

Abstract
DNA nanoparticles (DNA NPs), which self-assemble from DNA plasmids and poly-L-lysine (pLL)-polyethylene glycol (PEG) block copolymers, transfect several cell types in vitro and in vivo with minimal toxicity and immune response. To further enhance the gene transfer efficiency of DNA NP and control its tropism, we established a strategy to efficiently attach peptide ligands to DNA NPs. The non-covalent biotin–streptavidin (SA) interaction was used for ligand conjugation to overcome problems associated with covalent conjugation methods. A fusion protein of SA with the HIV-1 TAT peptide was cloned, expressed, purified and attached to biotinylated DNA NPs. A modified SA system with tetrameric structure but monovalent biotin binding capacity was adopted and shown to reduce the aggregation of biotinylated DNA NPs compared to neutravidin. Compared to unmodified DNA NPs, TAT modified DNA NPs significantly enhanced in vitro gene transfer, particularly at low DNA concentrations. Studies of cellular uptake and cellular distribution of the DNA NPs indicated that attaching TAT enhanced binding of DNA NPs to cell surface but not internalization at high DNA concentrations. In vivo studies showed that TAT modified DNA NPs mediated equal level of gene transfer to the mouse airways via the luminal route compared to unmodified DNA NPs.
Keywords: Non-viral gene therapy, polylysine-PEG block copolymer, ligand conjugation, biotin–streptavidin interaction, monovalent streptavidin
Introduction
High molecular weight poly-L-lysine (pLL) is one of the first polycation polymers that were used to form nanoparticulate polyelectrolyte complexes with DNA for non-viral gene therapy (Wu and Wu, 1987). The conjugation of polyethylene glycol (PEG) to pLL enhanced the in vivo performance of the pLL-DNA complex (Ward et al., 2002) and allowed shorter pLL peptides to be used to from stable nanoparticles with more favorable safety profiles (Kwok et al., 1999; Plank et al., 1996; Rimann et al., 2008). Among various pLL based gene carriers, one formulation consists of 30 lysine residues conjugated to a 10k Da PEG through an N-terminal cysteine (PEG-CK30) (Liu et al., 2003). DNA plasmids are condensed by PEG-CK30 into charge neutral, unimolecular DNA nanoparticles (DNA NPs) that are stable, non-toxic and capable of transgene delivery to several cell types in vitro and in vivo (Farjo et al., 2006; Liu et al., 2003; Yurek et al., 2009; Ziady et al., 2003a, 2003b; Konstan et al., 2004). Chen et al. showed that DNA NP binds to cell surface nucleolin which is essential for internalization and/or transport of the nanoparticles from cell surface to the nucleus (Chen et al., 2008). Subsequent study showed that the nucleolin-mediated cell entry pathway is a clathrin- and caveolin-independent non-degradative pathway that depends on lipid rafts (Chen et al., 2011).
The ease of manufacturing and production scaling up of DNA NP and its excellent safety profile make it a promising non-viral vector for clinical use. However, compared with viral vectors, the gene transfer efficiency of DNA still needs improvement. Although the nucleolin-mediated entry pathway seems to be a novel non-degradative pathway, its cellular uptake capacity remains untested and it also limits DNA NP mediated-gene transfer to those cell types that have substantial surface nucleolin expression. Surface modification of DNA NPs with targeting ligands may further enhance the gene transfer efficiency of DNA NP and allow control of its tropism in vivo. The initial success of DNA NPs in airway gene transfer aimed at treatment of cystic fibrosis (Ziady et al., 2003a, 2003b; Konstan et al., 2004) prompted us to test whether ligand modification could potentiate gene transfer in the airway and eventually benefit cystic fibrosis patients.
Covalent conjugation of targeting ligands to the end groups of PEG usually involves amine or thiol chemistry. Those methods are associated with problems such as lack of suitable reactive groups on the polymer, unstable intermediates, and inefficient coupling and purification. Our previous study also showed that disulfide linkage can be unstable in the extracellular environment (Sun and Davis, 2010). In contrast, non-covalent interactions between biotin and its binding proteins (avidin, streptavidin (SA), or neutravidin (NA)) are stable, highly specific and do not involve unstable intermediates. Different biotin-binding proteins have previously been used to conjugate proteins to the surface of nanoparticles (Townsend et al., 2007). A monovalent SA system was recently developed by Howarth and Ting (Howarth et al., 2006). In this system, the tetrameric structure of SA was kept intact, but the biotin-binding sites in three of the four subunits were inactivated by mutations, to give a SA with monovalent biotin-binding activity. The monovalent SA not only avoids the potential problem of aggregating biotinylated NPs that wild-type SA might have, but also retains a biotin-binding affinity equivalent to the original tetramer.
The TAT peptide, a protein transduction domain (PTD) from a transactivation factor of HIV-1 has been used to mediate cellular delivery of an assortment of cargos with various sizes including proteins, magnetic particles, poly(L-lactide) nanoparticles, lipoplexes and polyplexes (Josephson et al., 1999; Lewin et al., 2000; Torchilin et al., 2003; Suk et al., 2006; Peetla et al., 2009; Becker-Hapak et al., 2001). A TAT–SA fusion protein has been successfully constructed before to deliver biotinylated cargo into mammalian cells (Albarran et al., 2005), but has not been tested for gene delivery.
In this study, we investigated a non-covalent conjugation strategy in which a genetic fusion of the TAT peptide and monovalent SA is attached to biotinylated DNA NPs (Scheme 1). The use of ligand–SA fusion eliminates chemical conjugation and purification steps and allows accurate control over ligand stoichiometry and orientation. TAT is chosen as the model peptide ligand mainly because its ability to deliver cargos across many biological barriers. Fusion proteins and TAT modified DNA NPs were characterized in vitro. Intratracheal injection of DNA NPs into wild-type mice was also evaluated to test whether TAT could mediate gene delivery across the mucus layer.
Scheme 1.

(A) Synthesis of biotinylated DNA NPs and ligand conjugation through biotin-streptavidin (SA) interaction. (B) Construction and purification of monovalent ligand-SA fusion proteins. B, biotin binding activity.
Methods
Reagents
CK30 was synthesized at the Molecular Biotechnology Core of the Cleveland Clinic Lerner Research Institute (Cleveland, OH). Maleimide-PEG10kDa-biotin (MAL-PEG-biotin) and MAL-PEG was purchased from Rapp Polymere (Tübingen, Germany). SA-Alexa Fluor 647, biocytin-Alexa Fluor 488, 5′-phosphorylated oligonucleotides and TOP 10 chemically competent cells were obtained from Invitrogen (Carlsbad, CA). NA agarose beads, NA protein, bacterial protein extraction reagents (B-PER) and fluorescence biotin quantitation kit were purchased from Pierce/Thermo Scientific (Rockford, IL). DNA sequencing service was provided by ACGT, Inc. (Wheeling, IL). BL21(DE3)pLysS cells and Benzonase were provided by Novagen/EMD (Gibbstown, NJ). Isopropyl β-D-1-thiogalactopyranoside (IPTG) was obtained from Roche (Indianapolis, IN). Ni-NTA column was from Qiagen (Santa Clara, CA). pUL plasmid was a gift from Copernicus Therapeutics, Inc. (Cleveland, OH). pUL and pCMV-Luc plasmid DNA was purified from bacteria culture using EndoFree plasmid giga kit from Qiagen (Santa Clara, CA). All other reagents were purchased from Sigma (St. Louis, MO) unless specified otherwise.
Synthesis and purification of biotin-PEG-CK30
2 mM CK30 was mixed with equal volume of 4 mM MAL-PEG (with or without biotin) in 50 mM sodium phosphate (pH 7.0) with 5 mM EDTA. The reaction mixture was incubated overnight at room temperature on a rotating wheel. Polymers were purified by ion exchange chromatography as described previously (Sun and Davis, 2010).
Acetic acid-urea polyacrylamide gel electrophoresis (AU-PAGE) of biotin-PEG-CK30
AU-PAGE was performed according to a method previously described (Panyim and Chalkley, 1969) with minor modification. Briefly, 12% polyacrylamide gels containing 5 M urea and 5% acetic acid were pre-electrophoresed at 150 V for 1 h. Samples were solubilized in 5% acetic acid and diluted 2:1 with sample buffer (9 M urea in 5% acetic acid containing methylene blue as the tracking dye).
1H-NMR
1H-NMR spectra of the biotin-PEG-CK30 as well as control samples were obtained on a Varian Inova 600 NMR spectrometer (Varian Medical Systems, Palo Alto, CA) with samples dissolved in deuterium oxide (D2O) at a concentration of 2–5 mg/mL. The data were processed by MestReNova 6.0.2.
Formulation of DNA NP
PEGylated CK30 (with or without biotin) were used to compact plasmid DNA as described previously (Sun and Davis, 2010). Plasmids encoding a luciferase reporter gene under the control of CMV promoter (pCMV-Luc) or polyubiquitin C promoter (pUL) (Yurek et al., 2009) were used. All DNA NPs used in this study met or exceeded a series of qualification parameters (Sun and Davis, 2010).
Transmission electron microscopy (TEM)
Sample preparation and image acquisition were performed as described previously (Sun and Davis, 2010).
Fluorescent bead assay
Biotin-DNA NPs or DNA NPs without biotin (3 μg DNA in 100 μL 1×PBS) were either mixed with SA-fluorophore conjugates in 100 μL 1×PBS first (with Alexa Fluor 647, in 10-fold molar excess to the biotin in biotin-DNA NPs) and 8 μL NA agarose beads next or mixed with the same two reagents in reverse order. In control experiments, biotin-DNA NPs and SA-fluorophore conjugates were first blocked with free NA (0.3 mg/mL in 100 μL H2O) and free biotin (0.2 mg/mL in 100 μL 1×PBS), respectively, before mixing with other reagents. Biotin-PEG-CK30 was also used instead of DNA NPs as a negative control. The solution was sometimes diluted to make the final volume of all the different mixtures to be 300 μL. After incubation, the beads were washed three times with 500 μL 1×PBS and resuspended in a small volume of 1×PBS for visualization of fluorescence. Fluorescence and bright-field images of the beads were acquired using a Zeiss Axiovert 200M inverted microscope (Carl Zeiss, Peabody, MA) with a 10× objective.
Fluorescence biotin quantitation assay
Samples were diluted with 1×PBS to ensure that the final biotin concentration was within 1–6 μM. The concentration of biotin was measured in a microplate by comparing the fluorescence signal to a biocytin standard curve according to the manufacturer’s instruction.
Construction of TAT-SA gene
Wild-type (wt) and mutant (mt) SA genes carried in a pET-21a vector plasmid were obtained from addgene.org (addgene plasmid 20860 and 20859). The TAT-SA wt and mt genes were constructed by inserting a double stranded oligo encoding the TAT peptide to the Nde I site at the N-terminus of the SA wt or mt gene (forward: 5′ TAT GTA CGG TCG TAA AAA ACG TCG TCA GCG TCG TCG TGG 3′; reverse: 5′ TAC CAC GAC GAC GCT GAC GAC GTT TTT TAC GAC CGT ACA 3′). Clones were screened for the presence and direction of the insert using colony PCR with the following primer pairs: forward 5′ ATC TTC CCC ATC GGT GAT GT 3′ and TAT reverse oligo. Positive clones were further verified by diagnostic restriction digestion and sequencing.
Expression and purification of SA and TAT–SA fusion protein
pET-21a TAT-SA-wt and mt were transformed into BL21(DE3)pLysS cells in preparation for large-scale expression. Fusion proteins were expressed and purified as described with minor modifications (Howarth and Ting, 2008). Briefly, TAT-SA W (wild-type) and M (mutant) subunits were expressed separately, extracted as inclusion bodies using B-PER and then dissolved in 6 M guanidium hydrochloride pH 1.5. IPTG, at a final concentration of 1 mM, was used to induce the protein expression. 400 μL lysozyme (10 mg/mL in PBS) and 1 μL Benzonase (25 U/μL) were added to each 10 mL B-PER to help lyze the cells and reduce viscosity, respectively. W and M subunits were mixed at a 1:3 molar ratio and refolded via dilution into 1×PBS. Refolded fusion proteins were precipitated with ammonium sulfate, resuspended and dialyzed against 1×PBS. Monovalent TAT-SA was further purified on a Ni-NTA column. Fractions containing monovalent TAT-SA were pooled and dialyzed against 1×PBS.
Mass spectrometry analysis of purified SA and TAT-SA
Matrix-assisted laser desorption/ionization (MALDI)-time of flight (TOF) analysis was performed at the Case Western Reserve University Center for Proteomics and Bioinformatics. Briefly, purified SA or TAT-SA was mixed with matrix α-cyano-4-hydroxycinnamic acid with 0.1% trifluoroacetic acid. 1 μL of the mixture was then spotted onto the plate and analyzed.
Fluorescence microscopy
HeLa cells were seeded 24 h prior to the assay in 35 mm glass-bottomed dishes (MatTek, Ashland, MA) at initial density of 2.5 × 105 cells/dish. Biocytin-Alexa 488 was premixed with SA or TAT-SA at 1:1 molar ratio in 1 mL serum-free DMEM (final concentration 100 nM) and incubated at room temperature in the dark for 30 min. The cells were then incubated with the 1 mL mixture for 3.5 h followed by three washes with 1×DPBS. Biocytin-Alexa 488 alone in serum-free medium was also incubated with the cells as a control. For cellular distribution study, plasmids were labeled with tetramethylrhodamine (TM-Rhodamine) as described previously (Chen et al., 2008). The cells were incubated with TM-Rhodamine labeled DNA NPs for 3–4 h followed by three washes with 1×DPBS. Live cells incubated in 1 mL DMEM without phenol red were imaged using a Zeiss Axiovert 200 M inverted microscope with a 100× objective. The GFP filter set and the Cy3 filter set were used to image Alexa 488 and TM-Rhodamine, respectively.
Sedimentation assay
After incubation of biotinylated DNA NPs with NA or TAT-SA, 200 μL of DNA solution was centrifuged at 6000 rpm for 1 min. DNA in 100 μL of the supernatant was recovered by treatment with 1/10 volume of 2.5% trypsin at 37°C for 1 h. The recovered DNA was purified with one phenol/chloroform extraction and analyzed by 1% aga-rose gel electrophoresis. Sedimentation was quantified by comparing the band density of the sample before and after centrifugation.
In vitro gene transfer studies
Cells were seeded 24 h prior to transfection in 24-well plates at initial density of 0.8~1.2 × 105 cells/well. At the time of transfection, the medium in each well was replaced with 200 μL of fresh serum-free medium containing various amounts of DNA NPs (as indicated in the figure). TAT-SA- or SA-DNA NPs were prepared by mixing TAT-SA or SA with biotinylated DNA NPs in the serum free medium at proper molar ratios and incubated for 5 min at room temperature. After incubation for 6 h at 37°C, the cells were washed once with 1×DPBS and incubated in 1 mL of fresh complete medium for additional 42 h. The luciferase gene activity was then evaluated using the Luciferase Assay System (Promega, Madison, WI) and a Tropix Optocomp I luminometer (MGM Instruments, Hamden, CT). The results were expressed as relative luminescence units (RLU) normalized to total cellular protein content determined by DC protein assay kit (BioRad, Hercules, CA).
Flow cytometry
DNA NPs were labeled with YOYO-1 as described before (Sun and Davis, 2010). After transfection with YOYO-1 labeled DNA NPs, cells were trypsinized, washed once with 1×DPBS and resuspended in ice cold HBSS with 0.5% bovine serum albumin. Samples were analyzed by BD FACSAria flow cytometer (BD Biosciences, San Jose, CA). The total cell population was appropriately gated by forward and side scattering to exclude dead cells. A total of 10 000 events were recorded per sample.
In vivo gene transfer study
All animal experiments were performed in the Case Cystic Fibrosis Animal Core in accordance with guidelines approved by the Institutional Animal Care and Use Committee office of the Case Western Reserve University. Since effective CFTR gene transfer had been achieved in the human cystic fibrosis nasal epithelium using DNA NP (Konstan et al., 2004), further investigation of the airway approach was warranted. Mice seemed a suitable initial species in which to test enhancement of gene transfer by TAT conjugation because of prior success with gene transfer into mouse airways with the parent DNA NP (Ziady et al., 2003a), even though the correspondence with human airway anatomy is not exact. C57BL6 mice were operated on with a sterile surgical technique. pUL was used to prepare DNA NPs for better in vivo expression (Yurek et al., 2009). Different concentrations of DNA NPs or TAT-SA-DNA NPs (40 tetramers/DNA NP) in 100 μL saline were administered to wild-type mice intratracheally on day 1. On day 3 and 5, animals were sacrificed and mouse lungs and tracheas were harvested and frozen on dry ice. To extract luciferase, organs were thawed and homogenized on ice in freshly prepared 1×reporter lysis buffer (RLB, Promega, Madison, WI) using a Tissue Tearor homogenizer (BioSpec Products, Bartlesville, OK). Mouse lungs were homogenized in 1 mL 1×RLB while tracheas in 0.8 mL. Homogenized tissues were frozen on dry ice for 15 min and thawed in 37°C water bath for 5 min. After an additional freeze-thaw cycle, homogenates were centrifuged at 13 000 rpm 4°C for 10 min and the supernatants were collected. For luciferase activity analysis, 80 μL of luciferin solution were added to 20 μL of each sample and luminescence was detected as described for in vitro study.
Statistical analysis
The data in Figure 7, 8A and 9 were analyzed by unpaired t test (tails = 2) using Excel software.
Figure 7.
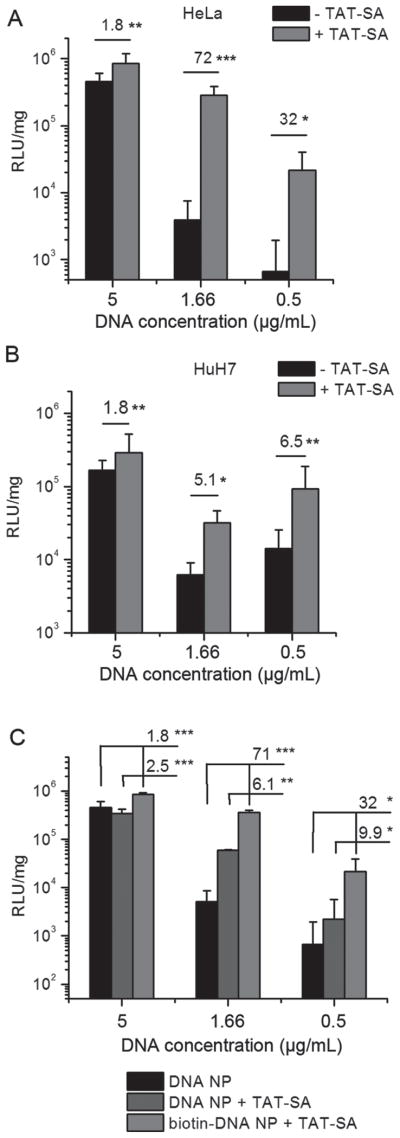
(A, B) In vitro gene transfer with lower concentrations of DNA NPs into HeLa (A) or HuH7 cells (B). For each well, 1, 0.3 or 0.1 μg of DNA was added to 200 μL medium to give the final DNA concentrations of 5, 1.66 or 0.5 μg/mL. Results are shown as mean ± SD (6–9 replicates from 2–3 independent experiments). (C) TAT-SA partially enhanced in vitro gene transfer when mixed with DNA NPs without biotin. In vitro gene transfer study was conducted using HeLa cells as described in A and B except that TAT-SA was mixed at the same molar ratio with DNA NPs with or without biotin. Results are mean ± SD (six replicates from two independent experiments). *P < 0.05; **P < 0.01; ***P < 0.001.
Figure 8.

(A) Cellular uptake of YOYO-1 labeled DNA NPs measured by flow cytometry. HeLa cells in 6 well plates were transfected with YOYO-1 labeled DNA NPs (20 μg/mL DNA) for 10 h. Results are shown as mean fluorescence of each sample with 10,000 events recorded (mean ± SD, n = 3). **P < 0.01; ***P < 0.001. (B) Cellular distribution of TM-Rhodamine labeled DNA NPs. HeLa cells grown in 35 mm glass-bottom dish were transfected with TM-Rhodamine labeled DNA NPs (10 μg/mL DNA) for 4 h. Cells were washed three times with 1×DPBS and images were acquired by a wide-field fluorescence microscope.
Figure 9.

In vivo gene transfer study. DNA NPs were injected intratracheally in wild-type mice at doses of 50, 10, 2, 0.5 or 0.1 μg DNA/mouse. Animals were sacrificed and luciferase activity of lungs and tracheas were assayed on day 5. Results are shown as mean ± SD. Each DNA NP group contains 4–5 mice.
Results
Synthesis and characterization of biotin-PEG-CK30
Biotin-PEG-CK30 was synthesized by reacting CK30 with heterobifunctional biotin-PEG-MAL and purified by ion exchange column. The conjugation of biotin to CK30 through PEG was confirmed by acetic acid-urea polyacrylamide gel electrophoresis (AU-PAGE) (Figure 1) and 1H-NMR (Figure 2). As shown in Figure 2A and 2B, in spite of the strong signals from the PEG chain, all the biotin signature peaks were detected in the spectrum of MAL-PEG-biotin. When the triblock polymer was tested, some of the biotin peaks were masked by the strong signal from the polylysine (Figure 2C). Nonetheless, five out of nine biotin peaks were detected. More importantly, a mixture of PEG-CK30 and free biotin was also tested after it underwent the same purification process for biotin-PEG-CK30. None of the biotin peaks was detected by 1H-NMR (Figure 2D), indicating that free biotins were removed during the purification step and the signals in Figure 2C came from biotins that are covalently attached to the polymer.
Figure 1.

AU-PAGE analysis of purified PEGylated CK30. Purified PEGylated CK30 with or without biotin and unPEGylated CK30 were analyzed by AU-PAGE and stained with Coomassie blue. Dimers of CK30 peptides crosslinked by disulfide bond were formed in solution over time. Dimers and oligomers of PEGylated peptides were also formed through non-covalent interaction.
Figure 2.

1H-NMR spectrum of free and conjugated biotin. Free biotin (A), MAL-PEG-biotin (B), purified biotin-PEG-CK30 (C), and PEG-CK30 mixed with free biotin and then purified with ion exchange column (D) were dissolved in deuterium oxide and the 600 MHz NMR spectrum for each sample was obtained. Signals from PEG and polylysine were labeled in the figure. Signals from biotin were indicated by arrows.
Preparation and characterization of biotin-DNA NPs
Purified biotin-PEG-CK30 was used to compact luciferase reporter DNA plasmids into biotin-DNA NPs (Scheme 1A). To demonstrate the availability of biotin on the surface of DNA NPs, the binding of SA-conjugated fluorophore to the biotin-DNA NPs was tested by a fluorescent beads assay. In this assay, the biotin-DNA NPs were immobilized on NA beads, which enabled a subsequent washing step and visualization by fluorescence microscopy. Beads were fluorescently decorated only when incubated with biotin-DNA NPs in the presence of the SA-fluorophore conjugate, but not with unmodified DNA NPs or 1×PBS (Figure 3A, 3E and 3G). Reversing the order of the addition of the fluorophore conjugates and the beads did not alter the results (Figure 3B and 3F). Fluorescence was not detected when biotin-DNA NPs were preincubated with free NA or when the fluorophore conjugates were pretreated with free biotin (Figure 3C and 3D). Free biotin-PEG-CK30 polymer failed to produce the fluorescence signal (Figure 3H), which indicates that multiple biotins are available and required on a single biotin-DNA NP for binding with both the fluorophore conjugates and the NA beads at the same time. Taken together, these results showed that the biotin on the DNA NPs was accessible and fully functional.
Figure 3.

Fluorescent beads assay to confirm conjugation of biotins to DNA NPs. Reagents used in each experiment and their order of mixing were indicated in letters and numbers in brackets on top of each image. After incubation of biotin-DNA NPs or controls with NA agarose beads and SA conjugated fluorophores, the beads were washed and imaged with fluorescence microscope with a 10× objective. Fluorescence and bright field images of the same field of the NA beads are shown side by side.
In addition to the qualitative assay, a fluorescence biotin quantitation system was used to determine how many biotin sites on average are available on each biotin-DNA NP. To ensure no free biotinylated polymer was present in the DNA NP solution, the sample was purified using a spin filter with a 100 kDa molecular weight cut-off membrane. The biotin concentrations of both unpurified and purified biotin-DNA NP samples were measured. The result showed that the purification step did not reduce the biotin/DNA ratio in purified biotin-DNA NP sample and there are 690 ± 27 biotin sites on each DNA NP.
Expression, purification and characterization of TAT–SA fusion protein
As shown in Figure 4A, to construct the monovalent TAT-SA gene, a synthesized double stranded oligo encoding TAT peptide was inserted at the N-terminus of the SA gene where an NdeI restriction site was found. Monovalent SA was prepared as described previously (Howarth and Ting, 2008). Monovalent TAT-SA was prepared using the same procedure. The TAT-SA W and M subunits were expressed separately and mixed at a molar ratio of 1:3. A mixture of tetramers with different subunit compositions were still formed after refolding (Scheme 1B). Since the TAT-SA W subunit has a C-terminal 6His tag, the mixture was further separated by nickel affinity chromatography according to the number of 6His tags they have (Scheme 1B). SDS-PAGE analysis showed that, with increasing imidazole concentrations, the tetramers were eluted in the order of M4, W1M3, W2M2 and W3M1 as indicated by the approximate ratios of the two subunits for each tetramer under denaturing conditions (Figure 4C). Eluted fractions containing TAT-SA W1M3 (simply referred to as TAT-SA below) were pooled, desalted and concentrated for subsequent use.
Figure 4.

(A) TAT-SA wt and mt genetic constructs. TAT sequence is inserted at the NdeI site at the N-terminus of the SA gene. The NdeI site is preserved at the N-terminus of the fusion gene after the insertion. The yellow triangles indicate three amino acid mutations in the TAT-SA mt gene. A BamHI site is interrupted by one of the mutations and used in diagnostic digestion to verify the identity of the two genes. TAT reverse oligo and an upstream sequencing primer are used for colony PCR. (B,C) SDS-PAGE of SA and TAT-SA. Samples were loaded onto 15% SDS-PAGE gels with or without boiling. (B) Different TAT-SA tetramers eluted from the Ni-NTA column with imidazole of the concentrations indicated in the figure. (C) SA and TAT-SA after one month storage at 4°C or −80°C.
The stability of monovalent SA and TAT-SA were tested with SDS-PAGE. After over one month storage of the purified protein in 1×PBS at 4°C or −80°C, they were analyzed under denaturing and non-denaturing conditions with 15% SDS-PAGE. As shown in Figure 4C, no degradation was detected under either storage condition, indicating that both SA and TAT-SA were stable. Both TAT-SA subunits migrated slower than SA subunits as expected (Figure 4C denaturing condition). However, TAT-SA tetramer had an apparent molecular weight not larger but smaller than SA tetramer (Figure 4B non-denaturing condition), which may be attributed to altered conformation and surface charge of the fusion protein.
The molecular weight of purified monovalent TAT-SA was determined by MALDI-TOF mass spectrometry. The measured molecular weight for the two subunits of TAT-SA were 15946.03 Da (M) and 15092.00 Da (W) which are in good agreement with the calculated theoretical mass of 15945.83 Da (M) and 15092.47 Da (W) (monoisotopic mass obtained online from ExPASy). The molecular weights of the subunits of monovalent SA were also confirmed. It is worth noting that the N-terminal methionine residue was retained by TAT-SA but removed from SA as indicated by the measured results.
To test whether monovalent TAT-SA is capable of delivering biotinylated cargo into cells, the TAT-SA mediated uptake of biocytin-conjugated Alexa Fluor 488 into HeLa cells was examined by fluorescence microscopy. As shown in Figure 5 top panel, after 3.5 h incubation, TAT-SA efficiently delivered the biotinylated fluorophore into the cells. When TAT-SA was replaced by SA, no fluorescence was detected (Figure 5 middle panel), suggesting that the cargo delivery is mediated by TAT. When biocytin-Alexa 488 alone was incubated with the cells, no fluorescence was detected inside the cells (Figure 5 bottom panel). This is expected because the conjugate is cell impermeant. These results showed that both the TAT and the SA moiety in TAT-SA are fully functional and the fusion protein is capable of delivering biotinylated cargos with small molecular weight into HeLa cells.
Figure 5.

Cellular delivery of biotinylated fluorophore by TAT-SA. HeLa cells cultured in 35 mm glass-bottomed dishes were incubated with a premix of biocytin-Alexa Fluor 488 with SA or TAT-SA (at 1:1 molar ratio) for 3.5 h. Cells were then washed with 1×DPBS and imaged live using a wide field fluorescence microscope. Cells were also incubated with biocytin-Alexa 488 alone as a control.
Characterization of TAT-SA-DNA NPs
TAT-SA was mixed with biotinylated DNA NPs to form DNA NPs with surface modified by TAT (TAT-SA-DNA NPs). The average number of TAT on each DNA NP was controlled by adjusting the molar ratio of TAT-SA to DNA. TAT-SA-DNA NPs with increasing density of TAT-SA were prepared and their colloidal stability was tested by sedimentation assay and compared to that of biotinylated DNA NPs mixed with NA (NA-DNA NPs). At high protein density, both TAT-SA and NA caused sedimentation of biotinylated DNA NPs as shown in Figure 6A. However, TAT-SA-DNA NPs were colloidally stable with 100 tetramers per DNA while NA-DNA NPs with the same ligand density were sedimented (Figure 6A). This result indicates that the monovalent design of TAT-SA, though did not eliminate aggregation completely, did reduce it; whereas NA or wild-type SA caused more aggregation presumably by cross-linking multiple biotinylated DNA NPs. This result also validates the usefulness of monovalent TAT-SA by which higher density of ligands could be conjugated compared to the use of NA to link biotinylated ligand and vector. No more than 100 TAT-SA per DNA were used to prepare TAT-SA-DNA NPs in the following experiment to ensure no aggregation of resultant DNA NPs.
Figure 6.

(A) Sedimentation assay. DNA NPs were mixed with TAT-SA or NA at molar ratios indicated in the figure. After incubation at room temperature for 30 min, sedimentation assay was performed by centrifugation at 6000 rpm for 1 min. DNA in the supernatant before and after centrifugation was compared. Results are mean ± SD (n = 3). (B) Transmission electron microscopy images of DNA NPs. Scale bar = 200 nm.
To further characterize TAT-SA-DNA NPs, their morphology was analyzed by TEM. As shown in Figure 6B, no difference in morphology was observed among DNA NPs, biotin-DNA NPs, TAT-SA-DNA NPs and SA-DNA NPs (biotinylated DNA NPs mixed with monovalent SA). In addition, attaching different number of ligands (from 5 to 100 TAT-SA per DNA) to DNA NPs did not cause significant change in morphology as observed by TEM (data not shown).
In vitro gene transfer studies with TAT-SA-DNA NPs
We then tested the effect of TAT modification on DNA NP mediated in vitro gene transfer. HeLa cells grown in 24-well plates were transfected with different DNA NPs at a relatively high concentration of 5 μg/mL. With optimization of incubation time and the number of ligand per DNA, a maximum of two-fold increase was observed when DNA NPs were replaced by TAT-SA-DNA NPs (data not shown). SA-DNA NPs failed to show the same effect.
Previous study of non-targeted DNA NPs suggests that they enter cells via a lipid raft mediated non-degradative pathway (Chen et al., 2011). Modification of DNA NPs with TAT may redirect them to endocytic compartment where they can be trapped, resulting in inefficient trans-gene expression. To test that hypothesis, chloroquine was added to the media before and during transfection to disrupt endosomes. Although the highest concentration of chloroquine the cell can tolerate was used (200 μM), no enhancement of in vitro gene transfer was observed for either DNA NPs or TAT-SA-DNA NPs (data not shown).
Huang et al. reported previously that the enhancing effect of TAT on gene transfer mediated by nanolipoparticles (NLPs) became more prominent as the concentration of DNA was lowered (Huang et al., 2005). We therefore repeated the in vitro transfection experiment with lower concentrations of DNA NPs and similar phenomenon was observed. In HeLa cells, when DNA concentration was lowered from 5 μg/mL to 0.5 μg/mL, the TAT mediated gene transfer enhancement increased to 32-fold. The highest increase (72-fold) was seen at 1.67 μg/mL (Figure 7A). Although the luciferase expression levels of both DNA NPs dropped, it was linear for TAT-SA-DNA NPs and almost exponential for control DNA NPs. In HuH7 cells, TAT mediated in vitro gene transfer also become more efficient at lower DNA concentrations, albeit with smaller fold change (Figure 7B).
To determine whether the biotin-SA interaction is indispensable for the TAT mediated gene transfer enhancement, we repeated the in vitro gene transfer experiment with an additional control of TAT-SA mixed with DNA NP without biotin. Interestingly, at lower DNA concentration, simply mixing the TAT-SA with the DNA NPs without any specific attachment (TAT-SA + DNA NPs) enhanced the in vitro gene transfer to a certain level (Figure 7C). This is probably due to non-specific interactions between TAT-SA and DNA NPs. However, this non-specific effect cannot fully account for the in vitro gene transfer enhancement observed when the TAT is attached to the DNA NPs through biotin-SA interaction. At lower DNA concentrations, TAT-SA-DNA NPs still mediated five- to nine-fold higher in vitro gene transfer compared to TAT-SA + DNA NPs (Figure 7C). Therefore, we concluded that attaching TAT to DNA NPs through biotin-SA interaction is necessary to achieve maximum enhancement of in vitro gene transfer.
Cellular uptake and cellular distribution
To further investigate the mechanism of TAT-SA-DNA NP mediated in vitro gene transfer, the cellular uptake of DNA NPs with or without TAT was compared by flow cytometry. The DNA plasmid was fluorescently labeled with YOYO-1 as described previously (Sun and Davis, 2010). YOYO-1 labeled DNA plasmids were condensed into DNA NPs and used to transfect HeLa cells. A high concentration of DNA NPs (20 μg/mL) was used to obtain detectable fluorescent signals. As shown in Figure 8A, cellular uptake of YOYO-1 labeled DNA NPs was detected as an increase of mean fluorescence from the background of the untreated control cells. When YOYO-1 TAT-SA-DNA NP was compared to YOYO-1 DNA NPs without TAT, only a small increase of mean fluorescence was observed. The small increase of cellular uptake is consistent with the mild increase of in vitro gene transfer by TAT-SA-DNA NPs at high concentrations. However, the reason why TAT failed to more efficiently deliver DNA NPs into cells at high DNA concentration is still not clear.
Although flow cytometry study provided quantitative data on cellular uptake, no information on cellular distribution was yielded. Fluorescence imaging study was therefore conducted with DNA NPs covalently labeled with TM-Rhodamine on the DNA plasmids. Only high concentration of DNA (10 μg/mL) was tested so that sufficient fluorescence signal could be detected. Images of live HeLa cells transfected with fluorescently labeled DNA NPs showed that more TAT-SA-DNA NPs were associated with the cells compared to DNA NPs without TAT (Figure 8B). However, most of the TAT-SA-DNA NPs appeared to bind to the cell surface without entering the cells (Figure 8B). Those surface bound DNA NPs could not be detected by flow cytometry because they were degraded during the trypsinization step and washed away. This result may explain why TAT-SA-DNA NPs did not work as efficiently as expected at high concentrations of DNA. But it further demonstrated that TAT was successfully conjugated to the DNA NP and was able to bind to the cell surface. The reason why surface bound DNA NPs were not internalized needs to be further investigated.
In vivo gene transfer study
To evaluate the effect of attaching TAT to DNA NPs on in vivo gene transfer, we monitored the expression of luciferase reporter gene in the airways of wild-type mice after intratracheal injection of DNA NPs. Luminal instead of systemic gene delivery was tested mainly because TAT mediated gene transfer lacks cell specificity. Different doses of DNA NPs or TAT-SA-DNA NPs were administered on day 1 and the luciferase activity of homogenized mouse lungs and tracheas was assayed on day 3 and 5. For both groups, luciferase expression persisted from day 3 to day 5 with a small increase on day 5 (data not shown). In general, expression in the lung was higher than that in the trachea. Surprisingly, lowering the dose of DNA NPs from 50 μg DNA to 0.5 μg did not result in significant decrease of reporter gene expression in either group (Figure 9). In the trachea, mean luciferase expression from TAT-SA-DNA NP group was mostly higher compared to DNA NP group (Figure 9). In the lung, luciferase expression mediated by TAT-SA-DNA NP peaked at 2 μg DNA/mouse (Figure 9). However, no statistically significant difference was found between the DNA NP group and the TAT-SA-DNA NP group.
Discussion
In this study, the biotin-SA interaction, instead of covalent conjugation, was used to attach targeting ligand to DNA NPs for several reasons. (1) This strategy enables surface modification of the DNA NPs without interfering with the DNA compaction. (2) The use of non-covalent interaction avoids the problems encountered by covalent conjugation. (3) The biotin-SA binding has a dissociation constant on the order of 10−14 M, which is one of the strongest non-covalent biological interactions.
Compared to previous studies using biotin–avidin interaction for ligand conjugation, our strategy using monovalent TAT-SA has several advantages. In several previous studies, NA was used to link biotinylated ligands with biotinylated vectors (Suk et al., 2006; Townsend et al., 2007). Aggregation caused by nanoparticle cross-linking was documented in one of those studies (Townsend et al., 2007). Although monovalent TAT-SA did not eliminate aggregation, it did reduce it so that more ligands could be conjugated to the DNA NPs. In another study, ligand–SA fusion protein was also used but as SA monomer, not tetramer (Kakimoto et al., 2010). There is evidence that the biotin-binding affinity of a SA monomer is greatly reduced compared to a tetramer (Howarth and Ting, 2008). The monovalent TAT-SA maintains its tetrameric structure so that its biotin binding activity is unaltered. Therefore, the conjugation of the monovalent TAT-SA to biotinylated vectors is more stable than that of ligand-SA monomer.
When DNA NPs are formulated with biotin-PEG-CK30 at N/P ratio of 2:1, the calculated number of biotin sites on each DNA NP is 720 (for 5.4 kb plasmid). Only 100 or less biotin sites are conjugated to TAT-SA, leaving the rest of the sites unoccupied. Those free biotin sites may interfere with gene transfer. One solution is to reduce the biotin sites on each DNA NP by mixing biotin-PEG-CK30 with PEG-CK30 during the formulation of DNA NPs. With this method, the number of biotin sites can be controlled so that all sites are occupied by TAT-SA. And if there are free polymers in the solution, this method can also reduce the concentration of free biotin-PEG-CK30 and improve the efficiency of conjugation. For those reasons, we prepared TAT-SA-DNA NPs without unoccupied biotins and conducted in vitro gene transfer study. At least at high DNA concentrations, no further improvement was found with those DNA NPs (data not shown). Therefore, we concluded that the extra-biotin sites on the DNA NPs have no significant effect on in vitro gene transfer and no further effort was made to reduce the number of those sites.
In vitro gene transfer study indicated that at high DNA concentrations, attaching TAT to DNA NPs only had a mild enhancing effect. Several efforts have been made to further improve the gene transfer efficiency of TAT-SA-DNA NPs at high DNA concentrations. Those efforts include varying the N/P ratio and length of PEG, and reducing the degree of PEGylation among others. However, none of them further improved the gene transfer efficiency (data not shown).
The results from the cellular uptake and cellular distribution studies, taken together with the current model of TAT mediated cellular delivery and the unique uptake mechanism of DNA NP may provide explanations for why TAT-SA-DNA NP only enhances gene transfer at low DNA concentrations in vitro. The cellular uptake process can be divided into two steps, binding and internalization. The result from the fluorescence microscopic study suggested more efficient binding of TAT-SA-DNA NPs to the cell surface (Figure 8B) than unmodified DNA NPs. However, efficient binding doesn’t necessarily lead to efficient internalization. Chen et al. previously demonstrated that cell surface nucleolin binds to and serves as the receptor for DNA NPs (Chen et al., 2008). A follow-up study revealed a lipid raft dependent uptake mechanism for unmodified DNA NPs (Chen et al., 2011). A number of studies suggest that lipid-raft-mediated endocytosis is also involved in TAT mediated delivery of high molecular weight cargo (Chu et al., 2004; Console et al., 2003; Duchardt et al., 2007). At high DNA concentrations, the lipid raft dependent internalization is operated at full capacity for both DNA NPs and TAT-SA-DNA NPs, so more efficient surface binding does not further enhance gene transfer. At low DNA concentrations, the internalization pathway is not saturated, so more efficient binding of TAT-SA-DNA NP leads to more efficient internalization. However, whether and to what extent nucleolin is involved needs to be further investigated.
For the in vivo gene transfer study, we tested a series of DNA concentrations to see whether the concentration effect on TAT mediated gene transfer observed in vitro applies in vivo. Unexpectedly, the luciferase expression in the airway from either TAT modified or unmodified DNA NPs did not decrease with decreasing doses of DNA NPs over an extended range from 50 μg to 0.5 μg DNA/ mouse. This indicates an unidentified rate limiting factor in vivo which is saturated at very low dosage of DNA NPs. The stability of the targeting fusion protein may be a rate limiting factor. A report on increased in vivo gene transfer after intratracheal instillation of a TAT-PEG-PEI complex in mice suggests TAT peptide remains functional in the mouse airway (Kleemann et al., 2005). The extensive use of antibody–SA fusion protein in pretargeted radioimmunotherapy indicates that SA is stable in the circulation (Schultz et al., 2000). Whether it is stable in the airway remains to be tested. Boylan et al. showed that DNA NPs are immobilized by mucoadhesive interactions in sputum that lines the airways of patients with cystic fibrosis, which might preclude the efficient delivery of the nanoparticles to the underlying epithelium (Boylan et al., 2012). If this is an important limiting factor, our result may indicate that modification with TAT may not facilitate a better penetration of DNA NPs through the mucus layer. However, it does not preclude the possibility that expression from DNA NPs could be enhanced by TAT modification at other sites such as brain or retina.
Although DNA NPs proved to be safe in humans (Konstan et al., 2004), the toxicity of newly introduced TAT and SA needs to be evaluated in vivo. A clinical study of an antibody-SA conjugate showed that though antibodies against SA were detected in the patients, no significant adverse events were observed (Breitz et al., 1999). If necessary, SA can be further engineered to have reduced immunogenicity (Meyer et al., 2001). In the above mentioned study of TAT-PEG-PEI, TAT conjugation even lowered toxicity both in vitro and in vivo (Kleemann et al., 2005). A study of the in vivo delivery of an enzyme by fusing it to TAT showed that antibodies were raised only against the enzyme, but not TAT (Toro and Grunebaum, 2006), suggesting the toxicity of TAT may not be an issue.
Conclusion
We describe a non-covalent method of efficient ligand conjugation to DNA NPs and observed that attaching TAT peptide to DNA NPs using this method significantly enhanced in vitro gene transfer at low DNA concentrations. However, TAT modification did not significantly increase in vivo gene transfer to airway epithelium at any concentration, suggesting that TAT cannot overcome the presumably mechanical barriers to delivery of large biomolecules across the airway mucus layer. Nonetheless, because of its versatility, this method can be used for attachment of other peptide ligands and may serve as a universal strategy for ligand conjugation to biotinylated gene and drug carriers.
Acknowledgments
This research was supported by the National Institutes of Health Grants R01DK058318, R21DK084371, and P30DK27651.
We thank Midori Hitomi in Electron Microscopy Facility and Kathleen Lundberg in Case Center for Proteomics for their technical assistance. We also thank Linas Padegimas at Copernicus Therapeutics Inc. for assistance with molecular cloning.
References
- Albarran B, To R, Stayton PS. A TAT-streptavidin fusion protein directs uptake of biotinylated cargo into mammalian cells. Protein Eng Des Sel. 2005;18:147–152. doi: 10.1093/protein/gzi014. [DOI] [PubMed] [Google Scholar]
- Becker-Hapak M, McAllister SS, Dowdy SF. TAT-mediated protein transduction into mammalian cells. Methods. 2001;24:247–256. doi: 10.1006/meth.2001.1186. [DOI] [PubMed] [Google Scholar]
- Boylan NJ, Suk JS, Lai SK, Jelinek R, Boyle MP, Cooper MJ, Hanes J. Highly compacted DNA nanoparticles with low MW PEG coatings: In vitro, ex vivo and in vivo evaluation. J Control Release. 2012;157:72–79. doi: 10.1016/j.jconrel.2011.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitz HB, Fisher DR, Goris ML, Knox S, Ratliff B, Murtha AD, Weiden PL. Radiation absorbed dose estimation for 90Y-DOTA-biotin with pretargeted NR-LU-10/streptavidin. Cancer Biother Radiopharm. 1999;14:381–395. doi: 10.1089/cbr.1999.14.381. [DOI] [PubMed] [Google Scholar]
- Chen X, Kube DM, Cooper MJ, Davis PB. Cell surface nucleolin serves as receptor for DNA nanoparticles composed of pegylated polylysine and DNA. Mol Ther. 2008;16:333–342. doi: 10.1038/sj.mt.6300365. [DOI] [PubMed] [Google Scholar]
- Chen X, Shank S, Davis PB, Ziady AG. Nucleolin-mediated cellular trafficking of DNA nanoparticle is lipid raft and microtubule dependent and can be modulated by glucocorticoid. Mol Ther. 2011;19:93–102. doi: 10.1038/mt.2010.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu CL, Buczek-Thomas JA, Nugent MA. Heparan sulphate proteoglycans modulate fibroblast growth factor-2 binding through a lipid raft-mediated mechanism. Biochem J. 2004;379:331–341. doi: 10.1042/BJ20031082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Console S, Marty C, García-Echeverría C, Schwendener R, Ballmer-Hofer K. Antennapedia and HIV transactivator of transcription (TAT) “protein transduction domains” promote endocytosis of high molecular weight cargo upon binding to cell surface glycosaminoglycans. J Biol Chem. 2003;278:35109–35114. doi: 10.1074/jbc.M301726200. [DOI] [PubMed] [Google Scholar]
- Duchardt F, Fotin-Mleczek M, Schwarz H, Fischer R, Brock R. A comprehensive model for the cellular uptake of cationic cell-penetrating peptides. Traffic. 2007;8:848–866. doi: 10.1111/j.1600-0854.2007.00572.x. [DOI] [PubMed] [Google Scholar]
- Farjo R, Skaggs J, Quiambao AB, Cooper MJ, Naash MI. Efficient non-viral ocular gene transfer with compacted DNA nanoparticles. PLoS ONE. 2006;1:e38. doi: 10.1371/journal.pone.0000038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howarth M, Chinnapen DJ, Gerrow K, Dorrestein PC, Grandy MR, Kelleher NL, El-Husseini A, Ting AY. A monovalent streptavidin with a single femtomolar biotin binding site. Nat Methods. 2006;3:267–273. doi: 10.1038/NMETHXXX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howarth M, Ting AY. Imaging proteins in live mammalian cells with biotin ligase and monovalent streptavidin. Nat Protoc. 2008;3:534–545. doi: 10.1038/nprot.2008.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z, Li W, MacKay JA, Szoka FC., Jr Thiocholesterol-based lipids for ordered assembly of bioresponsive gene carriers. Mol Ther. 2005;11:409–417. doi: 10.1016/j.ymthe.2004.10.013. [DOI] [PubMed] [Google Scholar]
- Josephson L, Tung CH, Moore A, Weissleder R. High-efficiency intracellular magnetic labeling with novel superparamagnetic-Tat peptide conjugates. Bioconjug Chem. 1999;10:186–191. doi: 10.1021/bc980125h. [DOI] [PubMed] [Google Scholar]
- Kakimoto S, Tanabe T, Azuma H, Nagasaki T. Enhanced internalization and endosomal escape of dual-functionalized poly(ethyleneimine)s polyplex with diphtheria toxin T and R domains. Biomed Pharmacother. 2010;64:296–301. doi: 10.1016/j.biopha.2009.06.017. [DOI] [PubMed] [Google Scholar]
- Kleemann E, Neu M, Jekel N, Fink L, Schmehl T, Gessler T, Seeger W, Kissel T. Nano-carriers for DNA delivery to the lung based upon a TAT-derived peptide covalently coupled to PEG-PEI. J Control Release. 2005;109:299–316. doi: 10.1016/j.jconrel.2005.09.036. [DOI] [PubMed] [Google Scholar]
- Konstan MW, Davis PB, Wagener JS, Hilliard KA, Stern RC, Milgram LJ, Kowalczyk TH, Hyatt SL, Fink TL, Gedeon CR, Oette SM, Payne JM, Muhammad O, Ziady AG, Moen RC, Cooper MJ. Compacted DNA nanoparticles administered to the nasal mucosa of cystic fibrosis subjects are safe and demonstrate partial to complete cystic fibrosis transmembrane regulator reconstitution. Hum Gene Ther. 2004;15:1255–1269. doi: 10.1089/hum.2004.15.1255. [DOI] [PubMed] [Google Scholar]
- Kwok KY, McKenzie DL, Evers DL, Rice KG. Formulation of highly soluble poly(ethylene glycol)-peptide DNA condensates. J Pharm Sci. 1999;88:996–1003. doi: 10.1021/js990072s. [DOI] [PubMed] [Google Scholar]
- Lewin M, Carlesso N, Tung CH, Tang XW, Cory D, Scadden DT, Weissleder R. Tat peptide-derivatized magnetic nanoparticles allow in vivo tracking and recovery of progenitor cells. Nat Biotechnol. 2000;18:410–414. doi: 10.1038/74464. [DOI] [PubMed] [Google Scholar]
- Liu G, Li D, Pasumarthy MK, Kowalczyk TH, Gedeon CR, Hyatt SL, Payne JM, Miller TJ, Brunovskis P, Fink TL, Muhammad O, Moen RC, Hanson RW, Cooper MJ. Nanoparticles of compacted DNA transfect postmitotic cells. J Biol Chem. 2003;278:32578–32586. doi: 10.1074/jbc.M305776200. [DOI] [PubMed] [Google Scholar]
- Meyer DL, Schultz J, Lin Y, Henry A, Sanderson J, Jackson JM, Goshorn S, Rees AR, Graves SS. Reduced antibody response to streptavidin through site-directed mutagenesis. Protein Sci. 2001;10:491–503. doi: 10.1110/ps.19901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panyim S, Chalkley R. High resolution acrylamide gel electrophoresis of histones. Arch Biochem Biophys. 1969;130:337–346. doi: 10.1016/0003-9861(69)90042-3. [DOI] [PubMed] [Google Scholar]
- Peetla C, Rao KS, Labhasetwar V. Relevance of biophysical interactions of nanoparticles with a model membrane in predicting cellular uptake: study with TAT peptide-conjugated nanoparticles. Mol Pharm. 2009;6:1311–1320. doi: 10.1021/mp900011h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plank C, Mechtler K, Szoka FC, Jr, Wagner E. Activation of the complement system by synthetic DNA complexes: a potential barrier for intravenous gene delivery. Hum Gene Ther. 1996;7:1437–1446. doi: 10.1089/hum.1996.7.12-1437. [DOI] [PubMed] [Google Scholar]
- Rimann M, Lühmann T, Textor M, Guerino B, Ogier J, Hall H. Characterization of PLL-g-PEG-DNA nanoparticles for the delivery of therapeutic DNA. Bioconjug Chem. 2008;19:548–557. doi: 10.1021/bc7003439. [DOI] [PubMed] [Google Scholar]
- Schultz J, Lin Y, Sanderson J, Zuo Y, Stone D, Mallett R, Wilbert S, Axworthy D. A tetravalent single-chain antibody-streptavidin fusion protein for pretargeted lymphoma therapy. Cancer Res. 2000;60:6663–6669. [PubMed] [Google Scholar]
- Suk JS, Suh J, Choy K, Lai SK, Fu J, Hanes J. Gene delivery to differentiated neurotypic cells with RGD and HIV Tat peptide functionalized polymeric nanoparticles. Biomaterials. 2006;27:5143–5150. doi: 10.1016/j.biomaterials.2006.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W, Davis PB. Reducible DNA nanoparticles enhance in vitro gene transfer via an extracellular mechanism. J Control Release. 2010;146:118–127. doi: 10.1016/j.jconrel.2010.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torchilin VP, Levchenko TS, Rammohan R, Volodina N, Papahadjopoulos-Sternberg B, D’Souza GG. Cell transfection in vitro and in vivo with nontoxic TAT peptide-liposome-DNA complexes. Proc Natl Acad Sci USA. 2003;100:1972–1977. doi: 10.1073/pnas.0435906100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toro A, Grunebaum E. TAT-mediated intracellular delivery of purine nucleoside phosphorylase corrects its defficiency in mice. J Clin Invest. 2006;116:2717–2726. doi: 10.1172/JCI25052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend SA, Evrony GD, Gu FX, Schulz MP, Brown RH, Jr, Langer R. Tetanus toxin C fragment-conjugated nanoparticles for targeted drug delivery to neurons. Biomaterials. 2007;28:5176–5184. doi: 10.1016/j.biomaterials.2007.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward CM, Pechar M, Oupicky D, Ulbrich K, Seymour LW. Modification of pLL/DNA complexes with a multivalent hydrophilic polymer permits folate-mediated targeting in vitro and prolonged plasma circulation in vivo. J Gene Med. 2002;4:536–547. doi: 10.1002/jgm.296. [DOI] [PubMed] [Google Scholar]
- Wu GY, Wu CH. Receptor-mediated in vitro gene transformation by a soluble DNA carrier system. J Biol Chem. 1987;262:4429–4432. [PubMed] [Google Scholar]
- Yurek DM, Fletcher AM, Smith GM, Seroogy KB, Ziady AG, Molter J, Kowalczyk TH, Padegimas L, Cooper MJ. Long-term transgene expression in the central nervous system using DNA nanoparticles. Mol Ther. 2009;17:641–650. doi: 10.1038/mt.2009.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziady AG, Gedeon CR, Miller T, Quan W, Payne JM, Hyatt SL, Fink TL, Muhammad O, Oette S, Kowalczyk T, Pasumarthy MK, Moen RC, Cooper MJ, Davis PB. Transfection of airway epithelium by stable PEGylated poly-L-lysine DNA nanoparticles in vivo. Mol Ther. 2003a;8:936–947. doi: 10.1016/j.ymthe.2003.07.007. [DOI] [PubMed] [Google Scholar]
- Ziady AG, Gedeon CR, Muhammad O, Stillwell V, Oette SM, Fink TL, Quan W, Kowalczyk TH, Hyatt SL, Payne J, Peischl A, Seng JE, Moen RC, Cooper MJ, Davis PB. Minimal toxicity of stabilized compacted DNA nanoparticles in the murine lung. Mol Ther. 2003b;8:948–956. doi: 10.1016/j.ymthe.2003.09.002. [DOI] [PubMed] [Google Scholar]