-induced translocation of NF-κB into the cell nucleus as assessed 30 min after addition of cytokines (representative of 3 independent experiments). (C) Zinc decreases cytokines induced NF-κB activation, as assessed 6 h after addition of cytokines by using an NF-κB luciferase reporter construct transfected in endothelial cells. ANOVA p<0.0001; ⁎p<0.05 vs. untreated; #p<0.05 vs. Cyt-treated cells. (D) Zinc inhibits cytokine-induced iNOS promoter activity as assessed 6 h after addition of cytokines in A549-iNOS reporter cells. 1-Way ANOVA p=0.0135, #T-test p<0.05 vs. Cyt-treated cells.
We further found that treatment with zinc decreased cytokine-induced transcriptional activation of an NF-κB-reporter plasmid, which was transiently transfected in the endothelial cells (Fig. 4C). To further confirm that zinc inhibits transactivation of iNOS promoter, we repeated these experiments by using a reporter cell line transfected with 1.3 kB of a human iNOS promoter in front of a luciferase reporter gene. We found that zinc decreased the cytokine-induced luciferase activity also in this cell line (Fig. 4D). Zinc itself did not affect luciferase basal activity or the viability of A549 cells as tested by neutral red assay (not shown).
Taken together these data show that zinc inhibits iNOS expression and activity via inhibition NF-κB-dependent transactivation of the iNOS promoter.
Discussion
In this study we found that an increase in intracellular “free” zinc concentration by addition of exogenous zinc results in a decreased NF-κB-dependent activation of iNOS expression in endothelial cells, thereby limiting high output NO production and cell activation, while promoting cell proliferation under pro-inflammatory conditions. Thus we found that 1. addition of non-toxic zinc concentrations to cultured primary endothelial cells increased intracellular “free” zinc concentrations as demonstrated by increased Zinquin fluorescence, and increases in mRNA levels of the zinc responsive gene Znt1, 2. similar to NOS inhibition, zinc reverses cytokine-dependent inhibition of endothelial cell proliferation, 3. zinc decreases cytokines-induced iNOS expression and high out NO synthesis, and 4. zinc decreases NF-κB-mediated transactivation of iNOS promoter. Taken together these results show that zinc limits iNOS-derived high-output NO production in endothelial cells by inhibiting NF-κB-mediated transactivation of iNOS promoter, pointing to a role of zinc as a regulator of iNOS activation in inflammation in endothelial cells.
Zinc-mediated inhibition of iNOS activity
Although in recent years both post-transcriptional as well as post-translational mechanisms regulating iNOS activity have been discovered [4], the regulation of iNOS expression remains the main regulatory step to control iNOS activity. Usually iNOS synthesizes NO continuously until the enzyme becomes degraded. We here found that zinc strongly decreases cytokine-induced iNOS expression (mRNA+protein) as well as NOS-dependent nitrite accumulation in the culture medium of the endothelial cells. Similar results were previously obtained with mouse keratinocytes [27], but the molecular mechanisms involved were not further investigated. As shown here, zinc directly inhibits iNOS catalytic activity in a concentration-dependent fashion (EC50≈75 µM), but would require local intracellular zinc concentrations of >50 µM. The inhibitory effects exerted by zinc on NOS catalytic activity are well known [25,26], and other divalent transition metals like copper exert similar effects at micromolar concentrations. Total cellular zinc concentrations are in the range of a few hundred micromolar, and it is distributed within sub-cellular compartments. However, the affinity of zinc for cytosolic zinc proteins is in the picomolar range, suggesting that the availability of free zinc ions within the cytoplasm is quite low [11]. In lysosomes and zincosomes the concentration of zinc may reach millimolar concentrations. Since iNOS is mainly localized within the cytoplasmic compartment of the cell, we may exclude a direct effect of zinc ions on the enzyme itself. Therefore, we conclude that the effects of zinc supplementation on iNOS activity within the endothelial cells are mainly dependent on the modulation of iNOS expression.
By using a reporter cell line stably transfected with the human iNOS promoter, we found that zinc supplementation directly inhibits the cytokine-mediated activation of the iNOS promoter. The sequence of human, rat and mouse iNOS promoters show a high grade of homology. The transcription factor NF-κB seems to be a central target for activators or inhibitors of iNOS expression in all three species. Pro-inflammatory stimuli including LPS, IL-1β, TNFα, in combination with INFγ, have been shown to induce iNOS expression in different cell types via activation of NF-κB [4]. By silencing the expression of p65, we found a 99% reduction of cytokine-induced up-regulation of iNOS, confirming that in our system NF-κB plays a central role in cytokine-mediated induction of iNOS expression. The rat promoter contains an upstream (−965 to −956 bp) and downstream (−107 to −98) NF-κB site, both of which are important for iNOS induction. In the rat promoter a third NF-κB site located at −901 to −892 bp with an opposite orientation, also defined as “the reverse NF-κB site”, is important for IL-1β-/INFγ-induced promoter activity [32].
We here show that zinc-mediated down regulation of iNOS expression is due to inhibition of NF-κB transactivation activity, and not NF-κB activation/translocation into the nucleus. The active transcription factor, which typically is the p50/p65 heterodimer [33], is released from an inhibitory cytosolic complex and translocates into the nucleus to bind to κB-responsive elements upstream of its target genes. Inhibition of NF-κB can occur by direct capture of NF-κB via protein–protein interactions [34], inhibition of NF-κB phosphorylation [35], inhibition of nuclear NF-κB-translocation [36], inhibition of NF-κB transactivation activity [37], or by increasing the expression of i-κB, the specific inhibitor of NF-κB [38].
We found that zinc inhibited NF-κB transactivation activity in cytokine-treated endothelial cells, as assessed by transient transfection of a κB-luciferase construct. While inhibition of the NF-κB signaling by zinc was shown in different tissues and cells, the mechanisms involved are cell/tissue specific and unknown for many cell types [39,40]. Zinc was shown to inhibit NF-κB activation/translocation in mononuclear cells [35,41], and other cell types [35,42,43] and to reduce the levels of activated NF-κB in diabetic CD1 mice [44], Severe zinc deficiency was shown to inhibit the translocation and transactivation capacity of NF-κB in neuronal cells and rat testes [45,46]. Zinc chelation has been shown to increase LPS-induced iNOS expression in RAW 264.7 cells [47]. In endothelial cells it was shown that zinc affects the expression of pro-inflammatory cytokines [48,49], while zinc deficiency exerts a permissive effect on the expression of inflammatory genes and exacerbates chronic inflammation in endothelial cells [48,49]. However, to the best of our knowledge this is the first time that zinc was shown to regulate iNOS expression via inhibition of NF-κB transactivation in primary endothelial cells. Future studies should be undertaken to investigate the molecular mechanisms responsible for zinc-mediated inhibition of transactivation activity of NF-κB into the nucleus.
Zinc and cell signaling: in vitro evidence and limitations
Because zinc ion fluctuations occur at such low concentrations and zinc interacts strongly with proteins, released zinc ions are now considered as potent intracellular signals [9,11]. Picomolar to low nanomolar concentrations of zinc ions inhibit enzymes involved in energy metabolism, signaling and mitochondrial respiration [50–54]. Here we found that zinc ion fluctuation in cytokine-treated cells induced by administration of non-toxic zinc concentrations, reduced the activation of the cells [55] and rescued their proliferation capability, leading to inhibition of iNOS expression and activity. Changes in intracellular free zinc concentrations and zinc ion fluctuations were induced here by exogenous administration of zinc ions to the culture medium, as demonstrated by analyzing increases in intracellular Zinquin fluorescence, and in the expression of zinc-responsive genes, including metallothionein-1 (MT-1) and zinc transporter-1 (ZnT1) [13,16]. Metallothioneins and zinc transporters contribute to rapidly “buffer” high intracellular zinc concentrations either by binding zinc (e.g. MT-1), or by transporting zinc outside of the cell (e.g. ZnT1) or into sub-cellular compartments, like lysosomes and zincosomes (e.g. via ZnT2 and/or other zinc transporters) [56], which also explains why cells are resistant to micromolar levels of zinc.
Should we consider the changes of intracellular “free” zinc, which are induced by adding micromolar concentrations of zinc to the culture media, physiological or superphysiological? Are the responses to these zinc fluctuations relevant for in vivo biology and/or pharmacology of zinc? The concentrations of zinc needed to increase the intracellular zinc concentrations in our endothelial culture system [12,13] are 5–20 fold higher as the zinc concentrations found in serum of healthy persons (about 12–15 µmol/l; 78–98 mg/dl) [57]. The effects of zinc supplementation to cultured cells are strongly dependent on the cell type under consideration, but also on the composition of the cell culture media, which may contain micromolar levels of zinc and millimolar concentrations of zinc-complexing molecules/chelators [58]. In our medium, we measured a total zinc concentrations of 2.28±0.32 µM, as assessed in 3 different media preparations. As pointed out by Bozym et al. [58], in media containing 5–10% v/v serum, albumin is present at a concentration of 0.3 mmol/l, and therefore will bind a significant portion of the available zinc with relatively high affinity (KD=0.1 µmol/l). The “free” zinc concentration is typically two to three orders of magnitude below the total added zinc, such that adding ~100 µmol/l total zinc typically resulted in ~100 nmol/l free zinc (pZn=7) being present, as assessed by different fluorimetric methods [58]. This means that in serum containing media the zinc levels are functionally buffered [58]. This is very similar to the in vivo situation were zinc is transported in the serum bound principally to albumin (70%), α-2-macroglobulin (18%), and other proteins such as transferrin or ceruloplasmin. A very small amount (i.e., 0.01%) is complexed with amino acids, especially histidine and cysteine [57,58]. As also pointed out by Bozym et al., care is particularly necessary if cells are cultured in serum-free media, since zinc buffering ligands may be inadequate. The same could be said if zinc chelators – like pyrithione – is used to transport zinc inside the cells. Both zinc-depleted media or chelators decrease the effective dosis of zinc used for experiments, but may also interfere with intracellular zinc equilibrium and with zinc signaling. The equilibrium between buffering and fluctuation of zinc ions will determine the borderline between physiology and pathophysiology [11].
Although we carefully characterized our model for: 1. concentration of zinc in media and sera; 2. increases in intracellular “free” zinc concentrations; 3 bioactivity of zinc, as shown by increases in the expression of the zinc responsive gene Znt1; 3. lack of overall toxicity, metabolic activity or proliferation, we cannot provide direct evidence that the effects exerted by zinc in vitro in this and other similar studies are also relevant for in vivo biology. This is one of the main limitations of cell culture models. The role of zinc in iNOS regulation in the endothelium should be addressed in future study in animal models, and in humans.
Are iNOS and zinc signaling pathways interdependent?
We and others have shown that iNOS-derived NO can nitrosate metallothionein [59] and thereby induce zinc release. Zinc released by NO may exert signaling functions leading to both cytoprotection as shown in endothelial cells by us [12,14] or in the lung [17,19], as well as to neurotoxicity in the brain [60], depending on the cellular compartment. By considering zinc-mediated inhibition of iNOS, it is tempting to speculate that zinc released by iNOS-derived NO may be part of the termination signals needed for the control of acute inflammatory reactions. Thus, under pro-inflammatory conditions, NF-κB activation leads to endothelial cell activation corresponding to the expression of inflammatory genes, including pro-inflammatory cytokines and iNOS, and contribute to the propagation of the inflammatory response. In activated endothelial cells expression of iNOS is induced and long-term high-output NO synthesis follows. iNOS-derived NO may then induce zinc release from zinc–sulfur clusters of intracellular proteins, and NF-κB might be a target of NO-mediated zinc redistribution [40,61]. If this would be the case, the NO-mediated intracellular zinc release may contribute to limit an excessive propagation of the inflammatory response by inhibiting or limiting endothelial cell activation. At present, this feedback loop connecting iNOS-mediated zinc release, and zinc mediated inhibition of iNOS expression should be considered a speculative observation, which should be addressed experimentally in future.
Summary and conclusions
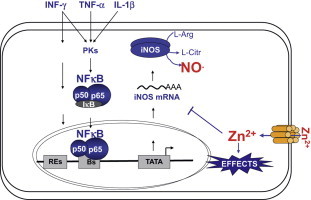
To summarize, we here have shown that zinc ions control iNOS expression and activity in endothelial cells via inhibition of NF-κB transactivation activity (Fig. 5). Interestingly, it has been shown that not only zinc supplementation inhibits NF-κB activation, but also that zinc deficiency exerts a permissive effect on the expression of inflammatory genes and exacerbates chronic inflammation [48,49]. Therefore, zinc signals and the maintenance of an adequate zinc status in the endothelium may be an important clinical strategy to prevent the development of endothelial dysfunction and cardiovascular diseases.
Fig. 5.
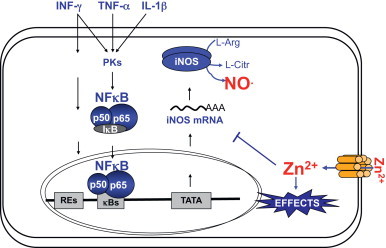
Potential feedback regulation of iNOS expression by zinc in endothelial cells. Pro-inflammatory cytokines induce iNOS expression. iNOS-derived NO induces zinc release from zinc clusters of intracellular proteins. We here show that zinc decreases NF-κB mediated iNOS expression.
Acknowledgements
We are grateful to the Susanne-Bunnenberg-Stiftung of the Düsseldorf Heart Center (to Prof. Malte Kelm, Department of Cardiology, Faculty of Medicine), to the Forschungskommission of the Medical Faculty of the University of Düsseldorf (to CVS and MCK) for financial support. We thank Marja Lenzen, Sophie Lecoturier and Tanja Wollersheim for expert technical assistance.
Appendix A. Supplementary material
Time-dependent increase in NOS-dependent nitrite accumulation in cytokine-treated cells as assessed by the Griess assay. ANOVA p<0.001. * p<0.05 vs. untreated.
References
- 1.Vita J.A. Endothelial function. Circulation. 2011;124:e906–e912. doi: 10.1161/CIRCULATIONAHA.111.078824. 22184047 [DOI] [PubMed] [Google Scholar]
- 2.Moncada S., Palmer R.M.J., Higgs E.A. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacological Reviews. 1991;43:109–142. 1852778 [PubMed] [Google Scholar]
- 3.Nathan C. Points of control in inflammation. Nature. 2002;420:846–852. doi: 10.1038/nature01320. 12490957 [DOI] [PubMed] [Google Scholar]
- 4.Kleinert H., Schwarz P.M., Förstermann U. Regulation of the expression of inducible nitric oxide synthase. Biological Chemistry. 2003;384:1343–1364. doi: 10.1515/BC.2003.152. 14669979 [DOI] [PubMed] [Google Scholar]
- 5.Förstermann U., Sessa W.C. Nitric oxide synthases: regulation and function. European Heart Journal. 2012;33:829–837. doi: 10.1093/eurheartj/ehr304. 21890489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Villanueva C., Giulivi C. Subcellular and cellular locations of nitric oxide synthase isoforms as determinants of health and disease. Free Radical Biology & Medicine. 2010;49:307–316. doi: 10.1016/j.freeradbiomed.2010.04.004. 20388537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hill B.G., Dranka B.P., Bailey S.M., Lancaster J.R., Jr., Darley-Usmar V.M. What part of no don’t you understand? Some answers to the cardinal questions in nitric oxide biology. Journal of Biological Chemistry. 2010;285:19699–19704. doi: 10.1074/jbc.R110.101618. 20410298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee M., Choy J.C. Positive feedback regulation of human inducible nitric-oxide synthase expression by Ras protein S-nitrosylation. Journal of Biological Chemistry. 2013;288:15677–15686. doi: 10.1074/jbc.M113.475319. 23599434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krezel A., Hao Q., Maret W. The zinc/thiolate redox biochemistry of metallothionein and the control of zinc ion fluctuations in cell signaling. Archives of Biochemistry and Biophysics. 2007;463:188–200. doi: 10.1016/j.abb.2007.02.017. 17391643 [DOI] [PubMed] [Google Scholar]
- 10.Kröncke K.D. Cellular stress and intracellular zinc dyshomeostasis. Archives of Biochemistry and Biophysics. 2007;463:183–187. doi: 10.1016/j.abb.2007.03.008. 17442256 [DOI] [PubMed] [Google Scholar]
- 11.Maret W. Molecular aspects of human cellular zinc homeostasis: redox control of zinc potentials and zinc signals. Biometals: An International Journal on the Role of Metal Ions in Biology, Biochemistry, and Medicine. 2009;22:149–157. doi: 10.1007/s10534-008-9186-z. 19130267 [DOI] [PubMed] [Google Scholar]
- 12.Cortese-Krott M.M., Suschek C.V., Wetzel W., Kröncke K.D., Kolb-Bachofen V. Nitric oxide-mediated protection of endothelial cells from hydrogen peroxide is mediated by intracellular zinc and glutathione. American Journal of Physiology – Cell Physiology. 2009;296:C811–C820. doi: 10.1152/ajpcell.00643.2008. 19193864 [DOI] [PubMed] [Google Scholar]
- 13.Cortese M.M., Suschek C.V., Wetzel W., Kröncke K.D., Kolb-Bachofen V. Zinc protects endothelial cells from hydrogen peroxide via Nrf2-dependent stimulation of glutathione biosynthesis. Free Radical Biology & Medicine. 2008;44:2002–2012. doi: 10.1016/j.freeradbiomed.2008.02.013. 18355458 [DOI] [PubMed] [Google Scholar]
- 14.Spahl D.U., Berendji-Grün D., Suschek C.V., Kolb-Bachofen V., Kröncke K.D. Regulation of zinc homeostasis by inducible NO synthase-derived NO: nuclear metallothionein translocation and intranuclear Zn2+ release. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:13952–13957. doi: 10.1073/pnas.2335190100. 14617770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oteiza P.I. Zinc and the modulation of redox homeostasis. Free Radical Biology & Medicine. 2012;53:1748–1759. doi: 10.1016/j.freeradbiomed.2012.08.568. 22960578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Berendji D., Kolb-Bachofen V., Meyer K.L., Grapenthin O., Weber H., Wahn V., Kröncke K.D. Nitric oxide mediates intracytoplasmic and intranuclear zinc release. FEBS Letters. 1997;405:37–41. doi: 10.1016/s0014-5793(97)00150-6. 9094420 [DOI] [PubMed] [Google Scholar]
- 17.Bernal P.J., Leelavanichkul K., Bauer E., Cao R., Wilson A., Wasserloos K.J., Watkins S.C., Pitt B.R., St Croix C.M. Nitric-oxide-mediated zinc release contributes to hypoxic regulation of pulmonary vascular tone. Circulation Research. 2008;102:1575–1583. doi: 10.1161/CIRCRESAHA.108.171264. 18483408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pearce L.L., Wasserloos K., St Croix C.M., Gandley R., Levitan E.S., Pitt B.R. Metallothionein, nitric oxide and zinc homeostasis in vascular endothelial cells. Journal of Nutrition. 2000;130:1467S–1470S. doi: 10.1093/jn/130.5.1467S. 10801961 [DOI] [PubMed] [Google Scholar]
- 19.Li H., Cao R., Wasserloos K.J., Bernal P., Liu Z.Q., Pitt B.R., St Croix C.M. Nitric oxide and zinc homeostasis in pulmonary endothelium. Annals of the New York Academy of Sciences. 2010;1203:73–78. doi: 10.1111/j.1749-6632.2010.05558.x. 20716286 [DOI] [PubMed] [Google Scholar]
- 20.St Croix C.M., Stitt M.S., Leelavanichkul K., Wasserloos K.J., Pitt B.R., Watkins S.C. Nitric oxide-induced modification of protein thiolate clusters as determined by spectral fluorescence resonance energy transfer in live endothelial cells. Free Radical Biology & Medicine. 2004;37:785–792. doi: 10.1016/j.freeradbiomed.2004.06.004. 15304254 [DOI] [PubMed] [Google Scholar]
- 21.St Croix C.M., Wasserloos K.J., Dineley K.E., Reynolds I.J., Levitan E.S., Pitt B.R. Nitric oxide-induced changes in intracellular zinc homeostasis are mediated by metallothionein/thionein. American Journal of Physiology – Lung Cellular and Molecular Physiology. 2002;282:L185–L192. doi: 10.1152/ajplung.00267.2001. 11792622 [DOI] [PubMed] [Google Scholar]
- 22.Stitt M.S., Wasserloos K.J., Tang X., Liu X., Pitt B.R., St Croix C.M. Nitric oxide-induced nuclear translocation of the metal responsive transcription factor, MTF-1 is mediated by zinc release from metallothionein. Vascular Pharmacology. 2006;44:149–155. doi: 10.1016/j.vph.2005.10.004. 16423564 [DOI] [PubMed] [Google Scholar]
- 23.Li H., Raman C.S., Glaser C.B., Blasko E., Young T.A., Parkinson J.F., Whitlow M., Poulos T.L. Crystal structures of zinc-free and -bound heme domain of human inducible nitric-oxide synthase. Implications for dimer stability and comparison with endothelial nitric-oxide synthase. Journal of Biological Chemistry. 1999;274:21276–21284. doi: 10.1074/jbc.274.30.21276. 10409685 [DOI] [PubMed] [Google Scholar]
- 24.Mitchell D.A., Erwin P.A., Michel T., Marletta M.A. S-nitrosation and regulation of inducible nitric oxide synthase. Biochemistry. 2005;44:4636–4647. doi: 10.1021/bi0474463. 15779890 [DOI] [PubMed] [Google Scholar]
- 25.Perry J.M., Marletta M.A. Effects of transition metals on nitric oxide synthase catalysis. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:11101–11106. doi: 10.1073/pnas.95.19.11101. 9736696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stuehr D.J., Griffith O.W. Mammalian nitric oxide synthases. Advances in Enzymology and Related Areas of Molecular Biology. 1992;65:287–346. doi: 10.1002/9780470123119.ch8. 1373932 [DOI] [PubMed] [Google Scholar]
- 27.Yamaoka J., Kume T., Akaike A., Miyachi Y. Suppressive effect of zinc ion on iNOS expression induced by interferon-γ or tumor necrosis factor-α in murine keratinocytes. Journal of Dermatological Science. 2000;23:27–35. doi: 10.1016/s0923-1811(99)00062-6. 10699762 [DOI] [PubMed] [Google Scholar]
- 28.Suschek C., Rothe H., Fehsel K., Enczmann J., Kolb-Bachofen V. Induction of a macrophage-like nitric oxide synthase in cultured rat aortic endothelial cells. IL-1beta-mediated induction regulated by tumor necrosis factor-alpha and IFN-gamma. Journal of Immunology (Baltimore, MD: 1950) 1993;151:3283–3291. 7690801 [PubMed] [Google Scholar]
- 29.Kleinert H., Euchenhofer C., Ihrig-Biedert I., Förstermann U. Glucocorticoids inhibit the induction of nitric oxide synthase II by down-regulating cytokine-induced activity of transcription factor nuclear factor-kappa B. Molecular Pharmacology. 1996;49:15–21. 8569701 [PubMed] [Google Scholar]
- 30.Rassaf T., Bryan N.S., Kelm M., Feelisch M. Concomitant presence of N-nitroso and S-nitroso proteins in human plasma. Free Radical Biology & Medicine. 2002;33:1590–1596. doi: 10.1016/s0891-5849(02)01183-8. 12446216 [DOI] [PubMed] [Google Scholar]
- 31.Slater T.F., Sawyer B., Sträuli U. Studies on succinate-tetrazolium reductase systems: III. Points of coupling of four different tetrazolium salts. III. Points of coupling of four different tetrazolium salts. Biochimica et Biophysica Acta. 1963;77:383–393. doi: 10.1016/0006-3002(63)90513-4. 14089413 [DOI] [PubMed] [Google Scholar]
- 32.Teng X., Zhang H., Snead C., Catravas J.D. A reverse nuclear factor-kappaB element in the rat type II nitric oxide synthase promoter mediates the induction by interleukin-1beta and interferon-gamma in rat aortic smooth muscle cells. General Pharmacology. 2000;34:9–16. doi: 10.1016/s0306-3623(99)00047-6. 10793263 [DOI] [PubMed] [Google Scholar]
- 33.Brigelius-Flohé R., Flohé L. Basic principles and emerging concepts in the redox control of transcription factors. Antioxidants & Redox Signaling. 2011;15:2335–2381. doi: 10.1089/ars.2010.3534. 21194351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mukaida N., Morita M., Ishikawa Y., Rice N., Okamoto S., Kasahara T., Matsushima K. Novel mechanism of glucocorticoid-mediated gene repression. Nuclear factor-kappa B is target for glucocorticoid-mediated interleukin 8 gene repression. Journal of Biological Chemistry. 1994;269:13289–13295. 8175759 [PubMed] [Google Scholar]
- 35.Liu M.J., Bao S., Gálvez-Peralta M., Pyle C.J., Rudawsky A.C., Pavlovicz R.E., Killilea D.W., Li C., Nebert D.W., Wewers M.D., Knoell D.L. ZIP8 regulates host defense through zinc-mediated inhibition of NF-kappaB. Cell Reports. 2013;3:386–400. doi: 10.1016/j.celrep.2013.01.009. 23403290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jeon Y.J., Han S.H., Lee Y.W., Yea S.S., Yang K.-H. Inhibition of NF-κB/Rel nuclear translocation by dexamethasone: mechanism for the inhibition of iNOS gene expression. IUBMB Life. 1998;45:435–441. doi: 10.1080/15216549800202822. [DOI] [PubMed] [Google Scholar]
- 37.Heyninck K., De Valck D., Vanden Berghe W., Van Criekinge W., Contreras R., Fiers W., Haegeman G., Beyaert R. The zinc finger protein A20 inhibits TNF-induced NF-κB-dependent gene expression by interfering with an RIP- or TRAF2-mediated transactivation signal and directly binds to a novel NF-κB-inhibiting protein ABIN. Journal of Cell Biology. 1999;145:1471–1482. doi: 10.1083/jcb.145.7.1471. 10385526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.De Vera M.E., Taylor B.S., Wang Q., Shapiro R.A., Billiar T.R., Geller D.A. Dexamethasone suppresses iNOS gene expression by upregulating I-κBα and inhibiting NF-κB. American Journal of Physiology. 1997;273:G1290–G1296. doi: 10.1152/ajpgi.1997.273.6.G1290. 9435553 [DOI] [PubMed] [Google Scholar]
- 39.Haase H., Rink L. Zinc signals and immune function. BioFactors (Oxford, England) 2014;40:27–40. doi: 10.1002/biof.1114. 23804522 [DOI] [PubMed] [Google Scholar]
- 40.Jeon K.I., Jeong J.Y., Jue D.M. Thiol-reactive metal compounds inhibit NF-kappa B activation by blocking I kappa B kinase. Journal of Immunology (Baltimore, MD: 1950) 2000;164:5981–5989. doi: 10.4049/jimmunol.164.11.5981. 10820281 [DOI] [PubMed] [Google Scholar]
- 41.Prasad A.S. Clinical, immunological, anti-inflammatory and antioxidant roles of zinc. Experimental Gerontology. 2008;43:370–377. doi: 10.1016/j.exger.2007.10.013. 18054190 [DOI] [PubMed] [Google Scholar]
- 42.Uzzo R.G., Leavis P., Hatch W., Gabai V.L., Dulin N., Zvartau N., Kolenko V.M. Zinc inhibits nuclear factor-kappa B activation and sensitizes prostate cancer cells to cytotoxic agents. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research. 2002;8:3579–3583. 12429649 [PubMed] [Google Scholar]
- 43.Kim C.H., Kim J.H., Moon S.J., Chung K.C., Hsu C.Y., Seo J.T., Ahn Y.S. Pyrithione, a zinc ionophore, inhibits NF-kappaB activation. Biochemical and Biophysical Research Communications. 1999;259:505–509. doi: 10.1006/bbrc.1999.0814. 10364448 [DOI] [PubMed] [Google Scholar]
- 44.Ho E., Quan N., Tsai Y.H., Lai W., Bray T.M. Dietary zinc supplementation inhibits NFkappaB activation and protects against chemically induced diabetes in CD1 mice. Experimental Biology and Medicine (Maywood, N.J.) 2001;226:103–111. doi: 10.1177/153537020122600207. 11446433 [DOI] [PubMed] [Google Scholar]
- 45.Oteiza P.I., Clegg M.S., Keen C.L. Short-term zinc deficiency affects nuclear factor-kappab nuclear binding activity in rat testes. Journal of Nutrition. 2001;131:21–26. doi: 10.1093/jn/131.1.21. 11208933 [DOI] [PubMed] [Google Scholar]
- 46.Mackenzie G.G., Zago M.P., Keen C.L., Oteiza P.I. Low intracellular zinc impairs the translocation of activated NF-kappa B to the nuclei in human neuroblastoma IMR-32 cells. Journal of Biological Chemistry. 2002;277:34610–34617. doi: 10.1074/jbc.M203616200. 12089148 [DOI] [PubMed] [Google Scholar]
- 47.Brieger A., Rink L., Haase H. Differential regulation of TLR-dependent MyD88 and TRIF signaling pathways by free zinc ions. Journal of Immunology (Baltimore, MD: 1950) 2013;191:1808–1817. doi: 10.4049/jimmunol.1301261. 23863901 [DOI] [PubMed] [Google Scholar]
- 48.Connell P., Young V.M., Toborek M., Cohen D.A., Barve S., McClain C.J., Hennig B. Zinc attenuates tumor necrosis factor-mediated activation of transcription factors in endothelial cells. Journal of the American College of Nutrition. 1997;16:411–417. doi: 10.1080/07315724.1997.10718706. 9322188 [DOI] [PubMed] [Google Scholar]
- 49.Hennig B., Meerarani P., Toborek M., McClain C.J. Antioxidant-like properties of zinc in activated endothelial cells. Journal of the American College of Nutrition. 1999;18:152–158. doi: 10.1080/07315724.1999.10718843. 10204831 [DOI] [PubMed] [Google Scholar]
- 50.Maret W., Jacob C., Vallee B.L., Fischer E.H. Inhibitory sites in enzymes: zinc removal and reactivation by thionein. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:1936–1940. doi: 10.1073/pnas.96.5.1936. 10051573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hogstrand C., Verbost P.M., Wendelaar Bonga S.E. Inhibition of human erythrocyte Ca2+-ATPase by Zn2+ Toxicology. 1999;133:139–145. doi: 10.1016/s0300-483x(99)00020-7. 10378480 [DOI] [PubMed] [Google Scholar]
- 52.Ye B., Maret W., Vallee B.L. Zinc metallothionein imported into liver mitochondria modulates respiration. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:2317–2322. doi: 10.1073/pnas.041619198. 11226237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gazaryan I.G., Krasnikov B.F., Ashby G.A., Thorneley R.N., Kristal B.S., Brown A.M. Zinc is a potent inhibitor of thiol oxidoreductase activity and stimulates reactive oxygen species production by lipoamide dehydrogenase. Journal of Biological Chemistry. 2002;277:10064–10072. doi: 10.1074/jbc.M108264200. 11744691 [DOI] [PubMed] [Google Scholar]
- 54.Haase H., Maret W. Intracellular zinc fluctuations modulate protein tyrosine phosphatase activity in insulin/insulin-like growth factor-1 signaling. Experimental Cell Research. 2003;291:289–298. doi: 10.1016/s0014-4827(03)00406-3. 14644152 [DOI] [PubMed] [Google Scholar]
- 55.Gerlier D., Thomasset N. Use of MTT colorimetric assay to measure cell activation. Journal of Immunological Methods. 1986;94:57–63. doi: 10.1016/0022-1759(86)90215-2. 3782817 [DOI] [PubMed] [Google Scholar]
- 56.Lichten L.A., Cousins R.J. Mammalian zinc transporters: nutritional and physiologic regulation. Annual Review of Nutrition. 2009;29:153–176. doi: 10.1146/annurev-nutr-033009-083312. 19400752 [DOI] [PubMed] [Google Scholar]
- 57.Gibson R.S., Hess S.Y., Hotz C., Brown K.H. Indicators of zinc status at the population level: a review of the evidence. British Journal of Nutrition. 2008;99(Suppl. 3):S14–S23. doi: 10.1017/S0007114508006818. 18598584 [DOI] [PubMed] [Google Scholar]
- 58.Bozym R.A., Chimienti F., Giblin L.J., Gross G.W., Korichneva I., Li Y., Libert S., Maret W., Parviz M., Frederickson C.J., Thompson R.B. Free zinc ions outside a narrow concentration range are toxic to a variety of cells in vitro. Experimental Biology and Medicine (Maywood, N.J.) 2010;235:741–750. doi: 10.1258/ebm.2010.009258. 20511678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kröncke K.D., Fehsel K., Schmidt T., Zenke F.T., Dasting I., Wesener J.R., Bettermann H., Breunig K.D., Kolb-Bachofen V. Nitric oxide destroys zinc–sulfur clusters inducing zinc release from metallothionein and inhibition of the zinc finger-type yeast transcription activator LAC9. Biochemical and Biophysical Research Communications. 1994;200:1105–1110. doi: 10.1006/bbrc.1994.1564. 8179589 [DOI] [PubMed] [Google Scholar]
- 60.Frederickson C.J., Koh J.-Y., Bush A.I. The neurobiology of zinc in health and disease. Nature Reviews Neuroscience. 2005;6:449–462. doi: 10.1038/nrn1671. 15891778 [DOI] [PubMed] [Google Scholar]
- 61.Shumilla J.A., Wetterhahn K.E., Barchowsky A. Inhibition of NF-[kappa]B Binding to DNA by chromium, cadmium, mercury, zinc, and arsenite in vitro: evidence of a thiol mechanism. Archives of Biochemistry and Biophysics. 1998;349:356–362. doi: 10.1006/abbi.1997.0470. 9448725 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Time-dependent increase in NOS-dependent nitrite accumulation in cytokine-treated cells as assessed by the Griess assay. ANOVA p<0.001. * p<0.05 vs. untreated.