

Abstract
Introduction
An increased understanding of cellular signaling pathways, like the JAK--STAT pathway, and the identification of the JAK2 V617F mutation in the classic Philadelphia chromosome-negative myeloproliferative neoplasms (MPNs), has generated great interest in the development of targeted JAK2 inhibitors. In a recently completed Phase I--II study, ruxolitinib, a selective orally available JAK1 and JAK2 inhibitor, has shown efficacy in patients with advanced myelofibrosis. Constitutive activation of the JAK--STAT pathway has also been implicated in other hematological malignancies suggesting a potential role of JAK kinase inhibitors in these malignancies.
Areas covered
This article reviews the chemistry, pharmacodynamics, pharmacokinetics, clinical efficacy, safety and tolerability of ruxolitinib. The literature for this article was retrieved from PubMed database searches using the keywords ‘ruxolitinib’, ‘INCB 018424’, ‘JAK2 inhibitors’ and ‘leukemia’.
Expert opinion
The JAK--STAT signaling pathway plays a vital role in leukemogenesis. Ruxolitinib, a potent JAK1 and JAK2 inhibitor, known to decrease spleen size and alleviate constitutional symptoms in myelofibrosis, represents a potentially promising agent for the treatment of leukemias by inhibiting the JAK--STAT signaling. Further studies of ruxolitinib, in patients with acute and chronic leukemias, are now needed to establish the clinical usefulness of this promising drug.
Keywords: INCB 018424, JAK2 inhibitors, leukemia, ruxolitinib
1. Introduction
Leukemias are a group of clonal stem cell disorders that can be broadly classified into myeloid and lymphoid lineages and further into acute and chronic subtypes. In adults, acute myeloid leukemia (AML) is the most common acute leukemia, accounting for approximately 80% of the cases [1]. The median age at diagnosis is 65 years. Most cases of AML are idiopathic, but a significant number of cases are secondary to prior chemotherapy or radiotherapy, or arise from prior disorders such as myelodysplastic syndromes (MDS) or myeloproliferative neoplasms (MPNs) [2]. Acute lymphoblastic leukemia (ALL), on the other hand, is more common in children [3–5]. The median age of adults with ALL at diagnosis is 39 years [6]. Chemotherapy remains the standard of care for the treatment of acute leukemias. Although chronic leukemias such as chronic myeloid leukemia (CML) have a more protracted course, they can transform into more aggressive, immediately life-threatening disorders requiring effective treatment [7]. Over the past several decades, improvements in chemotherapeutic agents and supportive care have resulted in significant progress in treating patients with acute and chronic leukemias. In addition, better description of the molecular abnormalities that occur in leukemic cells has resulted in the identification of several new agents with potential therapeutic activity in patients with hematological neoplasms including acute and chronic leukemias, MPNs and MDS [8]. Good examples are all-trans-retinoic acid for the treatment of acute promyelocytic leukemia and imatinib mesylate for CML. These agents, through targeting specific molecular aberrations important in the pathogenesis of the leukemia, have demonstrated great efficacy and a better toxicity profile than prior treatments. Despite this progress, the majority of adult patients with acute leukemia still die from the disease and its complications. For patients with AML who fail to achieve a complete remission (CR) with the first attempts at induction or who have relapsed disease, current therapy is inadequate with a true long-term cure rate of < 10% [9]. The same is true for patients with ALL [10]. Newer therapeutic strategies are therefore needed.
1.1 Present treatment guidelines
The primary goal of treatment in patients with leukemia is the eradication of disease from the bone marrow and restoration of normal hematopoiesis. Chemotherapy is the main modality of treatment. These differ depending on the type of leukemia. In AML, cytarabine- and anthracycline-based regimens are the mainstay of the treatment [11]. Depending on the age and patient selection, approximately 60 – 80% of the patients achieve a CR with such regimens [12–14]. In ALL, multiple induction regimens have been developed based on pediatric regimens. Most standard regimens consist of vincristine, l-asparaginase, an anthracycline and a glucocorticoid [15–21]. With standard protocols, 85% of the adults with ALL achieve CR with a median duration of remission of 15 months and ultimate cure rate of 30 – 40% [22,23]. However, acute leukemias remain challenging with a high incidence of relapse despite post-remission treatment. It is likely that we have reached the full potential of conventional cytotoxic chemotherapeutic agents used in the treatment of leukemias and that further progress is likely to be achieved only using risk-adapted approaches incorporating more disease-specific agents designed based on the molecular biology of the disease.
1.2 Compounds in development
This article is a review of the new JAK2 inhibitor, ruxolitinib, and its potential role in the treatment of leukemia (Box 1). Ruxolitinib is an inhibitor of the Janus kinase family of protein tyrosine kinases (JAKs) that is currently being evaluated for the treatment of MPNs. The discovery of a recurrent activating tyrosine kinase mutation known as JAK2 V617F in MPNs has raised the specter of the role of tyrosine kinases in the pathogenesis of MPNs and the potential for therapeutic efficacy of their inhibitors [24–26]. Humans express > 500 protein kinases broadly classified into tyrosine, serine or threonine protein kinases [27]. These are further classified into receptor and cytoplasmic protein kinases [27,28]. The Janus family of kinases is among the cytoplasmic protein tyrosine kinases and consists of JAK1, JAK2, JAK3 and Tyk2 [29]. These play a major role in the transmission of signals from cytokine and growth factor receptors into the nucleus [30,31]. JAK proteins activate several intracellular signaling proteins, among which STATs are the best defined [32]. The STAT transcription factors are coded by six known mammalian genes [32,33]. Their activation through tyrosine phosphorylation results in their dimerization and translocation into the nucleus where they activate specific genes [34–36]. STATs mediate diverse cellular events that affect cell growth, differentiation and apoptosis [37,38]. For example, STAT1 mediates the growth-inhibitory effects of gamma interferon, through induction of the CDKI p21waf1, whereas STAT5 mediates proliferative effects of IL-3 and GM-CSF [39,40]. Similarly, phosphorylation of STAT3 can result in both IL-6- and IL-10-induced growth arrest and GM-CSF- and IL-3-induced proliferation [40–42].
Box 1. Drug summary.
| Drug name (generic) | Ruxolitinib (INCB018424) |
| Phase | Currently in Phase III |
| Indication | Treatment of relapsed/refractory leukemia |
| Pharmacology description/mechanism of action | Selective inhibitor of JAK1 and JAK2 |
| Route of administration | Oral |
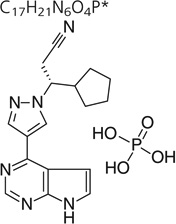
| Chemical structure | C17H21N6O4P* |
|
|
| Pivotal trial(s) | Patients with either primary or post-ET and post-PV myelofibrosis (NCT:00952289) |
| Patients with polycythemia that are hydroxyurea resistant, refractory, or intolerant (NCT:01243944) |
ET: Essential thrombocythemia; PV: Polycythemia vera.
Abnormal activation of JAK2 seems to play a vital role in various hematological malignancies. The V617F mutation occurs in the pseudokinase domain of JAK2 (amino acid 617, valine to phenylalanine) resulting in the impaired ability of mutated pseudokinase domain to negatively regulate the kinase domain (the active part of JAK2) [43,44]. The result is the unchecked JAK2 activation causing uncontrolled cytokine and growth factor signaling believed to play a major role in the pathophysiology of MPNs [24,26,45–46]. The JAK2 V617F mutation is seen in approximately 95% of the patients with polycythemia vera (PV) and in 50 – 60% patients with essential thrombocythemia (ET) and primary myelofibrosis (PMF) [24,26,45–47]. In addition to JAK2 V617F mutation, other mutations have also been discovered that abnormally activate JAK2. Recurring abnormalities in the short arm of chromosomes 9 and 12 are commonly seen (7 and 15%) in childhood ALLs [48,49]. Several studies have shown the presence of translocation t(9;12)(p24;p13) in childhood ALL that results in the fusion of the 3′ portion of JAK2 to the 5′ portion of TEL (gene encoding a member of the ETS transcription factor family). TEL-JAK2 constructs result in constitutive activation of the tyrosine kinase activity of JAK2 causing an IL3-independent cellular proliferation of the Ba/F3 hematopoietic cell line by STAT5 [50,51]. Recently, Ikezoe and colleagues have shown the constitutive expression of p-JAK2 in AML cells. They report the elevated levels of p-JAK2 to be directly correlated with high white blood cell count, low platelet count, lower CR rates and a poor overall survival in AML (both de novo and secondary). They have also provided evidence that the inhibition of JAK2 in such patients results in the downregulation of p-JAK2 levels. This causes a decline in the levels of p-STAT5 and p-STAT5-dependent activation of Bcl-xL, an anti-apoptotic protein resulting in an inhibition of clonogenic growth of AML cells [52]. In a separate study, the same group has shown that the inhibition of JAK2/STAT5 signaling stimulates cell cycling in CD34+/CD38− cells in association with the downregulation of p21waf1, sensitizing these cells to cytarabine-mediated growth inhibition [53]. Pradhan et al. described the overexpression of IL-27R (a type 1 cytokine receptor) on the surface of AML cells. In response to IL-27, the AML cells show high levels of various signaling proteins, including JAK1 and JAK2. Inhibition of JAK proteins induces cell cycle arrest and apoptosis in these cells [54].
Several studies have also demonstrated constitutive activation of JAK--STAT pathway in CML cells [55]. While resistance to the BCR--ABL tyrosine kinase inhibitors such as imatinib can arise from mutations in the drug-binding site, previous studies have demonstrated that cytokine signaling from the microenvironment can allow tumor cells to overcome drug inhibition [56–58]. Wang et al. has demonstrated that GM-CSF (which also signals using the JAK--STAT pathway) could induce resistance to the cytotoxic and cytostatic effects of nilotinib without impacting the ability of the compound to inhibit its target kinase [55]. Therefore, aberrant activation of the JAK--STAT pathway has been described in a variety of leukemias and its inhibition can be a goal for leukemia therapy.
A number of JAK2 inhibitors have been discovered and are currently being evaluated for their activity in hematological malignancies, in particular MPNs. It is important to recognize that the V617F mutation is localized outside the ATP-binding pocket of the JAK2 enzyme [25,59]. Hence ATP-competitive inhibitors of the enzyme are not likely to differentiate between the mutated and the wild-type JAK2 enzymes. Unlike the endogenous ABL kinase that has no indispensable function in hematopoiesis and its inhibition by BCR--ABL inhibitors in CML causes no mechanism-related adverse effects, inhibition of the wild-type JAK2 enzyme by JAK2 inhibitors results in the inhibition of normal hematopoiesis especially thrombopoietin signaling that requires JAK2 [25]. Myelosuppression, predominantly thrombocytopenia and anemia, is therefore an expected adverse effect of JAK2 inhibitors if administered at doses that aim to inhibit the mutant enzyme.
Reports of three JAK2 inhibitors have been published so far in peer-reviewed publications. As described in detail in this review, ruxolitinib, a selective JAK1 and JAK2 inhibitors, has been the first to be evaluated in PMF and post ET/PV myelofibrosis. This compound has demonstrated a ≥ 50% reduction in the size of palpable spleen as well as improvement of constitutional symptoms namely fatigue, weakness and weight loss, in patients with myelofibrosis [60]. CEP-701, a derivative of the indolocarbazole K252, is a staurosporine analog that inhibits JAK2. CEP-701 has been evaluated in patients with myelofibrosis with the JAK2 V617F mutation, at an established dose of 80 mg twice/day [61]. CEP-701 reduced the spleen size in a limited number of patients; its efficacy might have been compromised due to its poor tolerance, as majority of patients experience gastrointestinal toxicity (nausea, vomiting and/or diarrhea) [62]. TG101348 is a selective and potent inhibitor of JAK2 [63,64]. It has been evaluated in Phase I/II study in patients with myelofibrosis and has shown to cause reduction in the spleen size, marked reduction in platelet and leukocyte count and improvements in patients’ symptoms, but with significant myelosuppression and gastrointestinal toxicity [65].
2. Introduction to the compound
Ruxolitinib (INCB 018424) is a potent, selective and orally bioavailable inhibitor of JAK1 and JAK2 with an IC50 of 3.3 and 2.8 nM, respectively. Since abnormalities of the JAK--STAT pathways have been described in a variety of leukemias, the role of ruxolitinib in the treatment of several leukemias is currently ongoing (see Box 1).
2.1 Chemistry
Ruxolitinib or INCB 018424 phosphate is (R)-3-(4-(7H-pyrrolo[2, 3–d]pyrimidin-4-yl)-1H-pyrazol-1-yl)-3-cyclopentylpropanenitrile phosphate. The molecular formula of this compound is C17H21N6O4P and it has a molecular weight of 404.36 g/mol. The drug substance is a white to off-white powder. The drug has shown to be stable for up to 6 months at 40° C/75% RH and up to 12 months when stored at 25°C/60% RH. Stability studies for 5 and 25 mg capsule formulations were conducted at 25°C/60% RH and 40°C/75% RH. The capsule formulations have shown to be stable when stored up to 6 months at 40°C/75% RH and 9 months when stored at 25°C/60% RH.
2.2 Pharmacodynamics
The first-in-human study conducted evaluated single oral doses of ruxolitinib of 5, 10, 25, 50, 100 and 200 mg in healthy volunteers (INCB 018424-131) [66]. Following fasting, oral single-dose administration, ruxolitinib demonstrated dose- and time-dependent inhibition of cytokine-induced phosphorylated STAT3 (pSTAT3) with maximal inhibition occurring 1 – 2 h after the administration of all doses, coincident with the Cmax. Maximal mean inhibition of pSTAT3 ranged from approximately 40% at the lowest dose (5 mg) to greater than 90% inhibition at the highest dose (200 mg). Levels of pSTAT3 returned to control levels by 24 h in all instances. Similar effects on STAT3 phosphorylation were detected in patients with myelofibrosis who were treated with ruxolitinib in Phase I/II study (INCB018424-251) [60]. Prior to treatment with the drug, patients with myelofibrosis had elevated levels of pSTAT3 as well as elevated response to IL-6 stimulation. Following the administration of the drug for 28 days, there was normalization of JAK signaling as measured in whole blood by pSTAT3 levels. No pharmacological evidence of drug accumulation or drug resistance was observed.
2.3 Pharmacokinetics
Following fasting, oral single-dose administration of ruxolitinib capsules, ruxolitinib was absorbed rapidly, typically attaining peak plasma concentrations within 2 h after the administration of all doses [66]. After attaining the Cmax, ruxolitinib plasma concentrations declined in a multiphasic fashion with a mean terminal-phase disposition t1/2 of approximately 3 h for the five lowest doses. The mean terminal phase disposition t1/2 was slightly higher at 5 h for the highest dose of 200 mg. The oral dose clearance (CL/F) of ruxolitinib was low (~ 20 L/h) and dose independent (p = .895). Similarly, the evaluation of 14C-labeled ruxolitinib was conducted by Shilling et al. in six healthy human subjects by administering a single oral 25 mg dose [67]. The study showed rapid absorption of the parent drug and radioactivity attained peak concentrations within 1 – 2 h after administration. The parent drug plasma concentration and the radioactivity declined in a monophasic or biphasic fashion with a mean observed terminal half-life of 2.3 and 5.8 h, respectively. The mean Cmax and AUC0 – ∞ for the parent drug were 1093 nM and 3200 nM h, respectively. The recovery of the administered radioactivity was fairly rapid (> 70% within 24 h post-dose) with 74 and 22% recovered in the urine and feces, respectively. The parent drug was the predominant entity in the circulation, representing 58 – 74% of the total radioactivity up to an hour after administration. In vitro studies have also shown ruxolitinib-induced inhibition of IL-6 (IC50 = 281 nM) and proliferation of JAK2 V617F + Ba/F3 cells (IC50 = 127 nM) [68].
2.4 Clinical efficacy
2.4.1 Role in myelofibrosis
Verstovsek and colleagues conducted a Phase I/II study evaluating the role of ruxolitinib in myelofibrosis [60]. The Phase I dose escalation part of the study used a starting dose of 25 mg twice/day followed by escalation to 50 mg twice/day. Once daily dosing from 25 to 200 mg was also evaluated. Thrombocytopenia was identified as the main dose-limiting toxicity. The maximum tolerated doses were identified as 25 mg twice/day and 100 mg once/day. A dose-dependent suppression of phosphorylated signal transducer and activator of transcription 3 (STAT3) was noted, irrespective of the JAK2 mutation status. Additional doses of ruxolitinib were also studied. A 15 mg twice-daily starting dose followed by individualized dose titration was established as the most effective and safest dosing regimen. Besides the reduction in splenomegaly and alleviation of debilitating symptoms like fatigue, weight loss, night sweats and pruritus were the major clinical benefits (Table 1).
Table 1.
Clinical trials with Ruxolitinib in hematological malignancies [71].
| Phase | Disease | Status |
|---|---|---|
| I | Myelofibrosis with low platelets (50,000 – 100,000) | Accruing patients |
| II | Myelofibrosis: sustained release formulation | Accruing patients |
| I/II | Myelofibrosis | Complete [60] |
| II | Polycythemia vera and essential thrombocythemia | Accrual complete; data to be published |
| III (COMFORT-I) | Myelofibrosis | Accrual complete; data to be published |
| III (COMFORT-II) | Myelofibrosis | Accrual complete; data to be published |
| II | Advance hematological malignancies; AML, ALL, MDS, CML blast crisis | Accruing patients |
| I | Acute leukemia | Accruing patients |
| III | Polycythemia vera | Accruing patients |
| I | CML, leukemia, MDS, MDS/MPN, childhood solid tumor | Accruing patients |
ALL: Acute lymphoblastic leukemia; AML: Acute myeloid leukemia; CML: Chronic myeloid leukemia; MDS: Myelodysplastic syndromes; MPN: Myeloproliferative neoplasms.
2.4.2 Role in other hematological malignancies
Ravandi et al. conducted a Phase II study using oral ruxolitinib in patients with relapsed/refractory AML, ALL, MDS (including chronic myelomonocytic leukemia (CMML)) and blast phase or tyrosine kinase refractory CML [69]. Ruxolitinib was administered at a dose of 25 mg by mouth twice/day, the maximum tolerated dose obtained from the Phase I dose escalation study in patients with myelofibrosis. Patients were evaluated after one full cycle of therapy (28 days) for response. In responding patients or those with stable disease, therapy was to be continued at the same dose until disease progression. Treatment was to be held in patients with grade 3 or 4 toxicity until they recover to grade 1 or lower and then be restarted at a lower dose of 10 or 15 mg by mouth twice/day. In the event that there was no recovery in 2 weeks, the treatment was to be completely stopped. Patients were then reevaluated after two cycles of therapy and, at the discretion of the treating physician, those who had progressive disease were either removed from the study or their dose escalated to 50 mg twice/day.
A total of 23 patients enrolled in the study with a median age of 68 years. Eight patients had de novo AML, 8 patients had AML secondary to underlying MPN, 2 patients had ALL, 4 had MDS (including 2 patients with CMML) and 1 had CML. JAK2 V617F was detected in eight patients, six of whom had secondary AML (post MF). A total of eight patients (35%) showed clinical benefit (complete remission (CR), partial remission (PR) and stable disease (SD)) to therapy with ruxolitinib, four with secondary AML, three with MDS and one with Philadelphia + ALL. Out of the eight patients showing clinical benefit, one patient achieved CR, one achieved PR, while the remaining six patients had SD over a median of four cycles (range 2 – 8) of therapy. Five of these eight patients had JAK2 V617Fmutation. Data on allele burden post-therapy were available only in three patients, with no significant decline in the percentage of cells with mutated JAK2 V617F after treatment. The majority of patients evaluated, and particularly those with JAK2 V617F mutation, demonstrated elevated baseline pSTAT3 levels measured in whole blood. The levels of pSTAT3 declined substantially 2 h post dosing with ruxolitinib indicating its on-target effect on the JAK--STAT pathway. The drug was overall well tolerated with only four patients developing grade 3 – 4 toxicity while on therapy. This study suggests the potential activity of ruxolitinib in patients with leukemia.
More recently, Eghtedar et al. reported an update of the study now including a total of 38 patients: 18 with secondary AML with underlying MPN, 10 with de novo AML, 5 with MDS (including 4 patients with CMML), 3 with ALL and 2 with CML [70]. JAK2 V617F was noted in 12 patients: 7 with secondary AML, 3 with de novo AML and 2 with MDS. Clinical benefit was noted in 15 patients (39%): 2 with CR, 1 with PR and 12 with SD. The three patients with CR and PR (11%) had post-MPN AML and two patients had JAK2 V617F. A decrease in the spleen size was noted in all the three patients on therapy. Patients with SD included seven with secondary AML, four with MDS and one with de novo AML. There was no suppression of the JAK2 V617F noted in the study. The toxicity profile remained unchanged. Both the original and the updated study showed benefit predominantly in patients with leukemia arising from MPNs.
2.5 Safety and tolerability
Non-hematological toxicity was seen infrequently (5% of the patients). The drug was overall well tolerated with only four patients developing grade 3 – 4 toxicity while on therapy, one with thrombocytopenia, one with neutropenia, one with elevated AST/ALT and one with CNS hemorrhage. On study extension, the toxicity profile remained unchanged with thrombocytopenia remaining the most common toxicity (8% of the total study group).
2.6 Conclusion
Preliminary evidence suggests a potential therapeutic role of ruxolitinib in the treatment of hematological malignancies other than MPNs. Achievement of responses in a minority of patients suggests that the drug may have activity in these disorders. Factors other than JAK2 V617F mutation such as high circulating cytokines or silencing of negative regulators of JAK signaling are known to result in dysregulated JAK signaling. Activation of JAK--STAT pathway and response to ruxolitinib were reported in myelofibrosis patients who were positive or negative for JAK2 V617F mutation and hence lack of correlation between the response and the expression of mutated JAK2 is not surprising.
3. Expert opinion
Despite the progress in leukemia therapy, most adult patients with leukemia still die from disease progression or from therapy-associated complications. This emphasizes the need for better, more targeted and less toxic treatments. The discovery of the JAK family of enzymes and the JAK2 V617F mutation seen in MPNs has generated interest in the development of JAK2-targeted therapies. Constitutive activation of the JAK--STAT pathway has been reported in various leukemias as well, suggesting that targeted therapies directed at this pathway may show clinical benefit. Several JAK2 inhibitors are in development. Ruxolitinib is the first oral JAK1 and JAK2 inhibitor evaluated in primary myelofibrosis and post ET/PV myelofibrosis. In our view, ruxolitinib represents a potential agent for relapsed/refractory leukemias particularly those with a prior history of MPN. A starting dose of 15 mg twice/day with individualized dose escalation was found to be the most effective and the safest dose of ruxolitinib. The drug produced rapid and sustained reduction of splenomegaly with alleviation of constitutional symptoms, like fatigue, weight loss and improvement of performance status [60]. Thrombocytopenia, an expected adverse effect of the drug, was the main dose-limiting toxicity. Non-hematological toxicity was infrequent, seen in 5% patients and of low grade [69]. No direct effect on malignant clone was described in patients treated with ruxolitinib so far, probably due to its lack of specificity for JAK2 V617F mutation and limitations in how much the drug can be given due to undesirable myelosuppression. Further studies of ruxolitinib in patients with acute and chronic leukemias are needed to establish clinical usefulness of this drug.
Acknowledgments
F Ravandi has received research funding from Incyte and honoraria from Novartis. S Verstovsek has received research funding from Incyte. H Kantarjian has research funding from Novartis.
Footnotes
Declaration of interest
K Naqvi has nothing to disclose.
Bibliography
- 1.Yamamoto JF, Goodman MT. Patterns of leukemia incidence in the United States by subtype and demographic characteristics, 1997–2002. Cancer Causes Control. 2008;19(4):379–390. doi: 10.1007/s10552-007-9097-2. [DOI] [PubMed] [Google Scholar]
- 2.Lowenberg B, Downing JR, Burnett A. Acute myeloid leukemia. N Engl J Med. 1999;341(14):1051–1062. doi: 10.1056/NEJM199909303411407. [DOI] [PubMed] [Google Scholar]
- 3.Greenlee RT, Hill-Harmon MB, Murray T, Thun M. Cancer statistics, 2001. CA Cancer J Clin. 2001;51(1):15–36. doi: 10.3322/canjclin.51.1.15. [DOI] [PubMed] [Google Scholar]
- 4.Cortes JE, Kantarjian HM. Acute lymphoblastic leukemia. A comprehensive review with emphasis on biology and therapy. Cancer. 1995;76(12):2393–2417. doi: 10.1002/1097-0142(19951215)76:12<2393::aid-cncr2820761203>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 5.Reis LA, Smith MA, Gurney JG, et al. Cancer incidence and survival among children and adolescents: United States SEER Program 1975–1995. National Cancer Institute, Bethesda. 1999 [Google Scholar]
- 6.Soslow RA, Baergen RN, Warnke RA. B-lineage lymphoblastic lymphoma is a clinicopathologic entity distinct from other histologically similar aggressive lymphomas with blastic morphology. Cancer. 1999;85(12):2648–2654. [PubMed] [Google Scholar]
- 7.Jabbour E, Kantarjian H, O’Brien S, et al. Sudden blastic transformation in patients with chronic myeloid leukemia treated with imatinib mesylate. Blood. 2006;107(2):480–482. doi: 10.1182/blood-2005-05-1816. [DOI] [PubMed] [Google Scholar]
- 8.Estey EH, Thall PF. New designs for phase 2 clinical trials. Blood. 2003;102(2):442–448. doi: 10.1182/blood-2002-09-2937. [DOI] [PubMed] [Google Scholar]
- 9.Ravandi F, Kantarjian H, Giles F, Cortes J. New agents in acute myeloid leukemia and other myeloid disorders. Cancer. 2004;100(3):441–454. doi: 10.1002/cncr.11935. [DOI] [PubMed] [Google Scholar]
- 10.Faderl S, Jeha S, Kantarjian HM. The biology and therapy of adult acute lymphoblastic leukemia. Cancer. 2003;98(7):1337–1354. doi: 10.1002/cncr.11664. [DOI] [PubMed] [Google Scholar]
- 11.Farag SS, Ruppert AS, Mrozek K, et al. Outcome of induction and postremission therapy in younger adults with acute myeloid leukemia with normal karyotype: a cancer and leukemia group B study. J Clin Oncol. 2005;23(3):482–493. doi: 10.1200/JCO.2005.06.090. [DOI] [PubMed] [Google Scholar]
- 12.Stone RM, Mayer RJ. Treatment of the newly diagnosed adult with de novo acute myeloid leukemia. Hematol Oncol Clin North Am. 1993;7(1):47–64. [PubMed] [Google Scholar]
- 13.Mayer RJ, Davis RB, Schiffer CA, et al. Intensive postremission chemotherapy in adults with acute myeloid leukemia. Cancer and Leukemia Group B. N Engl J Med. 1994;331(14):896–903. doi: 10.1056/NEJM199410063311402. [DOI] [PubMed] [Google Scholar]
- 14.Bishop JF. The treatment of adult acute myeloid leukemia. Semin Oncol. 1997;24(1):57–69. [PubMed] [Google Scholar]
- 15.Laport GF, Larson RA. Treatment of adult acute lymphoblastic leukemia. Semin Oncol. 1997;24(1):70–82. [PubMed] [Google Scholar]
- 16.Hoelzer D. Treatment of acute lymphoblastic leukemia. Semin Hematol. 1994;31(1):1–15. [PubMed] [Google Scholar]
- 17.Preti A, Kantarjian HM. Management of adult acute lymphocytic leukemia: present issues and key challenges. J Clin Oncol. 1994;12(6):1312–1322. doi: 10.1200/JCO.1994.12.6.1312. [DOI] [PubMed] [Google Scholar]
- 18.Copelan EA, McGuire EA. The biology and treatment of acute lymphoblastic leukemia in adults. Blood. 1995;85(5):1151–1168. [PubMed] [Google Scholar]
- 19.Hoelzer D, Gokbuget N. New approaches to acute lymphoblastic leukemia in adults: where do we go? Semin Oncol. 2000;27(5):540–559. [PubMed] [Google Scholar]
- 20.Takeuchi J, Kyo T, Naito K, et al. Induction therapy by frequent administration of doxorubicin with four other drugs, followed by intensive consolidation and maintenance therapy for adult acute lymphoblastic leukemia: the JALSG-ALL93 study. Leukemia. 2002;16(7):1259–1266. doi: 10.1038/sj.leu.2402526. [DOI] [PubMed] [Google Scholar]
- 21.Storring JM, Minden MD, Kao S, et al. Treatment of adults with BCR-ABL negative acute lymphoblastic leukaemia with a modified paediatric regimen. Br J Haematol. 2009;146(1):76–85. doi: 10.1111/j.1365-2141.2009.07712.x. [DOI] [PubMed] [Google Scholar]
- 22.Kantarjian HM, O’Brien S, Smith T, et al. Acute lymphocytic leukaemia in the elderly: characteristics and outcome with the vincristine-adriamycin-dexamethasone (VAD) regimen. Br J Haematol. 1994;88(1):94–100. doi: 10.1111/j.1365-2141.1994.tb04982.x. [DOI] [PubMed] [Google Scholar]
- 23.Kantarjian HM, Walters RS, Keating MJ, et al. Experience with vincristine, doxorubicin, and dexamethasone (VAD) chemotherapy in adults with refractory acute lymphocytic leukemia. Cancer. 1989;64(1):16–22. doi: 10.1002/1097-0142(19890701)64:1<16::aid-cncr2820640104>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 24.Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352(17):1779–1790. doi: 10.1056/NEJMoa051113. [DOI] [PubMed] [Google Scholar]
- 25.Verstovsek S. Therapeutic potential of JAK2 inhibitors. Hematology Am Soc Hematol Educ Program. 2009:636–642. doi: 10.1182/asheducation-2009.1.636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.James C, Ugo V, Le Couedic JP, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434(7037):1144–1148. doi: 10.1038/nature03546. [DOI] [PubMed] [Google Scholar]
- 27.Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature. 2001;411(6835):355–365. doi: 10.1038/35077225. [DOI] [PubMed] [Google Scholar]
- 28.Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103(2):211–225. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- 29.Campbell PJ, Green AR. The myeloproliferative disorders. N Engl J Med. 2006;355(23):2452–2466. doi: 10.1056/NEJMra063728. [DOI] [PubMed] [Google Scholar]
- 30.Vainchenker W, Dusa A, Constantinescu SN. JAKs in pathology: role of Janus kinases in hematopoietic malignancies and immunodeficiencies. Semin Cell Dev Biol. 2008;19(4):385–393. doi: 10.1016/j.semcdb.2008.07.002. [DOI] [PubMed] [Google Scholar]
- 31.Ghoreschi K, Laurence A, O’Shea JJ. Janus kinases in immune cell signaling. Immunol Rev. 2009;228(1):273–287. doi: 10.1111/j.1600-065X.2008.00754.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Darnell J., Jr STATs and gene regulation. Science (Wash DC) 1997;277:1630–1635. doi: 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- 33.Rane SG, Reddy EP. JAKs, STATs and Src kinases in hematopoiesis. Oncogene. 2002;21(21):3334–3358. doi: 10.1038/sj.onc.1205398. [DOI] [PubMed] [Google Scholar]
- 34.Darnell J, Jr, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science (Wash DC) 1994;264:1415–1421. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- 35.Fu XY, Schindler C, Improta T, et al. The proteins of ISGF-3, the interferon alpha-induced transcriptional activator, define a gene family involved in signal transduction. Proc Natl Acad Sci USA. 1992;89(16):7840–7843. doi: 10.1073/pnas.89.16.7840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Khan KD, Shuai K, Lindwall G, Jr, Bothwell AL, et al. Induction of the Ly-6A/E gene by interferon alpha/beta and gamma requires a DNA element to which a tyrosine-phosphorylated 91-kDa protein binds. Proc Natl Acad Sci USA. 1993;90(14):6806–6810. doi: 10.1073/pnas.90.14.6806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schindler C. STATs as activators of apoptosis. Trends Cell Biol. 1998;8(3):97–98. doi: 10.1016/s0962-8924(98)01233-1. [DOI] [PubMed] [Google Scholar]
- 38.Mui AL. The role of STATs in proliferation, differentiation, and apoptosis. Cell Mol Life Sci. 1999;55(12):1547–1558. doi: 10.1007/s000180050394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chin Y, Kitagawa M, Su WC, et al. Cell growth arrest and induction of cyclin-dependent kinase inhibitor p21 WAF1/CIP1 mediated by STAT1. Science (Wash DC) 1996;272:719–722. doi: 10.1126/science.272.5262.719. [DOI] [PubMed] [Google Scholar]
- 40.Mui AL, Wakao H, Kinoshita T, et al. Suppression of interleukin-3-induced gene expression by a C-terminal truncated Stat5: role of Stat5 in proliferation. EMBO J. 1996;15(10):2425–2433. [PMC free article] [PubMed] [Google Scholar]
- 41.O’Farrell AM, Liu Y, Moore KW, Mui AL. IL-10 inhibits macrophage activation and proliferation by distinct signaling mechanisms: evidence for Stat3-dependent and -independent pathways. EMBO J. 1998;17(4):1006–1018. doi: 10.1093/emboj/17.4.1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chaturvedi P, Reddy MV, Reddy EP. Src kinases and not JAKs activate STATs during IL-3 induced myeloid cell proliferation. Oncogene. 1998;16(13):1749–1758. doi: 10.1038/sj.onc.1201972. [DOI] [PubMed] [Google Scholar]
- 43.Levine RL, Gilliland DG. Myeloproliferative disorders. Blood. 2008;112(6):2190–2198. doi: 10.1182/blood-2008-03-077966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Saharinen P, Takaluoma K, Silvennoinen O. Regulation of the Jak2 tyrosine kinase by its pseudokinase domain. Mol Cell Biol. 2000;20(10):3387–3395. doi: 10.1128/mcb.20.10.3387-3395.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Baxter EJ, Scott LM, Campbell PJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365(9464):1054–1061. doi: 10.1016/S0140-6736(05)71142-9. [DOI] [PubMed] [Google Scholar]
- 46.Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7(4):387–397. doi: 10.1016/j.ccr.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 47.Zhao R, Xing S, Li Z, et al. Identification of an acquired JAK2 mutation in polycythemia vera. J Biol Chem. 2005;280(24):22788–22792. doi: 10.1074/jbc.C500138200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Collaborative study of karyotypes in childhood acute lymphoblastic leukemias. Groupe Francais de Cytogenetique Hematologique. Leukemia. 1993;7(1):10–19. [PubMed] [Google Scholar]
- 49.Raimondi SC. Current status of cytogenetic research in childhood acute lymphoblastic leukemia. Blood. 1993;81(9):2237–2251. [PubMed] [Google Scholar]
- 50.Lacronique V, Boureux A, Valle VD, et al. A TEL-JAK2 fusion protein with constitutive kinase activity in human leukemia. Science (Wash DC) 1997;278:1309–1312. doi: 10.1126/science.278.5341.1309. [DOI] [PubMed] [Google Scholar]
- 51.Peeters P, Raynaud SD, Cools J, et al. Fusion of TEL, the ETS-variant gene 6 (ETV6), to the receptor-associated kinase JAK2 as a result of t(9;12) in a lymphoid and t(9;15;12) in a myeloid leukemia. Blood. 1997;90(7):2535–2540. [PubMed] [Google Scholar]
- 52.Ikezoe T, Kojima S, Furihata M, et al. High levels of p-JAK2 are associated with adverse clinical outcome and can be a molecular target in individuals with acute myelogenous leukemia. Int J Cancer. doi: 10.1002/ijc.25910. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 53.Ikezoe T, Yang J, Nishioka C, et al. Inhibition of signal transducer and activator of transcription 5 by the inhibitor of janus kinases stimulates dormant human leukemia CD34(+)/CD38(−) cells and sensitizes them to antileukemia agents. Int J Cancer. 2011;128(10):2317–2325. doi: 10.1002/ijc.25806. [DOI] [PubMed] [Google Scholar]
- 54.Pradhan A, Lambert QT, Reuther GW. Transformation of hematopoietic cells and activation of JAK2-V617F by IL-27R, a component of a heterodimeric type I cytokine receptor. Proc Natl Acad Sci USA. 2007;104(47):18502–18507. doi: 10.1073/pnas.0702388104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang Y, Cai D, Brendel C, et al. Adaptive secretion of granulocyte-macrophage colony-stimulating factor (GM-CSF) mediates imatinib and nilotinib resistance in BCR/ABL+ progenitors via JAK-2/STAT-5 pathway activation. Blood. 2007;109(5):2147–2155. doi: 10.1182/blood-2006-08-040022. [DOI] [PubMed] [Google Scholar]
- 56.Hariharan IK, Adams JM, Cory S. bcr-abl oncogene renders myeloid cell line factor independent: potential autocrine mechanism in chronic myeloid leukemia. Oncogene Res. 1988;3(4):387–399. [PubMed] [Google Scholar]
- 57.Jiang X, Lopez A, Holyoake T, et al. Autocrine production and action of IL-3 and granulocyte colony-stimulating factor in chronic myeloid leukemia. Proc Natl Acad Sci USA. 1999;96(22):12804–12809. doi: 10.1073/pnas.96.22.12804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang X, Ren R. Bcr-Abl efficiently induces a myeloproliferative disease and production of excess interleukin-3 and granulocyte-macrophage colony-stimulating factor in mice: a novel model for chronic myelogenous leukemia. Blood. 1998;92(10):3829–3840. [PubMed] [Google Scholar]
- 59.Cross NC, Daley GQ, Green AR, et al. BCR-ABL1-positive CML and BCR-ABL1-negative chronic myeloproliferative disorders: some common and contrasting features. Leukemia. 2008;22(11):1975–1989. doi: 10.1038/leu.2008.231. [DOI] [PubMed] [Google Scholar]
- 60.Verstovsek S, Kantarjian H, Mesa RA, et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Engl J Med. 2010;363(12):1117–1127. doi: 10.1056/NEJMoa1002028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hexner EO, Serdikoff C, Jan M, et al. Lestaurtinib (CEP701) is a JAK2 inhibitor that suppresses JAK2/STAT5 signaling and the proliferation of primary erythroid cells from patients with myeloproliferative disorders. Blood. 2008;111(12):5663–5671. doi: 10.1182/blood-2007-04-083402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Moliterno AR, Roboz GJ, Carroll M, et al. An open-label study of CEP-701 in patients with JAK2 V617F-positive polycythemia vera and essential thrombocytosis. ASH Annual Meeting Abstracts. Blood. 2008;112(11):99. [Google Scholar]
- 63.Geron I, Abrahamsson AE, Barroga CF, et al. Selective inhibition of JAK2-driven erythroid differentiation of polycythemia vera progenitors. Cancer Cell. 2008;13(4):321–330. doi: 10.1016/j.ccr.2008.02.017. [DOI] [PubMed] [Google Scholar]
- 64.Wernig G, Kharas MG, Okabe R, et al. Efficacy of TG101348, a selective JAK2 inhibitor, in treatment of a murine model of JAK2V617F-induced polycythemia vera. Cancer Cell. 2008;13(4):311–320. doi: 10.1016/j.ccr.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 65.Pardanani A, Gotlib JR, Jamieson C, et al. Safety and efficacy of TG101348, a selective JAK2 inhibitor, in myelofibrosis. J Clin Oncol. 2011;29(7):789–796. doi: 10.1200/JCO.2010.32.8021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shi JG, Chen X, McGee RF, et al. The pharmacokinetics, pharmacodynamics, and safety of orally dosed INCB018424 phosphate in healthy volunteers. J Clin Pharmacol. doi: 10.1177/0091270010389469. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 67.Shilling AD, Nedza FM, Emm T, et al. Metabolism, excretion, and pharmacokinetics of [14C]INCB018424, a selective Janus tyrosine kinase 1/2 inhibitor, in humans. Drug Metab Dispos. Nov;38(11):2023–2031. doi: 10.1124/dmd.110.033787. [DOI] [PubMed] [Google Scholar]
- 68.Quintas-Cardama A, Vaddi K, Liu P, et al. Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: therapeutic implications for the treatment of myeloproliferative neoplasms. Blood. 2010;115(15):3109–3117. doi: 10.1182/blood-2009-04-214957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ravandi F, Verstovsek S, Estrov Z, et al. Significant Activity of the JAK2 inhibitor, INCB018424 in patients with secondary, post-myeloproliferative disorder (MPD) acute myeloid leukemia (sAML): results of an exploratory phase II study. ASH Annual Meeting Abstracts. Blood. 2009;114(22):261. abstract no. 631. [Google Scholar]
- 70.Eghtedar A, Verstovsek S, Cortes JE, et al. Phase II study of the JAK2 inhibitor, INCB018424, in patients with refractory leukemias including post-myeloproliferative disorder (MPD) acute myeloid leukemia (sAML). ASH Annual Meeting Abstracts. Blood. 2010;116(21):225. abstract no. 509. [Google Scholar]
- 71. Available from: http://www.clinicaltrials.gov.