

Abstract
The subarctic region is highly responsive and vulnerable to climate change. Understanding the structure of subarctic soil microbial communities is essential for predicting the response of the subarctic soil environment to climate change. To determine the composition of the bacterial community and its relationship with soil properties, we investigated the bacterial community structure and properties of surface soil from the moist acidic tussock tundra in Council, Alaska. We collected 70 soil samples with 25-m intervals between sampling points from 0–10 cm to 10–20 cm depths. The bacterial community was analyzed by pyrosequencing of 16S rRNA genes, and the following soil properties were analyzed: soil moisture content (MC), pH, total carbon (TC), total nitrogen (TN), and inorganic nitrogen (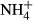 and
and 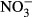 ). The community compositions of the two different depths showed that Alphaproteobacteria decreased with soil depth. Among the soil properties measured, soil pH was the most significant factor correlating with bacterial community in both upper and lower-layer soils. Bacterial community similarity based on jackknifed unweighted unifrac distance showed greater similarity across horizontal layers than through the vertical depth. This study showed that soil depth and pH were the most important soil properties determining bacterial community structure of the subarctic tundra soil in Council, Alaska.
). The community compositions of the two different depths showed that Alphaproteobacteria decreased with soil depth. Among the soil properties measured, soil pH was the most significant factor correlating with bacterial community in both upper and lower-layer soils. Bacterial community similarity based on jackknifed unweighted unifrac distance showed greater similarity across horizontal layers than through the vertical depth. This study showed that soil depth and pH were the most important soil properties determining bacterial community structure of the subarctic tundra soil in Council, Alaska.
Keywords: soil bacteria, soil depth, pH, Alaska, pyrosequencing, tussock tundra
Introduction
The Arctic region is currently receiving much attention because global warming is predicted to be the greatest and most rapid at high latitudes (IPCC, 2007). Evidence collected from the past few decades indicates that warming is already underway in the Arctic (ACIA, 2005; Chapin et al., 2005). Arctic permafrost soils contain a significant amount of soil carbon (Schlesinger, 1997; Ping et al., 2008; Tarnocai et al., 2009), and the warming effect is making soil organic carbon more vulnerable (Grosse et al., 2011). A warmer climate will cause carbon stored in the soil to be released into the atmosphere via microbial decomposition (Bardgett et al., 2008; Schuur et al., 2009). According to current climate change projections, studying microbial processes in the subarctic region is important because the area is highly responsive and vulnerable to climate change (Christensen et al., 1998; Anisimov & Fitzharris, 2001).
Understanding the soil microbial community structure is essential to elucidate microbial processes. The bacterial communities have been characterized in various arctic soil environments including subarctic regions using culture-independent techniques, such as DGGE, T-RFLP, clone libraries, and next-generation sequencing (Männistö et al., 2007; Steven et al., 2007; Wallenstein et al., 2007; Lauber et al., 2009; Margesin et al., 2009; Campbell et al., 2010; Chu et al., 2010; Larose et al., 2010; Schütte et al., 2010; Yergeau et al., 2010; Coolen et al., 2011). In general, these investigations have shown that bacterial communities in the Arctic are similar in structure and diversity to bacterial communities of other biomes at the phylum level (Chu et al., 2010). However, few studies have examined the Arctic bacterial community structure at a lower taxonomic level (Männistö et al., 2007; Steven et al., 2007; Campbell et al., 2010; Larose et al., 2010), and such studies may reveal important differences in the actual functional groups of bacteria present in the Arctic.
Several reports have shown the relationship between bacterial communities and various environmental factors. For example, bacterial community composition is related to vegetation type, the quality of soil organic matter, geographical region, and environmental factors such as temperature, water, nutrient availability, soil pH, etc. (Fierer & Jackson, 2006; Wallenstein et al., 2007; Coolen et al., 2011). Currently, soil pH is considered as the most important factor influencing microbial community structure (Lauber et al., 2009; Chu et al., 2010; Shen et al., 2013; Bartram et al., 2014).
In the present study, we investigated the bacterial community structure and compositional patterns in subarctic tundra soils located in Council, Alaska, and explored the relationships between bacterial community and soil properties. For this investigation, we obtained a large amount of bacterial sequence data from soil samples through pyrosequencing to examine the bacterial community at a deep phylogenetic level. We then compared the bacteria present in the soil samples to determine which taxa were dominant in the community and how the bacterial community structure changed with soil properties.
Materials and methods
Ethics statement
Access for the study site was approved by the International Arctic Research Center (IARC) of the University of Alaska Fairbanks (UAF).
Site description and sampling design
The study site is located in Council, on the Seward Peninsular in Northwest Alaska (64°51′N, 163°39′W), which is a subarctic region. The site is c. 30 m above sea level, and the annual mean air temperature and precipitation are −3.1 ± 1.4 °C and 258 mm, respectively (climate data were obtained from the International Arctic Research Center of the University of Alaska, Fairbanks). At the time of sampling (mid-August 2011), the depth of the active layer was c. 50–70 cm. The sampling site is composed of moist acidic tussock tundra, and the dominant vegetation was cotton grass (Eriophorum vaginatum) or tussock, blueberry (Vaccinium uliginosum), and lichen and moss (Sphagnum spp.) beds.
Thirty-six sampling points were selected over an area of c. 300 × 50 m. The points were spaced at 25-m intervals, resulting in a latticework of 12 points × 3 points. Before acquiring soil samples, we removed the aboveground vegetation and litter layer, and cleaned the shovel with 70% ethanol to prevent contamination between samples. At each site, soil samples were collected from a depth of 0–10 cm (upper-layer soil) and a depth of 10–20 cm (lower-layer soil). The collected soil samples were wrapped in autoclaved foil, placed in ziplock bags, and transported to the laboratory in a frozen state. The soils in sites 4 and 35 were saturated with water, and only the upper-layer soil was collected. Overall, 70 soil samples were collected for this study. The collected soil samples were stored at −20 °C until DNA extraction and soil analysis.
Physical and chemical properties of soil
Gravimetric MC was determined by measuring the difference in weight between the field-moist soil samples and the same soil samples dried at 105 °C for 48 h. For inorganic nitrogen (N) analyses, c. 7 g of fresh soil was immediately set aside after sampling and kept frozen until extraction. Inorganic N (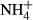 and
and 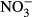 ) was extracted using a 2 M KCl solution and filtered through Whatman #42 paper. The filtrate was analyzed with an Auto-analyzer (Quaatro; Seal Analytical, Inc.). The remaining soil was air-dried and sieved through 2 mm mesh for further analyses. Soil pH was determined in a 1 : 10 soil : water (w/v) solution (Thomas, 1996). Soil was ground and passed through a 53 μm sieve to determine TC and TN content. TC and TN contents were measured by combustion (950 °C; FlashEA 1112; Thermo Fisher Scientific). Total inorganic carbon was negligible in all soil samples.
) was extracted using a 2 M KCl solution and filtered through Whatman #42 paper. The filtrate was analyzed with an Auto-analyzer (Quaatro; Seal Analytical, Inc.). The remaining soil was air-dried and sieved through 2 mm mesh for further analyses. Soil pH was determined in a 1 : 10 soil : water (w/v) solution (Thomas, 1996). Soil was ground and passed through a 53 μm sieve to determine TC and TN content. TC and TN contents were measured by combustion (950 °C; FlashEA 1112; Thermo Fisher Scientific). Total inorganic carbon was negligible in all soil samples.
PCR amplification and pyrosequencing
To extract genomic DNA from the soil samples, the soils were subsampled from the bulk soil, freeze-dried, and homogenized. Genomic DNA was extracted from 0.5 g of the homogenized soil samples using a FastDNA® SPIN kit for soil (MP Biomedicals) and a QuickPrep adapter (MP Biomedicals), according to the manufacturer's recommended protocol. The total DNA was quantified by Hoechst dye 33258 staining using a spectrophotometer with excitation and emission at 350 and 460 nm, respectively (Wallac EnVision 2013 Multilabel Reader, Perkin Elmer). The extracted DNA was stored at −20 °C until further analysis.
Genomic DNA extracted from the soil was amplified by PCR using the adapter-multiplex identifier–primer combinations targeting the V1–V3 regions (27F–518R) of bacterial 16S rRNA gene (Supporting Information, Table S4). The PCR reaction mixture (50 μL) contained 25 μL of master mix (DreamTaq™ Green PCR Master Mix [2×]), 1.4 μL of the forward and reverse primers (20 pmol of each primer), 1 μL of template DNA (1 ng μL−1), and 22.6 μL of deionized distilled water (DDW). The PCR program was as follows: an initial denaturation step at 95 °C for 3 min followed by 30 cycles of denaturation at 95 °C for 30 s, annealing at 56 °C for 30 s, and extension at 72 °C for 90 s, with a final extension at 72 °C for 7 min. All samples were amplified in triplicate, pooled in equal amounts, and purified using the QIAquick PCR Purification Kit (Qiagen). The PCR products were quantified with a NanoDrop. DNA sequencing was performed by DNALink (Seoul, Korea) using a GS-FLX 454 pyrosequencer (Roche).
Processing of pyrosequencing data
PCR amplicon pyrosequencing data were processed using the qiime software package, ver. 1.7 (Caporaso et al., 2010a). Briefly, raw flowgrams (sff files) were filtered and noise and chimeras were removed using ampliconnoise software, ver. 1.27 (Quince et al., 2011), using the platform option for FLX Titanium sequence data implemented in qiime. Sequences with an average (± SD) length of 378 ± 45 bp were clustered based on operational taxonomic units (OTUs) at 97% similarity using uclust (Edgar, 2010). OTUs were assigned to taxa using the RDP Classifier method (Wang et al., 2007) with a training set based on the Greengenes database (release 13.5; Werner et al., 2011). Sequence alignments for phylogenetic reconstruction were generated using pynast software (Caporaso et al., 2010b) and the Greengenes database (DeSantis et al., 2006). Using additional downstream tools in qiime, a phylogenetic tree was built from the aligned sequences using fasttree 2.1 (Price et al., 2010), and a pairwise beta-diversity distance matrix for a randomly selected subset of 700 sequences was generated for all samples based on the unweighted unifrac phylogenetic distance metric (Lozupone et al., 2006).
To facilitate diversity comparisons among bacterial communities, we estimated diversity indices, including the Chao1, Shannon, and Simpson indices, for a randomly selected subset of 700 sequences from each sample to avoid effects of different sample sizes (Kirchman et al., 2010).
The 454 FLX Titanium flowgrams have been deposited in the National Center for Biotechnology Information (NCBI) Sequence Read Archive database (accession number, SRP026166).
Statistical analysis
Statistical analyses were performed using r (version 3.0.0; The R Foundation for Statistical Computing) and primer-e v6 (Clarke & Gorley, 2006). Analysis of similarity (anosim) with 999 permutations and nonmetric multidimensional scaling (nmds) were conducted to compare bacterial community structure. Classification and regression tree (cart) analyses were conducted using rpart in the r software package (CP value set at 0.001) to determine which environmental variables explained the deviance of the dominant bacterial groups. A Mantel test was used to determine which physical and chemical properties of soil were significantly correlated with the bacterial community. To assess how the bacterial community changed with sampling distance (c. 22–427 m), a distance-decay relationship analysis, which assumes that community similarity will decrease with increasing geographical distance, was performed, as described in Martiny et al. (2011) with some modification. Briefly, the rate of distance-decay of the bacterial communities was calculated as the slope of a linear least squares regression on the relationship between geographic distance (m) versus the jackknifed unweighted unifrac distance of bacterial similarity, which is a qualitative metric of beta-diversity and is unaffected by the presence of duplicate sequences.
Results
Physical and chemical properties of soil
The soil at the study site was acidic and moist. The soil pH ranged from 3.90 to 5.02 (Table 1, Table S1). The upper-layer soil pH was slightly more acidic than the lower-layer soil pH (Table 1). Gravimetric MC was > 100%, except at site 17. The upper-layer soil contained higher MC than the lower-layer soil at most sites. In the upper-layer soil, the average TC and TN contents were 40% and 1.5%, respectively. The lower-layer soil had a lower TC content (36%), but the same TN content as the upper-layer soil. Therefore, the carbon and nitrogen (C/N) ratio was higher in the upper-layer soil. The ammonium (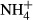 ) concentration was higher than the nitrate (
) concentration was higher than the nitrate (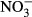 ) concentration at both depths and at all sites. Ammonium concentrations ranged from 8.6 to 93.1 μg g−1 soil, but the nitrate concentrations were negligible.
) concentration at both depths and at all sites. Ammonium concentrations ranged from 8.6 to 93.1 μg g−1 soil, but the nitrate concentrations were negligible.
Table 1.
The summary of physical and chemical properties of the subarctic tundra soil samples
| pH | TC (%) | TN (%) | C/N | MC (%) |
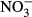 (μg N g−1 soil) (μg N g−1 soil) |
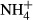 (μg N g−1 soil) (μg N g−1 soil) |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Upper | Lower | Upper | Lower | Upper | Lower | Upper | Lower | Upper | Lower | Upper | Lower | Upper | Lower | |
| Mean | 4.35 | 4.53* | 39.94 | 35.94* | 1.50 | 1.54 | 28.52 | 24.15* | 628.2 | 438.4* | 0.80 | 0.75 | 32.53 | 29.06 |
| SD | 0.29 | 0.28 | 6.81 | 12.35 | 0.46 | 0.58 | 9.05 | 6.56 | 222.5 | 252.6 | 0.48 | 0.53 | 22.25 | 17.91 |
| CV (%) | 6.61 | 6.25 | 17.04 | 34.36 | 30.38 | 37.52 | 31.71 | 27.14 | 35.4 | 57.6 | 59.18 | 70.56 | 68.40 | 61.61 |
| MAX | 5.02 | 5.01 | 43.87 | 48.55 | 2.23 | 2.38 | 66.30 | 56.93 | 1070.3 | 1201.6 | 3.19 | 3.29 | 93.08 | 91.55 |
| MIN | 3.90 | 3.96 | 2.10 | 1.85 | 0.09 | 0.08 | 19.31 | 18.26 | 53.4 | 32.1 | 0.27 | 0.21 | 8.62 | 9.79 |
TC, total carbon; TN, total nitrogen; C/N, a ratio of carbon to nitrogen; MC, moisture content; SD, standard deviation; CV, coefficient of variation.
Denotes significant differences (P < 0.05) of soil properties between the upper and lower-layers.
The soil properties of site 17 were completely different from those of the other sites (Fig. 1). This site contained lower MC, TC, and TN contents, and higher soil pH because it was primarily composed of mineral layers rather than organic layers, which comprised the other sampling sites.
Fig. 1.
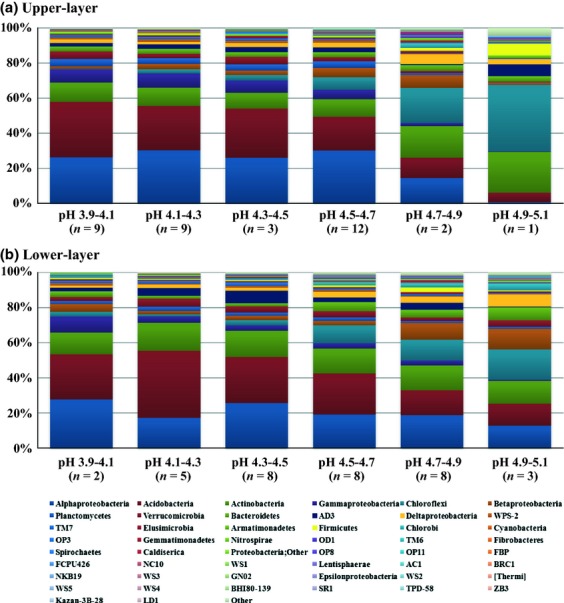
Relative abundance of phyla in the soil bacterial communities in the upper (a) and the lower-layer soils (b) separated according to pH.
General description of sequencing results
We obtained 91 742 good quality 16S rRNA gene sequences (V1–V3 region) from all the soil samples. On average, we obtained 1311 sequences (range, 718–1944 sequences) per sample. When we compared the soil bacterial communities using the same number of reads (700 sequences per sample), bacterial abundance and bacterial diversity were significantly higher in the upper-layer soils, as indicated by the Chao1 (P < 0.001), Shannon (P < 0.001), and Simpson (P < 0.05) indices (Table S2). OTUs accounted for 25.1–43.7% of the diversity, according to the Chao1 index (data not shown).
Bacterial community structure within and between sites
The classifiable sequences comprised members of 50 phyla, including candidate phyla. The dominant phyla were Acidobacteria, Alphaproteobacteria, and Actinobacteria, which accounted for more than 40% of the bacterial sequences in all soil samples (Fig. 1). In addition, sequences of Betaproteobacteria, Gammaproteobacteria, Chloroflexi, Deltaproteobacteria, Bacteroidetes, Verrucomicrobia, Planctomycetes, Chlorobi, Firmicutes, Elusimicrobia, Nitrospira, Armatimonadetes (former candidate division OP10), Gemmatimonadetes, Cyanobacteria, Spirochetes, Fibrobacteres, Caldiserica (former candidate division OP5), Lentisphaerae, and Epsilonproteobacteria were also identified at relatively low abundances, as well as members of 27 candidate phyla and several unclassified bacteria (Fig. 1).
In general, the bacterial community structures in the upper and lower-layer soils were different. Alphaproteobacteria, Gammaproteobacteria, and Planctomycetes were more abundant in the upper-layer soils, whereas Actinobacteria, Betaproteobacteria, Chloroflexi, and AD3 were more abundant in the lower-layer soils (Fig. S2). At the family level, Methylocystaceae, Acetobacteraceae, Sinobacteraceae, and Ellin6513 were more abundant in the upper-layer soil, whereas Gallionellaceae, Solibacteraceae, Intrasporangiaceae, and Ellin6529 were more abundant in the lower-layer soil (Fig. S2). The 16S rRNA gene sequences of the dominant OTUs, which accounted for more than 1% of the total sequences, were identified (Table 2). Only one OTU (OTU_1) showed > 97% sequence similarity with cultured bacteria, and most of the dominant OTUs have yet to be cultured (Table 2). The dominant bacterial OTUs accounted for 9.7% and 15.0% of the total sequences in the upper and lower-layer soil samples, respectively. Among the 11 dominant OTUs, three accounted for over 1% of total sequence in both soil layers. The nonmetric multidimensional scaling (nmds) plots indicated that the bacterial communities showed greater similarity across horizontal layers than through vertical depth (Fig. 2a). This pattern was confirmed by a significant anosim value (r = 0.338, P < 0.001) between the two depths. Bacterial communities were similar between sampling sites (Fig. 3). There was significant correlation between bacterial community similarity and sampling distance in the lower-layer soils (P < 0.05; Fig. 3); however, the relationship was not observed in upper-layer soils (P > 0.05).
Table 2.
A list of dominant OTUs which were accounted for over 1% among total reads through eztaxon-e* database
| Relative abundance (%) | ||||||
|---|---|---|---|---|---|---|
| OTU no. | The closest species (accession no.) | Detection source† | Pairwise similarity (%) | Lineage | Upper | Lower |
| 1 | Afipia broomeae (KB375282) | Human | 99.5 | Alphaproteobacteria | 3.61 | 2.38 |
| 2 | Pseudolabrys sp. (EU937836) | Biofilm | 98.5 | Alphaproteobacteria | 1.44 | 2.91 |
| 3 | Telmatobacter sp. (AJ292586) | Polychlorinated biphenyl-polluted soil | 98.8 | Acidobacteria | 1.38 | 1.87 |
| 4 | EU150278_s in Steroidobacter_f (EU150278) | Soil | 100 | Gammaproteobacteria | 2.04 | 0.62 |
| 5 | Koribacter sp. (AY913298) | Forest | 98.8 | Acidobacteria | 0.01 | 1.88 |
| 6 | Koribacter sp. (GQ339162) | Iron(II)-rich seep | 99.3 | Acidobacteria | 0.54 | 1.41 |
| 7 | Koribacter sp. (EU150193) | Soil from spruce fir forest | 99.8 | Acidobacteria | 1.24 | 0.40 |
| 8 | Oryzihumus sp. (4P001838)‡ | ND | 98.3 | Actinobacteria | 0.51 | 1.19 |
| 9 | Granulicella sp. (FJ466102) | Volcanic deposit | 99.8 | Acidobacteria | 0.97 | 1.13 |
| 10 | EU861899_s in Solirubrobacterales (EU861899) | Meadow surface soil | 100 | Actinobacteria | 0.69 | 1.10 |
| 11 | Gallionella sp. (4P002107)‡ | ND | 99.3 | Betaproteobacteria | 0.43 | 1.08 |
eztaxon-e database (Kim et al., 2012; http://eztaxon-e.ezbiocloud.net/).
Data for detection sources were from NCBI or publications.
Accession number was from eztaxon-e.
Fig. 2.
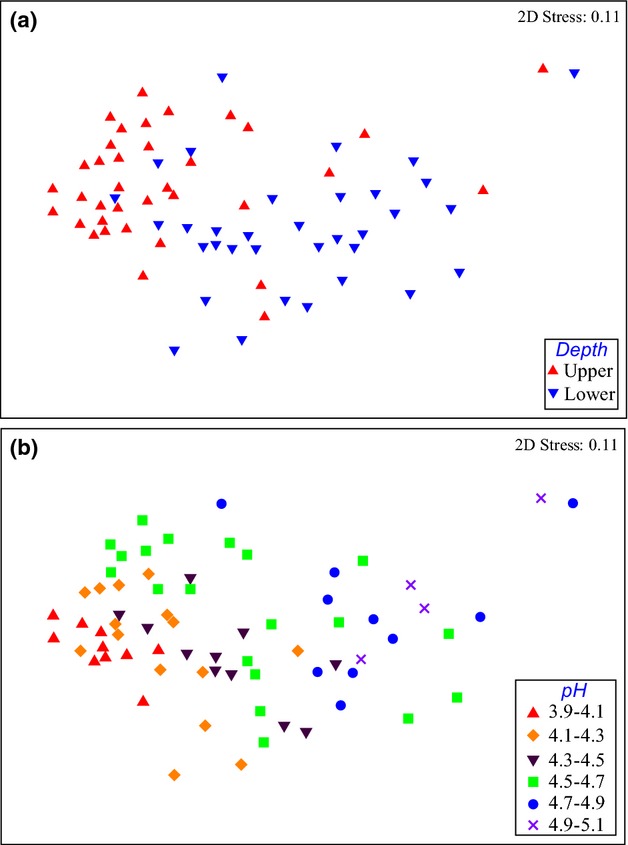
nmds plots derived from phylogenetic similarity based on jackknifed unweighted unifrac distances between soil samples, with symbols coded by depth (a) and pH (b).
Fig. 3.
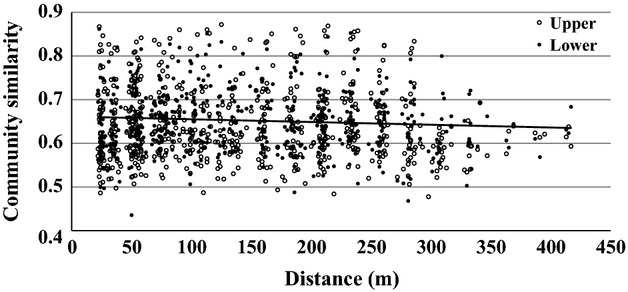
Distance-decay analysis of the relationship between geographic distance and bacterial community distance based on jackknifed unweighted unifrac distance in both layers. The slope was significant in the lower-layer soils (P < 0.05; y = −6E−05x + 0.6615, solid line).
Association between bacterial community structure and physical and chemical properties of soil
Significant associations were detected between soil bacterial community and physical and chemical properties of soil. In general, pH showed the highest correlation (r = 0.392, P < 0.001) with bacterial community composition in all soil samples (Table 3, Fig. 2b). Acidobacteria, Alphaproteobacteria and Gammaproteobacteria decreased with increasing soil pH, whereas Chloroflexi, Betaproteobacteria, Bacteroidetes, and Deltaproteobacteria increased with increasing soil pH (Fig. 1). Similar results were observed in the upper and lower-layer soils. At the class or order level, the bacterial community structure changed along the pH gradient, and some taxa showed opposite responses to pH compared with that at the phylum level (Fig. S1). For example, uncultured iii1-8 and Acidobacteria-6 of the Acidobacteria, Rhizobiales of the Alphaproteobacteria, and Legionellales of the Gammaproteobacteria increased with increasing soil pH, whereas Ktedonobacteria of the Chloroflexi decreased with increasing soil pH.
Table 3.
The significant correlations between physicochemical properties of soil and bacterial communities
| Soil physical and chemical properties | All soil samples (n = 70) | Upper-layer (n = 36) | Lower-layer (n = 34) | |||
|---|---|---|---|---|---|---|
| r | P | r | P | r | P | |
| pH | 0.392 | 0.001 | 0.393 | 0.001 | 0.395 | 0.001 |
| C/N | 0.112 | 0.021 | 0.148 | 0.054 | 0.213 | 0.025 |
| MC | 0.212 | 0.001 | 0.122 | 0.094 | 0.257 | 0.005 |
| TC | 0.196 | 0.003 | 0.168 | 0.062 | 0.116 | 0.137 |
| TN | 0.171 | 0.005 | 0.323 | 0.002 | 0.137 | 0.077 |
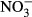 |
0.001 | 0.375 | 0.020 | 0.392 | −0.039 | 0.656 |
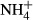 |
0.035 | 0.191 | 0.167 | 0.016 | −0.044 | 0.691 |
The Spearman's rank correlations (r) and significance (P) were determined by Mantel tests.
C/N, a ratio of carbon and nitrogen; MC, moisture content; TC, total carbon; TN, total nitrogen.
Significant correlation (P < 0.05) values are in bold.
Besides soil pH, C/N ratio (r = 0.112, P < 0.05), MC (r = 0.212, P < 0.001), TC (r = 0.196, P < 0.005), and TN (r = 0.171, P < 0.005) showed significant correlation with the overall soil bacterial community composition (Table 3). However, different soil properties were associated with the bacterial community structure in the two soil layers; in the upper-layer soils, TN (r = 0.323, P < 0.005) and 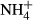 (r = 0.167, P < 0.05) were significantly associated with the community composition, whereas C/N ratio (r = 0.213, P < 0.05) and MC (r = 0.257, P < 0.005) were significantly associated with the community composition in the lower-layer soils (Table 3).
(r = 0.167, P < 0.05) were significantly associated with the community composition, whereas C/N ratio (r = 0.213, P < 0.05) and MC (r = 0.257, P < 0.005) were significantly associated with the community composition in the lower-layer soils (Table 3).
To identify the most influential soil properties, correlations between physical and chemical properties of soil and the dominant groups were determined using cart analysis (Fig. 4). The soil characteristics affecting each dominant group differed between the soil depths. Soil pH was the best predictor for the presence of Acidobacteria in both soil layers. The presence of Alphaproteobacteria was related to pH in the upper-layer soils and was related to TN in the lower-layer soils. Actinobacteria was related to pH in the upper-layer soils and was related to C/N ratio in the lower-layer soils (Fig. 4).
Fig. 4.

cart analysis to describe the main properties for the dominant phyla Acidobacteria, Alphaproteobacteria, and Actinobacteria in the upper (a) and the lower-layer soil (b) samples.
Discussion
We investigated the bacterial community structure and its relationships with soil properties in subarctic tundra soils in Council, Alaska. The results showed that the bacterial community in the moist acidic tussock tundra soil was very diverse and that soil depth and pH were the properties that were most influential in predicting the bacterial community structure.
The similarity of the bacterial community was more different through the vertical depth (10 cm) than across the horizontal layers (> 25 m). Soil depth is one of the major parameters influencing microbial community. Waldrop & Harden (2008) showed that microbial biomass and activity significantly decreased at the surface following wildfire; however, the effects of wildfire on those changes were not obvious at 20 cm depth. Furthermore, while long-term warming significantly decreased the evenness of bacterial communities at the surface organic layer soils, the effect of warming was relatively minor in the mineral layer (Deslippe et al., 2012). Previous studies indicated that this vertical variation was due to numerous soil properties that change with soil depth, such as pH, nutrient and water availability, plants, soil structure, oxygen, and temperature (Fierer et al., 2003; Ström et al., 2003; Kobabe et al., 2004; Hansel et al., 2008). In this study, we also found that there were significant differences in soil pH, MC, TC concentration, and C/N ratio between the two soil depths (P < 0.05, Table 1), whereas no obvious trends in soil properties were observed among soils obtained at the same depth.
Among the three major groups, the relative abundances of Alphaproteobacteria decreased with depth, although those of Acidobacteria and Actinobacteria were similar in both soil layers (Fig. S2). This observation corresponded with other observations of bacterial community composition changes with soil depth (Eilers et al., 2012; Frank-Fahle et al., 2014). Alphaproteobacteria prefer nutrient-rich environments (Nemergut et al., 2010; Thomson et al., 2010; Goldfarb et al., 2011). Moreover, Fierer et al. (2012) showed the increase in the relative abundance for Alphaproteobacteria with additional N input. The decomposition degree of plant and moss differed at different soil depths. The lower C/N ratio in the lower-layer soil reflected more decomposition in the lower-layer than in the upper-layer (Table 1). Therefore, labile materials that provide nutrients for bacteria might be more abundant in the upper-layer soils. In addition, the concentrations of TC, TN, and 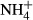 were higher in the upper-layer soils than in the lower-layer soils (Table S1). The environment in the upper-layer would favor Alphaproteobacteria propagation.
were higher in the upper-layer soils than in the lower-layer soils (Table S1). The environment in the upper-layer would favor Alphaproteobacteria propagation.
Soil pH was significantly correlated with bacterial community structure in both layers (Table 3; Fig. 2b). Specifically, Acidobacteria, Alphaproteobacteria, and Gammaproteobacteria decreased with increasing soil pH, whereas Betaproteobacteria and Chloroflexi increased with increasing pH (Fig. 1). Even when we excluded the dominant groups (Alphaproteobacteria, Acidobacteria, and Actinobacteria) from the statistical analyses, the minor groups also showed significant correlations with soil pH (P < 0.001) in both upper- and lower-layer soils (Table S3). These results corresponded with other studies of Arctic and subarctic soils (Männistö et al., 2007; Chu et al., 2010). Männistö et al. (2007) showed that the soil pH of parent materials had greater influence on bacterial community structure than changes in soil temperature in the subarctic region. Chu et al. (2010) compared bacterial community structure on a global scale, and concluded that bacterial community composition in arctic soil was strongly influenced by local environmental factors associated with soil acidity than by other factors. Moreover, soil pH is known as a strong driver shaping bacterial community structure in various soil ecosystems, including a wide range of soils in North and South America and agricultural soil in Scotland (Fierer & Jackson, 2006; Lauber et al., 2009; Bartram et al., 2014).
At lower taxonomic levels, some taxa showed responses to pH opposite to that observed at the phylum level. For example, Acidobacteria-6 and iii1-8 (Acidobacteria), and Legionellales (Gammaproteobacteria) increased with increasing soil pH, whereas Ktedonobacteria (Chloroflexi) decreased with increasing soil pH (Fig. S1). Our results agreed with other studies to show the relationship between soil pH and the lower taxonomic levels of bacteria (Jones et al., 2009; Bartram et al., 2014). Although these taxa were not the dominant groups, it is noteworthy to examine the different responses of bacterial groups at the lower taxonomic levels because not all members of the same phylum behaved in the same way.
The dominant bacterial phyla in the moist acidic tussock tundra soil in this study were Acidobacteria, Alphaproteobacteria, and Actinobacteria. It is consistent with other studies of various subarctic and Arctic soils, such as tundra soil from Nunavut, the Toolik Lake area, and spanning the Arctic region (Neufeld & Mohn, 2005; Wallenstein et al., 2007; Campbell et al., 2010; Chu et al., 2010; Nemergut et al., 2010; Schütte et al., 2010). Bacterial composition, including minor groups, was also similar to that of other Arctic soils (Zhou et al., 1997; Neufeld & Mohn, 2005; Wallenstein et al., 2007; Campbell et al., 2010; Chu et al., 2010; Nemergut et al., 2010; Schütte et al., 2010). Moreover, the three dominant phyla are also dominant in soils from nonpolar areas (Fierer & Jackson, 2006; Will et al., 2010; Li et al., 2012; Shen et al., 2013). According to a review by Janssen (2006), these phyla are ubiquitous, and they are the most abundant phyla in soils from various ecosystems. However, soil bacterial community in Hess Creek, Alaska showed different community structure as Actinobacteria, Proteobacteria, and Chloroflexi were dominant (Mackelprang et al., 2011). Because Chloroflexi increased with increasing soil pH, the abundant Chloroflexi in Hess Creek can be attributed to higher soil pH range of 6.43–6.52 in this area (Mackelprang et al., 2011).
The higher resolution of pyrosequencing data allowed us to look into the information on the potential ecological roles of bacteria in the subarctic tundra soil. At the species level, OTU_1 (99.5% sequence similarity with Afipia broomeae) and OTU_2 (98.5% sequence similarity with Pseudolabrys sp.) were predominant in this study (Table 2). They belonged to Rhizobiales (Alphaproteobacteria) which are known to fix nitrogen as plant root symbionts. The genus Steroidobacter of Gammaproteobacteria can reduce nitrate to dinitrogen monoxide and further to dinitrogen (Fahrbach et al., 2008). Gallionella of Betaproteobacteria is characterized by its oxidation of Fe (II) (Hedrich et al., 2011). Methane-consuming bacteria which belong to the family Methylocystaceae (Alphaproteobacteria) were detected in this study as well (Fig. S2). These results provide some information on the ecological roles of bacteria in tundra soil.
The distance-decay relationships showed that bacterial community similarity decreased with increasing sampling distance in lower-layer soils, whereas no significant relationship was detected in the upper-layer soils (Fig 4). Decreasing bacterial community similarity with distance can be explained by increasing differences in environmental properties (Nekola & White, 1999). Although it may not be direct evidence of dissimilarity in environmental properties across distance, we observed greater variation in soil properties (TC and TN concentrations and MC) in the lower-layer soils than in the upper-layer soils (Table 1). There is currently no consensus on the biogeographical patterns of bacterial communities. Several studies showed that bacterial community similarity decreased with increasing sampling distance. Monroy et al. (2012) reported that bacterial community composition changed with geographic distance (1–200 km range); however, they could not explain this relationship using measured soil properties. Stres et al. (2013) also observed an increasing pattern of bacterial community dissimilarity with distance in topographically complex high-altitude slopes in the Himalaya (1–1200 m range). However, other studies reported no biogeographical pattern in microbial community similarity, including bacteria, on either a local or a global scale (Ritz et al., 2004; Chu et al., 2010; Queloz et al., 2011). We also found contrasting results for the relationship between bacterial community similarity and sampling distance between the two depths; no differences were observed in the upper layer, whereas increasing dissimilarity with distance was observed in the lower layer. Therefore, additional research is needed to determine the relationships between geological distance and bacterial community structure.
In summary, soil depth and pH were the most influential soil properties to determine the bacterial community structure in subarctic tundra soil in Council, Alaska. Although various plants covered the top soil, the bacterial communities were relatively similar across the horizontal layers compared with the communities through the vertical depth. Bacterial communities were more significantly correlated with soil pH than the other measured soil properties. These results indicated that the bacterial communities in this study differed at the two different soil depths and were relatively stable against various soil properties except soil pH. Moreover, we found that certain phylogenetic groups at lower taxonomic levels showed a different response to pH from that at the phylum level. This indicated the necessity of analyzing bacterial communities at lower taxonomic levels such as species, which actually perform various functions in the environment. More metagenomic and transcriptomic studies are needed to understand the community structure of other soil microorganisms such as archaea and fungi, and the ecological functions of microorganisms in tundra soil.
Acknowledgments
This study was supported by a grant from the National Research Foundation of Korea funded by the Korean Government (MSIP) (NRF-2011-0021067) (PN13082, KOPRI). Fieldwork permits were granted by the UAF. We are grateful to the editor and anonymous reviewers for providing constructive comments that substantially improved the manuscript.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Table S1. The physical and chemical properties of the subarctic soil samples.
Table S2. Summary statistics of pyrosequencing 16S rRNA gene sequences of soil samples.
Table S3. The significant correlations between physicochemical properties of soil and bacterial minor groups*.
Table S4. The list of barcode sequences in this study.
Fig. S1. Bacterial community structures at class level of Acidobacteria (a, b) and Chloroflexi (c, d), and at order level of Alphaproteobacteira (e, f) and Gammaproteobacteria (g, h) with soil pH category.
Fig. S2. Hierarchical classifications of 16S rRNA gene sequences in the eight main bacterial communities.
References
- ACIA. Arctic Climate Impact Assessment. Cambridge, UK: Cambridge University Press; 2005. [Google Scholar]
- Anisimov OA. Fitzharris B. Polar regions (Arctic and Antarctic) In: White KS, editor; McCarthy OFCJ, Leary NA, Dokken DJ, editors. Climate Change 2001: Impacts, Adaptation, and Vulnerability. Contribution of Working group II to the Third Assessment Report of the Intergovernmental Panel on Climate Change. Cambridge, UK: Cambridge University Press; 2001. pp. 801–841. [Google Scholar]
- Bardgett RD, Freeman C. Ostle NJ. Microbial contributions to climate change through carbon cycle feedbacks. ISME J. 2008;2:805–814. doi: 10.1038/ismej.2008.58. [DOI] [PubMed] [Google Scholar]
- Bartram AK, Jiang X, Lynch MD, Masella AP, Nicol GW, Dushoff J. Neufeld JD. Exploring links between pH and bacterial community composition in soils from the Craibstone Experimental Farm. FEMS Microbiol Ecol. 2014;87:403–415. doi: 10.1111/1574-6941.12231. [DOI] [PubMed] [Google Scholar]
- Campbell BJ, Polson SW, Hanson TE, Mack MC. Schuur EAG. The effect of nutrient deposition on bacterial communities in Arctic tundra soil. Environ Microbiol. 2010;12:1842–1854. doi: 10.1111/j.1462-2920.2010.02189.x. [DOI] [PubMed] [Google Scholar]
- Caporaso JG, Kuczynski J, Stombaugh J, et al. qiime allows analysis of high-throughput community sequencing data. Nat Methods. 2010a;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Bittinger K, Bushman FD, DeSantis TZ, Andersen GL. Knight R. pynast: a flexible tool for aligning sequences to a template alignment. Bioinformatics. 2010b;26:266–267. doi: 10.1093/bioinformatics/btp636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapin FS, Sturm M, Serreze MC, et al. Role of land-surface changes in Arctic summer warming. Science. 2005;310:657–660. doi: 10.1126/science.1117368. [DOI] [PubMed] [Google Scholar]
- Christensen TR, Jonasson S, Michelsen A, Callaghan TV. Harström M. Environmental controls on soil respiration in the Eurasian and Greenlandic Arctic. J Geophys Res. 1998;103:29015–29021. [Google Scholar]
- Chu H, Fierer N, Christian LL, Caporaso JG, Knight R. Grogan R. Soil bacterial diversity in the Arctic is not fundamentally different from that found in other biomes. Environ Microbiol. 2010;12:2998–3006. doi: 10.1111/j.1462-2920.2010.02277.x. [DOI] [PubMed] [Google Scholar]
- Clarke KR. Gorley RN. primer v6: User Manual/Tutorial. Plymouth: Primer-E Ltd; 2006. [Google Scholar]
- Coolen MJL, van de Giessen J, Zhu EY. Wuchter C. Bioavailability of soil organic matter and microbial community dynamics upon permafrost thaw. Environ Microbiol. 2011;13:2299–2314. doi: 10.1111/j.1462-2920.2011.02489.x. [DOI] [PubMed] [Google Scholar]
- DeSantis TZ, Jr, Hugenholtz P, Keller K, et al. NAST: a multiple sequence alignment server for comparative analysis of 16S rRNA genes. Nucleic Acids Res. 2006;34:W394–W399. doi: 10.1093/nar/gkl244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deslippe JR, Hartmann M, Simard SW. Mohn WW. Long-term warming alters the composition of Arctic soil microbial communities. FEMS Microbiol Ecol. 2012;82:303–315. doi: 10.1111/j.1574-6941.2012.01350.x. [DOI] [PubMed] [Google Scholar]
- Edgar RC. Search and clustering orders of magnitude faster than blast. Bioinformatics. 2010;26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- Eilers KG, Debenport S, Anderson S. Fierer N. Digging deeper to find unique microbial communities: the strong effect of depth on the structure of bacterial and archaeal communities in soil. Soil Biol Biochem. 2012;50:58–65. [Google Scholar]
- Fahrbach M, Kuever J, Remesch M, Huber BE, Kampfer P, Dott W. Hollender J. Steroidobacter denitrificans gen. nov., sp. nov., a steroidal hormone-degrading gammaproteobacterium. Int J Syst Evol Microbiol. 2008;58:2215–2223. doi: 10.1099/ijs.0.65342-0. [DOI] [PubMed] [Google Scholar]
- Fierer N. Jackson RB. The diversity and biogeography of soil bacterial communities. P Natl Acad Sci USA. 2006;103:626–631. doi: 10.1073/pnas.0507535103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierer N, Schimel J. Holden P. Variations in microbial community composition through two soil depth profiles. Soil Biol Biochem. 2003;35:167–176. [Google Scholar]
- Fierer N, Lauber CL, Ramirez KS, Zaneveld J, Bradford MA. Knight R. Comparative metagenomic, phylogenetic and physiological analyses of soil microbial communities across nitrogen gradients. ISME J. 2012;6:1007–1017. doi: 10.1038/ismej.2011.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank-Fahle BA, Yergeau E, Greer CW, Lantuit H. Wagner D. Microbial functional potential and community composition in permafrost-affected soils of the NW Canadian Arctic. PLoS One. 2014;9:e84761. doi: 10.1371/journal.pone.0084761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfarb KC, Karaoz U, Hanson CA, Santee CA, Bradford MA, Treseder KK, Wallenstein MD. Brodie EL. Differential growth responses of soil bacterial taxa to carbon substrates of varying chemical recalcitrance. Front Microbiol. 2011;2:1–10. doi: 10.3389/fmicb.2011.00094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosse G, Harden J, Turetsky M, et al. Vulnerability of high-latitude soil organic carbon in North America to disturbance. J Geophys Res. 2011;116:G00K06. [Google Scholar]
- Hansel CM, Fendorf S, Jardine PM. Francis CA. Changes in bacterial and archaeal community structure and functional diversity along a geochemically variable soil profile. Appl Environ Microbiol. 2008;74:1620–1633. doi: 10.1128/AEM.01787-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedrich S, Schlömann M. Johnson DB. The iron-oxidizing proteobacteria. Microbiology. 2011;157:1551–1564. doi: 10.1099/mic.0.045344-0. [DOI] [PubMed] [Google Scholar]
- IPCC. Climate Change 2007. Geneva, Switzerland: Intergovernmental Panel on Climate Change; 2007. [Google Scholar]
- Janssen PH. Identifying the dominant soil bacterial taxa in libraries of 16S rRNA and 16S rRNA genes. Appl Environ Microbiol. 2006;72:1719–1728. doi: 10.1128/AEM.72.3.1719-1728.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RT, Robeson MS, Lauber CL, Hamady M, Knight R. Fierer N. A comprehensive survey of soil acidobacterial diversity using pyrosequencing and clone library analyses. ISME J. 2009;3:442–453. doi: 10.1038/ismej.2008.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim OS, Cho YJ, Lee K, et al. Introducing eztaxon-e: a prokaryotic 16S rRNA Gene sequence database with phylotypes that represent uncultured species. Int J Syst Evol Microbiol. 2012;62:716–721. doi: 10.1099/ijs.0.038075-0. [DOI] [PubMed] [Google Scholar]
- Kirchman DL, Cottrell MT. Lovejoy C. The structure of bacterial communities in the western Arctic Ocean as revealed by pyrosequencing of 16S rRNA genes. Environ Microbiol. 2010;12:1132–1143. doi: 10.1111/j.1462-2920.2010.02154.x. [DOI] [PubMed] [Google Scholar]
- Kobabe S, Wagner D. Pfeiffer E-M. Characterisation of microbial community composition of a Siberian tundra soil by fluorescence in situ hybridization. FEMS Microbiol Ecol. 2004;50:13–23. doi: 10.1016/j.femsec.2004.05.003. [DOI] [PubMed] [Google Scholar]
- Larose C, Berger S, Ferrari C, Navarro E, Dommergue A, Schneider D. Vogel TM. Microbial sequences retrieved from environmental samples from seasonal Arctic snow and meltwater from Svalbard, Norway. Extremophiles. 2010;14:205–212. doi: 10.1007/s00792-009-0299-2. [DOI] [PubMed] [Google Scholar]
- Lauber CL, Hamady M, Knight R. Fierer N. Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale. Appl Environ Microbiol. 2009;75:5111–5120. doi: 10.1128/AEM.00335-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Khafipour E, Krause DO, Entz MH, de Kievit TR. Fernando WD. Pyrosequencing reveals the influence of organic and conventional farming systems on bacterial communities. PLoS One. 2012;7:e51897. doi: 10.1371/journal.pone.0051897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone C, Hamady M. Knight R. unifrac-an online tool for comparing microbial community diversity in a phylogenetic context. BMC Bioinformatics. 2006;7:371. doi: 10.1186/1471-2105-7-371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackelprang R, Waldrop MP, DeAngelis KM, et al. Metagenomic analysis of a permafrost microbial community reveals a rapid response to thaw. Nature. 2011;480:368–371. doi: 10.1038/nature10576. [DOI] [PubMed] [Google Scholar]
- Männistö MK, Tiirola M. Häggblom MM. Bacterial communities in Arctic fields of Finnish Lapland are stable but highly pH-dependent. FEMS Microbiol Ecol. 2007;59:452–465. doi: 10.1111/j.1574-6941.2006.00232.x. [DOI] [PubMed] [Google Scholar]
- Margesin R, Jud M, Tscherko D. Schinner F. Microbial communities and activities in alpine and subalpine soils. FEMS Microbiol Ecol. 2009;67:208–218. doi: 10.1111/j.1574-6941.2008.00620.x. [DOI] [PubMed] [Google Scholar]
- Martiny JBH, Eisen JA, Penn K, Allison SD. Horner-Devine MC. Drivers of bacterial β-diversity depend on spatial scale. P Natl Acad Sci USA. 2011;108:7850–7854. doi: 10.1073/pnas.1016308108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monroy F, van der Putten WH, Yergeau E, Mortimer SR, Duyts H. Bezemer TM. Community patterns of soil bacteria and nematodes in relation to geographic distance. Soil Biol Biochem. 2012;45:1–7. [Google Scholar]
- Nekola JC. White PS. Distance decay of similarity in biogeography and ecology. J Biogeogr. 1999;26:867–878. [Google Scholar]
- Nemergut DR, Cleveland CC, Wieder WR, Washenberger CL. Townsend AR. Plot-scale manipulations of organic matter inputs to soils correlate with shifts in microbial community composition in a lowland tropical rain forest. Soil Biol Biochem. 2010;42:2153–2160. [Google Scholar]
- Neufeld JD. Mohn WW. Unexpectedly high bacterial diversity in Arctic tundra relative to boreal forest soils, revealed by serial analysis of ribosomal sequence tags. Appl Environ Microbiol. 2005;71:5710–5718. doi: 10.1128/AEM.71.10.5710-5718.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ping CL, Michaelson GJ, Jorgenson MT, Kimble JM, Epstein H, Romanovsky VE. Walker DA. High stocks of soil organic carbon in the North American Arctic region. Nat Geosci. 2008;1:616–619. [Google Scholar]
- Price MN, Dehal PS. Arkin AP. fasttree 2-approximately maximum-likelihood trees for large alignments. PLoS One. 2010;5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Queloz V, Sieber TN, Holdenrieder O, McDonald BA. Grünig CR. No biogeographical pattern for a root-associated fungal species complex. Glob Ecol Biogeogr. 2011;20:160–169. [Google Scholar]
- Quince C, Lanzen A, Davenport RJ. Turnbaugh PJ. Removing noise from pyrosequenced amplicons. BMC Bioinformatics. 2011;12:38. doi: 10.1186/1471-2105-12-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team. R, a Language and Environment for Statistical Computing. Vienna, Austria: R 21. Foundation for Statistical Computing; 2006. [Google Scholar]
- Ritz K, McNicol JW, Nunan N, et al. Spatial structure in soil chemical and microbiological properties in an upland grassland. FEMS Microbiol Ecol. 2004;49:191–205. doi: 10.1016/j.femsec.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Schlesinger WH. Biogeochemistry: An Analysis of Global Change. San Diego, CA: Academic Press; 1997. [Google Scholar]
- Schütte UME, Abdo Z, Foster J, Ravel J, Bunge J, Solheim B. Forney LJ. Bacterial diversity in a glacier foreland of the high Arctic. Mol Ecol. 2010;19:54–66. doi: 10.1111/j.1365-294X.2009.04479.x. [DOI] [PubMed] [Google Scholar]
- Schuur EAG, Vogel JG, Crummer KG, Lee H, Sickman JO. Osterkamp TE. The effect of permafrost thaw on old carbon release and net carbon exchange from tundra. Nature. 2009;459:556–559. doi: 10.1038/nature08031. [DOI] [PubMed] [Google Scholar]
- Shen C, Xiong J, Zhang H, Feng Y, Lin X, Li X, Liang W. Chu H. Soil pH drives the spatial distribution of bacterial communities along elevation on Changbai Mountain. Soil Biol Biochem. 2013;57:204–211. [Google Scholar]
- Steven B, Briggs G, McKay CP, Pollard WH, Greer CW. Whyte LG. Characterization of the microbial diversity in a permafrost sample from the Canadian high Arctic using culture-dependent and culture-independent methods. FEMS Microbiol Ecol. 2007;59:513–523. doi: 10.1111/j.1574-6941.2006.00247.x. [DOI] [PubMed] [Google Scholar]
- Stres B, Sul WJ, Murovec B. Tiedje JM. Recently deglaciated high-altitude soils of the Himalaya: diverse environments, heterogenous bacterial communities and long-range dust inputs from the upper troposphere. PLoS One. 2013;9:e76440. doi: 10.1371/journal.pone.0076440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ström L, Ekberg A, Mastepanov M. Christensen TB. The effect of vascular plants on carbon turnover and methane emissions from a tundra wetland. Glob Chang Biol. 2003;9:1185–1192. [Google Scholar]
- Tarnocai C, Canadell JG, Schuur EAG, Kuhry P, Mazhitova G. Zimov S. Soil organic carbon pools in the northern circumpolar permafrost region. Global Biogeochem Cycles. 2009;23:1–11. [Google Scholar]
- Thomas GW. Soil pH and soil acidity. In: Sparks DL, editor. Methods of Soil Analysis. Part 3. Chemical Methods. Madison, WI: Soil Science Society of America; 1996. pp. 475–490. [Google Scholar]
- Thomson B, Ostle N, McNamara N, Bailey M, Whiteley A. Griffiths R. Vegetation affects the relative abundances of dominant soil bacterial taxa and soil respiration rates in an upland grassland soil. Microb Ecol. 2010;59:335–343. doi: 10.1007/s00248-009-9575-z. [DOI] [PubMed] [Google Scholar]
- Waldrop MP. Harden JW. Interactive effects of wildfire and permafrost on microbial communities and soil processes in an Alaskan black spruce forest. Glob Chang Biol. 2008;14:2591–2602. [Google Scholar]
- Wallenstein MD, McMahon S. Schimel J. Bacterial and fungal community structure in Arctic tundra tussock and shrub soils. FEMS Microbiol Ecol. 2007;59:428–435. doi: 10.1111/j.1574-6941.2006.00260.x. [DOI] [PubMed] [Google Scholar]
- Wang Q, Garrity GM, Tiedje JM. Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner JJ, Koren O, Hugenholtz P, et al. Impact of training sets on classification of high-throughput bacterial 16s rRNA gene surveys. ISME J. 2011;6:94–103. doi: 10.1038/ismej.2011.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will C, Thürmer A, Wollherr A, Nacke H, Herold N, Schrumpf M, Gutknecht J, Wubet T, Buscot F. Daniel R. Horizon-specific bacterial community composition of German grassland soils, as revealed by pyrosequencing-based analysis of 16S rRNA genes. Appl Environ Microbiol. 2010;76:6751–6759. doi: 10.1128/AEM.01063-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yergeau E, Hogues H, Whyte LG. Greer CW. The functional potential of high Arctic permafrost revealed by metagenomic sequencing, qPCR, and microarray analyses. ISME J. 2010;4:1206–1214. doi: 10.1038/ismej.2010.41. [DOI] [PubMed] [Google Scholar]
- Zhou JZ, Davey ME, Figueras JB, Rivkina E, Gilichinsky D. Tiedje JM. Phylogenetic diversity of a bacterial community determined from Siberian tundra soil DNA. Microbiology. 1997;143:3913–3919. doi: 10.1099/00221287-143-12-3913. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. The physical and chemical properties of the subarctic soil samples.
Table S2. Summary statistics of pyrosequencing 16S rRNA gene sequences of soil samples.
Table S3. The significant correlations between physicochemical properties of soil and bacterial minor groups*.
Table S4. The list of barcode sequences in this study.
Fig. S1. Bacterial community structures at class level of Acidobacteria (a, b) and Chloroflexi (c, d), and at order level of Alphaproteobacteira (e, f) and Gammaproteobacteria (g, h) with soil pH category.
Fig. S2. Hierarchical classifications of 16S rRNA gene sequences in the eight main bacterial communities.