

Abstract
Despite major refinements in cancer therapy drugs, our progress at increasing the cure rates of most cancers has been hampered by high relapse rates. A possible biological explanation of the high frequency of relapse and resistance to currently available drugs has been provided by the cancer stem cell (CSC) proposition. Basically, the CSC theory hypothesizes the presence of a hierarchically organized, relatively rare population of cells that is responsible for tumor initiation, self-renewal and maintenance, mutation accumulation and therapy resistance. Since first postulated by John Dick, multiple reports have provided support for this hypothesis by isolating (more or less) rare cell populations, where the ability to initiate tumors in vivo has been demonstrated. Most progress and stronger data supporting this theory are found predominantly in myelogenous leukemias, whose study has benefited from over half-a-century progress in our understanding of the normal hierarchical organization of hematopoiesis. This review, however, also analyzes the advancement in the quantitative and functional analysis of solid tumor stem cells and in the analysis of the tumor microenvironment as specialized, nurturing niches for CSCs. Overall, this review intends to briefly summarize most of the evidences that support the CSC theory and the apparent contradictions, if not skepticism from the scientific community, about its validity for all forms of cancer, or alternatively on just a few cancers initiated by a limited number of somatic or germinal mutations.
Cancer is a multigenic complex disease that broadly represents uncontrolled proliferation, blockage in cellular differentiation and metastasis. During a significant part of the 20th century, cancer biologists have explained the process of cancer initiation and progression through a stochastic model. A cell or group of cells become tumorigenic after an initial germinal or somatic mutation, and subsequent cumulative somatic mutations, resulting in proliferative and survival advantage of selected cell clones. Predominant clones with the ability to produce identical cancer cells maintain the tumor and retain the ability to initiate other tumors after transplantation. This model assumed that: cancer is composed of a clone or group of clones with similar growth rates and represents a homogenous morphological pattern; and, all cancer cells could initiate and propagate the tumor by themselves.
Despite its extensive use, this model had two important limitations based on other observational and experimental data. First, tumors were morphologically, phenotypically, and functionally heterogenous. Not all cells looked alike, and patterns of differentiation in tumors could be distinguished. Second, the stochastic model predicted that every cell can initiate a tumor. This prediction was at odds with the observation that a large number of cells were needed to transfer the tumor. The long-term explanation was that, unlike aggressive hematological tumors, the probability of cell cycle entry in most solid tumor cells is low, and only a small fraction of cells is cycling at a given time point.
In the last years, an alternative model of cancer evolution has been proposed. This new model establishes the existence of a hierarchical order where a tissue-specific, programmed or reprogrammed cell, the so-called “cancer stem cell,” acquires or retains the properties of self-renewal, multi-lineage differentiation, and most importantly, tumor initiation, in vitro and in vivo. This population of rare cells is the only one with the ability to initiate and maintain the tumor, allow the tumor propagation, colonize distant sites, or transplant the tumor into other living beings. The other cells forming the tumor would be transient amplifying cells and mature cells with limited or no ability to initiate and/or maintain the tumor. This hierarchical model of cell organization, which is the basis for the definition of a cancer stem cell (CSC), is not novel. It is believed to be the paradigm during embryonic development, and in different tissues, for example, in high-turnover tissues such as the epithelium and the hematopoietic system, with a hierarchical system based on the existence of a stem cell that has self-renewal and multi-potential differentiation ability. Furthermore, this hierarchical model has also been extrapolated to many different adult tissues. This review will focus on the recent advances in the CSC model and on future developments to a feasible clinical targeting of the human CSC.
The CSC Model
The CSC hypothesis suggests that cancer propagation is usually driven by subpopulations of cancer cells having stem cell properties, regardless of whether they arise from a normal stem cell or not. The working definition of a CSC is therefore, a cell with “tumor-initiating” ability. This definition is based on the principles of stem cell biology, originally developed for hematopoiesis, of clonal assays, and prospective cell purification of putative tumor-initiating populations with a recognizable immunophenotype. Similarly to normal stem cells, CSCs would also form cancer tissue by undergoing epigenetic changes, and differentiate into phenotypically diverse progeny, that ultimately form functionally diverse sets of non-tumorigenic cancer cells which compose the bulk of the cells in a tumor. This effect of causation, based on an adapted Koch’s third postulate, is the basis of the CSC-initiated tumor theory (Dick, 2008). Despite strong evidence supporting a stem cell origin for some cancers, it is important to acknowledge a number of caveats.
First, there is no direct evidence that tumorigenic cells significantly differ from non-tumorigenic cells as a result of epigenetic rather than genetic differences (Dick, 2008; Visvader and Lindeman, 2008; Rosen and Jordan, 2009). Our current understanding of carcinogenesis is that cumulative mutations develop during cancer progression (Gilliland and Griffin, 2002). In theory, these mutations could result in more than one cell population able to maintain and propagate the tumor, by acquiring a self-renewal program. This theory is supported by the finding of tumors in which more than one phenotypically distinct population can initiate tumors in vivo (Hermann et al., 2007). Whether these mutations share commonalities in their epigenetic programs has not been sufficiently addressed. Second, a lineage committed progenitor cell can also function like a CSC by acquiring self-renewal properties, and behave as a cell of origin of disease transformation (Jamieson et al., 2004). Whether these two types of cells share similar molecular genetic signatures that are necessary for tumor progression is questionable. One source of doubt comes from re-programming studies where embryonic stem cells, with similar functional properties to induced pluripotent stem cells, were found to diverge in their epigenetic programs (Meissner and Jaenisch, 2006; Hanna et al., 2009). Third, the conclusion based on an adapted Koch’s third postulate, in which transplanted CSC can recapitulate the heterogeneity of the tumors from which they derive, is based on limited analyses of only two or three surface markers (Visvader and Lindeman, 2008; Zhou et al., 2009). It has not yet been determined whether there is also genetic heterogeneity within the primary tumors, which is not recapitulated after CSC transplantation. Therefore, it remains possible that the functional and phenotypic diversity within these cancers is underestimated and partially genetically determined. Fourth, a fundamental question in the CSC hypothesis, relates to the “scarcity” of the CSC component in the tumor. The method of transplantation of putative CSC in immunodeficient mice has been used as the gold standard to estimate and quantify CSC function in vivo. Using this method, it is possible to underestimate the frequency of human cancer cells with tumorigenic potential in some cancers (Kelly et al., 2007; Rosen and Jordan, 2009). Also primary tumors and their counterparts developed in immunodeficient mice differ considerably. There are only a few studies using tumors initiated by a single cell and propagated in limiting dilution assays, where the self-renewal ability of the tumor-initiating cells has been demonstrated (Quintana et al., 2008). Seeding and tumor development in animals varies widely from tumor to tumor, as demonstrated in acute myelogenous leukemia (AML) (Hope et al., 2004; Jin et al., 2009), suggesting that xenotransplantation may underestimate the frequency of tumor-sustaining cells in vivo. Interestingly, a recent report shows that gender-associated factors in immunodeficient recipient mice (NOD/SCID or NOD/SCID/IL-2Rgc null) may also play a decisive role in the survival, proliferation, and engraftment, but not homing, of human hematopoietic stem cells HSC in a standard xenograft model in vivo (Notta et al., 2010). Therefore, there is a serious need to re-evaluate the evidences supporting the hierarchical model using assays that allow better engraftment of human cancer cells. Nevertheless, there are distinctive subpopulations of cells residing within tumors with stem cell-like properties, which can be isolated using a variety of cell-surface antigens using flow cytometry. This has allowed researchers to characterize the specific signaling pathways in these groups of cells as compared with the bulk tumor cells and their normal counterparts (Table 1).
TABLE 1.
Immunophenotypic characterization of human CSC
| Human cancer type | CSC immunophenotype | Tumor cells-expressing CSC marker (%) | Site of transplantation; # of CSC injected | Recipient mice | Refs. | |
|---|---|---|---|---|---|---|
| Leukemia | CD34+/CD38− (AML) | Intravenous | NOD/SCID | Bonnet and Dick (1997) | ||
| Leukemia | CD34+/CD38−/CD19+ t(12;21) (ALL) | 1–4 | Intravenous | NOD/SCID | Castor et al. (2005) | |
| Leukemia | CD34+/CD38−/CD19 t(9;22) (CML-BC) | Intravenous | NOD/SCID | Castor et al. (2005) and Jamieson et al. (2004) | ||
| Breast carcinoma | CD44+/CD24−/low/Lin | 11–35 | Mammary fat pad; 200 | NOD/SCID | Al-Hajj et al. (2003) | |
| Breast carcinoma | ALDH1+ | 3–10 | Mammary fat pad; 500 | NOD/SCID | Ginestier et al. (2007) | |
| Brain tumor | CD133+ | 6–29 | Brain; 100 | NOD/SCID | Singh et al. (2004) | |
| Glioblastoma | CD133+ | 2–3 | Brain; 500 | nu/nu | Bao et al. (2006a) | |
| Colon carcinoma | CD133+ | 1.8–25 | Kidney capsule; 200 | NOD/SCID | O’Brien et al. (2007) | |
| Colon carcinoma | EpCAMhi/CD44+ | 0.03–38 | Subcutaneous; 200 | NOD/SCID | Dalerba et al. (2007) | |
| Head and neck cancer | CD44+ | 0.1–42 | Subcutaneous; 5 × 103 | Rag2/γ−/− DKO NOD/SCID | Prince et al. (2007) | |
| Pancreas | CD133+ | 1–3 | Pancreas; 500 | NMRI-nu/nu | Hermann et al. (2007) | |
| Pancreas | CD44+/CD24+/ESA+ | 0.2–0.8 | Pancreas; 100 | NOD/SCID | Li et al. (2007) | |
| Lung carcinoma | SP-C+CCA+ | Kim et al. (2005) | ||||
| Lung carcinoma | CD133+ | 0.32–22 | Subcutaneous; 104 | NOD/SCID | Eramo et al. (2008) | |
| Liver cancer | CD90+ | 0.03–6 | Liver; 5 × 103 | SCID/Beige | Yang et al. (2008) | |
| Melanoma | ABCB5+ | 1.6–2.0 | Subcutaneous; 106 Subcutaneous; 102–107 |
NOD/SCID NOD/SCID Il2rg−/− |
Schatton et al. (2008) Quintana et al. (2008) |
|
| Prostate |
|
Collins et al. (2005) |
ALDH, aldehyde dehydrogenase; CML-BC, chronic myelogenous leukemia-blast crisis; EpCAM, epithelial cell adhesion molecule; ESA, epithelial-specific antigen; NOD/SCID, non-obese diabetic-severe combined immunodeficient; Rag2/γ−/− DKO, Rag2 common cytokine receptor γ-chain double knockout.
CSC in Hematological Malignancies
The first evidence of CSC came from pioneering studies on human AML, where it was shown that CD34+ CD38− AML stem cells are organized as a hierarchy that originates from a primitive HSC (Bonnet and Dick, 1997; Dick, 2008). Xenotransplantation is instrumental in supporting human HSC engraftment, allowing quantitative assessment and purification of normal HSCs. The disease pattern observed in the non-obese diabetic severe combined immune (NOD/SCID) deficient mice is reflected in the clinical features of the human disease, pointing to the reliability of this xenograft model, compared with the older method of subcutaneous implantations in nude mice to mimic the human disease. Despite their myelomonocytic morphology, and low CD34+ and CD34+CD38− populations, leukemic stem cells (LSCs) were predominantly found in CD34+CD38− fractions. Some exceptions were reported in following years (Taussig et al., 2008), however, a significantly high proportion of AML patients show LSC activity in the CD34+CD38− population fraction, suggesting a hierarchical model in human AML. This fraction is not functionally homogenous, it may be comprised of distinct hierarchically arranged LSC subpopulations (Bonnet and Dick, 1997), including quiescent LSC.
The existence of a LSC population was subsequently observed for other human leukemias, including acute lymphoblastic leukemia (ALL), and chronic myelogenous leukemia (CML) presenting blast crisis (Jamieson et al., 2004; Castor et al., 2005). Two parameters seem to influence the phenotypic and functional properties of LSC. The first is the cell of origin. In chronic phase CML and some types of AML, the LSC compartment seems to be relatively well defined and demonstrates biological properties reminiscent of a normal hematopoietic hierarchy. In other leukemias, like ALL, t(12;21) containing ETV6-RUNX1 in particular, LSC are predominantly enriched in the CD34+CD38−CD19+ fraction (Castor et al., 2005). Interestingly, p210 and p190 BCR-ABL ALLs represent distinct tumor biological and clinical entities. The major breakpoint BCR-ABL fusions, encoding p210-BCR-ABL, originate in the HSC (Sengupta and Banerjee, 2007; Sengupta et al., 2007; Thomas et al., 2007), whereas minor BCR-ABL fusions, encoding p190-BCR-ABL usually have a B-cell progenitor origin (Calabretta and Perrotti, 2004). Similarly, the transformed leukemia-initiating stem cells in both p190 and p210 BCR-ABL+ ALLs, but not in p210-BCR-ABL+ CML, have a committed B progenitor phenotype, suggesting multi-clonal involvement of HSC in BCR-ABL+ hematological malignancies (Castor et al., 2005). Using a stem cell leukemia (Scl) promoter-driven binary transgenic mouse model of p210-BCR-ABL, we have recently shown that deficiency of hematopoietic system-specific Rac2 GTPase causes functional exhaustion of the CML HSC/P pool in vivo (Sengupta et al., 2010). We have demonstrated that absence of Rac2 GTPase prolongs survival of HSC-initiated, inducible Scl/p210-BCR-ABL mice, it induces apoptosis and, unlike in normal HSC/P (Gu et al., 2003; Cancelas et al., 2005), impairs LSC/P proliferation in vivo. However, this defect was not due to impaired interaction with hematopoietic microenvironment as reflected by its unaltered adhesion, migration, and homing to recipient organs, suggesting, Rac2-deficiency exhausts the LSC pool in vivo through impairment of oncogene-induced proliferation and survival signals (Sengupta et al., 2010).
The second factor that seems to influence the phenotypic and functional properties of LSC is the mutation-specific, self-renewal program induced by different mutations that are responsible for leukemogenesis. For instance, leukemias arising from MLL translocations, and/or some lymphoid leukemias, may be more variable and show much less similarity to normal hematopoiesis (Somervaille et al., 2009), with LSC frequencies much higher than would be predictable from hematopoietic-like hierarchical models. In a recent study, using syngenic mouse lines, it has been shown that the Wnt/β-catenin pathway is required for the development of leukemia stem cells in AML induced either by coexpression of the Hoxa9 and Meis1a oncogenes or by the fusion oncoprotein MLL-AF9 (Wang et al., 2010). Accordingly, Wnt/β-catenin signaling pathway is required for the self-renewal of LSCs that are derived from either HSC or more differentiated granulocyte–macrophage progenitors (GMP).
In another study, using syngenic mouse transplantation model, CD34+c-kit+FcgammaRIII/II+Gr1+/lo, a committed myeloid progenitor compartment, has been identified as the cancer-initiating cell in acute promyelocytic leukemia (APL) (Guibal et al., 2009). APL is characterized by a block in differentiation and accumulation of promyelocytes in the bone marrow and blood. The majority of APL patients harbor the t(15:17) translocation leading to expression of the fusion protein promyelocytic-retinoic acid receptor alpha (RAR-α). Treatment with retinoic acid (ATRA) leads to degradation of promyelocytic-retinoic acid receptor alpha protein and disappearance of leukemic cells; however, 30% of APL patients relapse after treatment. One possible explanation for relapse is the persistence of CSC in hematopoietic organs after treatment. It has been shown that APL-initiating cells down-regulate the transcription factor CCAAT/enhancer binding protein alpha (C/EBPalpha) possibly through a methylation-dependent mechanism, suggesting that C/EBPalpha deregulation contributes to transformation of APL cancer-initiating cells (Guibal et al., 2009).
However, three potential problems arise in the current studies using CSC-initiated leukemia models. First, syngeneic mouse models provide mechanistic systems to understand basic biology, but may not adequately reflect the genetic diversity/instability found in human disease, and it is unclear that experimental therapies can be effectively modeled in syngeneic systems. Second, xenograft models permit the analysis of primary human LSC, but may be limited with regard to providing an authentic microenvironment in which to study biology and drug response. LSC frequency in AML can vary over many orders of magnitude, depending on the subtype of AML, degree of sophistication of the immunodeficient animal model or the use of intravenous or intrafemoral administration of tumor cells, and can range from 1 in 100 to less than 1 in 106. Third, the extent to which results in any model system (either syngeneic or xenogeneic) can actually predict the outcome in human leukemia patients. While the biological sophistication of many systems has greatly improved in the past decade, a major challenge for the future is to determine whether such models will provide a superior means by which to design new therapeutic regimens.
Despite the limitations noted above, the available evidence suggests that more specific targeting of LSC will lead to improved therapeutic outcomes. This observation is true irrespective of the details inherent to any particular form of leukemia. Even if LSC are highly variable, abundant, or unstable, they are no less relevant as a potential drug target. Several exciting lines of investigation from pre-clinical studies have been recently reported that may provide better eradication of LSC. These approaches include the use of monoclonal antibodies to specific epitopes on LSC, and small molecules designed to block self-renewal pathways and/or leukemia-specific survival mechanisms.
CSC in Solid Tumors
Almost a decade after the identification of AML tumor-initiating cells (Bonnet and Dick, 1997), and the emergence of LSC, mounting data indicates that CSCs also exist in multiple solid tumors (Visvader and Lindeman, 2008). A number of cell surface markers have been used to isolate putative CSCs, including CD133, CD44, CD24, epithelial cell adhesion molecule (EpCAM), etc. (Al-Hajj et al., 2003; Bao et al., 2006a; Hermann et al., 2007; Eramo et al., 2008). Common cell surface markers, in particular CD133 and CD44 (hyaluronic (H)-CAM), have been predominantly used to characterize CSCs in multiple solid tumors. Hoechst exclusion by side population (SP) is also considered as a CSC-specific property. However, most of these markers are also expressed by normal adult stem cells, suggesting a need for novel cell surface markers, specific for the tumor-selective stem cell population.
The first solid malignancy from which CSCs were identified and isolated was a breast carcinoma where the CD44+CD24−/lo cell population was considered to have the tumor-initiating capacity (Al-Hajj et al., 2003). CD44 (H-CAM), a widely expressed adhesion molecule, either alone or in combination with other membrane markers has been used to identify CSC subsets in a variety of cancers, including head and neck (CD44+), colon (CD44+/epithelial-specific antigen (ESA)+), and pancreas (CD44+/CD24+/ESA+) (Table 1). Interestingly, bone marrow insertion-1 (Bmi1), a polycomb family gene involved in self-renewal (Lessard and Sauvageau, 2003), was differentially overexpressed in CD44+ cells versus CD44− cells from head and neck (Prince et al., 2007), and sonic hedgehog, a signal associated with progenitor proliferation (Zhao et al., 2009) and early initiation of pancreatic cancer (Morton et al., 2007), was also found to be overexpressed in CD44+/CD24+/ESA+ cells compared with the non-tumor-initiating cell fractions (Dalerba et al., 2007). CD44 function in tumorigenesis is further discussed below.
Subsequently, CD133 (prominin-1, a five-transmembrane domain glycoprotein) was found to mark CSCs in different types of brain tumors, including glioblastoma multiforme, pediatric medulloblastoma, and ependymomas (Singh et al., 2004; Bao et al., 2006a). In addition, CD133 was used in identifying CSCs in colorectal and pancreatic carcinomas (Hermann et al., 2007; O’Brien et al., 2007). CD44+α2 integrin+β1 integrinhi CD133+ population has been found to be enriched as putative human prostate CSC with an extensive proliferative capacity in vitro (Collins et al., 2005). Furthermore, high numbers of CD133+EpCAM+ cells isolated from fresh lung tumor specimens were capable of generating tumor xenografts upon subcutaneous injection (Dalerba et al., 2007). CD133 was first described as a cell surface antigen present on HSC/P and neural progenitors (Uchida et al., 2000; Hess et al., 2004). There is no clear understanding of the function of CD133, and even though CD133 expression can be used for CSC enrichment, there is no direct evidence that every CD133+cell represents a CSC. For instance, glioblastomas may also be propagated from CD133− cells, and CD133− glioblastoma cells do have in vivo tumorigenic properties that are similar to CD133+ cells in nude mice. This suggests that more phenotypic and functional heterogeneity may exist within the identifiable CSC compartment.
The use of SP has also been applied to the isolation and characterization of CSC. This assay, developed by Margaret Goodell and Richard Mulligan (Goodell et al., 1996, 1997), exploits the high levels of expression of drug-resistance transporters on the membrane of stem cells. SP cells have been shown to enrich in CSC in musculoskeletal sarcomas, and their frequency correlates with the aggressiveness of the tumor (Kim et al., 2005). One of the major components of the drug-resistance phenotype is the expression of adensosine-5′-triphosphate-binding cassette (ABC) proteins. Further support for the use of a drug-resistance phenotype in melanoma has been found using the expression of one of the ABC proteins, ABCB5 (Schatton et al., 2008). ABCB5+ melanoma cells were capable of being serially transplanted, showing a capacity for self-renewal. ABCB5+ cell-generated tumors were differentially sensitive to in vivo administration of an anti-ABCB5 antibody capable of inducing antibody-dependent cell-mediated cytotoxicity.
Finally, peripheral nerve human neurofibromas were identified as being initiated by human epidermal growth factor receptor (EGFR)+/p75+ cells in neurosphere cultures and in xenogeneic transplantation assays. EGFR+/p75+ neurospheres are enriched in EGF-dependent sphere-forming cells and contain glial-like progenitors that differentiate into neurons and stromal lineages in vitro and form benign neurofibroma-like lesions in nude mice, suggesting that the expansion of an EGFR-expressing early glial progenitor contributes to neurofibroma formation (Williams et al., 2008; Wu et al., 2008).
CSC–Microenvironment Interaction
CSCs may hijack normal stem cell niche for their proliferation, survival, and possibly for establishment of secondary niches during ulterior phases of disease evolution. The evidence for CSC–niche interaction comes from two recent reports showing the dependence of human AML stem cells and murine CML stem cells and progenitors on CD44 (H-CAM), a plasma membrane bound hyaluronic acid receptor, expressed on both normal and CSCs (Jin et al., 2006; Krause et al., 2006). Anti-CD44 antibody-treated NOD/SCID mice xenotransplanted with primary human AML cells, and BCR-ABL-transduced CML stem cells and progenitors derived from CD44 mutant donor mice, both have impaired and delayed development of leukemia in vivo. This indicates that CD44 expression may be essential for the adequate homing and engraftment of the CSCs to their niche. In addition, CD44 blockade has also been shown to prevent the formation of local and metastatic tumor nodules initiated by CD90+CD44+hepatocellular carcinoma cells in the lung of the xenografted immunodeficient mice (Yang et al., 2008), suggesting the dependence of CSC on CD44 signaling in the tumor microenvironment. In another example, lymphoblastic leukemic cell growth has been shown to disrupt normal hematopoietic progenitor niches and create abnormal stem cell factor (SCF)-rich microenvironments that sequester transplanted human HSC. In this model of xenografted lymphoblastic leukemia, normal CD34+ cells declined in number over time and failed to mobilize into the peripheral circulation in response to cytokine stimulation. Neutralization of SCF secreted by leukemic cells inhibited CD34+ cell migration into malignant niches, normalized CD34+ cell numbers, and restored CD34+ cell mobilization in leukemic mice, suggesting that the leukemic microenvironment causes normal progenitor dysfunction by displacing normal progenitors from their niches and that therapeutic inhibition of the normal progenitor interaction with tumor niches may be a therapeutic tool (Colmone et al., 2008).
The involvement of tumor microenvironment has also been proposed in other solid tumor mouse models. In a tumor metastasis model of lung carcinoma, vascular endothelial growth factor (VEGF) secreted by primary tumor cells has been shown to induce selective expression of matrix metalloproteinase 9 (MMP9) in lung endothelial cells and macrophages, thereby resulting in the formation of a localized CSC–niche in vivo (Hiratsuka et al., 2002). In a more recent work, the concept of “pre-metastatic niche” has been introduced that may support the development and maintenance of human breast carcinoma and lung cancers (Kaplan et al., 2005). According to this model, the existence of a novel type of CSC–niche was found that was reconstituted with bone marrow-derived cells (BMDC), which progressively developed into lung carcinoma or melanoma. The transplanted α4β1 integrin+ BMDC, expressing VEGF receptor (VEGFR1), colonized at lung that subsequently created a group of cells, known as “pre-metastatic niche.”
Although there is substantial evidence for an instructive role of the tumor microenvironment, the existence and architecture of the CSC–niche remain elusive. Nevertheless, aberrant CSC–niches may result in disease, as exemplified by an altered HSC–niche leading to the development of myeloproliferative disease and myelodysplasia followed by emergence of secondary leukemia, respectively (Walkley et al., 2007; Raaijmakers et al., 2010). Mesenchymal cells predominantly contribute to the stroma of normal and malignant tissues. It has been reported that deletion of Dicer1, a double stranded RNA-specific ribonuclease, specifically in mouse osteoprogenitors, but not in mature osteoblasts, disrupts the integrity of hematopoiesis (Raaijmakers et al., 2010). Interestingly, Dicer1 deletion results in reduced expression of Sbds, the gene mutated in Schwachman–Bodian–Diamond syndrome—a human bone marrow failure and leukemia pre-disposition condition (Raaijmakers et al., 2010). Deletion of Sbds in mouse osteoprogenitors also phenocopies bone marrow dysfunction with myelodysplasia, suggesting perturbation of specific mesenchymal subsets of stromal cells can disorder differentiation, proliferation, and apoptosis of heterologous cells, and disrupt tissue homeostasis. Therefore, primary stromal dysfunction can result in secondary neoplastic disease, supporting the concept of niche-induced oncogenesis. Altered HSC–niche interaction has also been shown in retinoblastoma (Rb)-deficient hematopoietic microenvironment that induces myeloproliferation (MPD) (Walkley et al., 2007). Widespread inactivation of Rb in the murine hematopoietic system resulted in massive mobilization of HSC to extramedullary sites and differentiation (Walkley et al., 2007). A schema depicting the putative role(s) of the LSC niches in leukemia development is presented in Figure 1. However, this phenotype was not intrinsic to HSC, rather it was secondary to Rb-dependent interaction between myeloid-derived cells and the stem cell microenvironment, suggesting perturbation in the extrinsic stem cell niche can favor towards abnormal proliferation of the stem cell compartment. Moreover, glioblastoma and medulloblastoma CSCs also appear to be maintained by signals from an aberrant vascular niche that mimics the normal stem cell niche. Parallel findings showed that freshly isolated CD133+ CSC-enriched cells, but not CD133−glioblastoma cells, can form highly vascular tumors in the brains of immunocompromised mice (Bao et al., 2006b; Calabrese et al., 2007). In a different study, it has been found that proliferation of CD133+ colon cancer cells depend on interleukin (IL)-4 signaling in a paracrine manner (Todaro et al., 2007). Interestingly, blocking IL-4 signaling can significantly enhance the sensitivity of CD133+ cells against chemotherapy (Todaro et al., 2007). Reactive oxygen species (ROS) can also affect the interaction between stem cells and their microenvironment (Hosokawa et al., 2007). Oxidative stress can affect the self-renewal of long-term HSC by acting through the p38 MAPK pathway (Ito et al., 2006). Interestingly, 5-fluorouracil (5-FU) treatment causes a shift of the mouse LSK-SP fraction in the bone marrow to non-SP compartment. However, treatment with the antioxidant N-acetyl-L-cysteine (NAC) inhibits this transition and maintains the SP fraction for a longer period of time. This suggests that ROS can induce cell cycle, thereby allowing them to escape from the microenvironment.
Fig. 1.
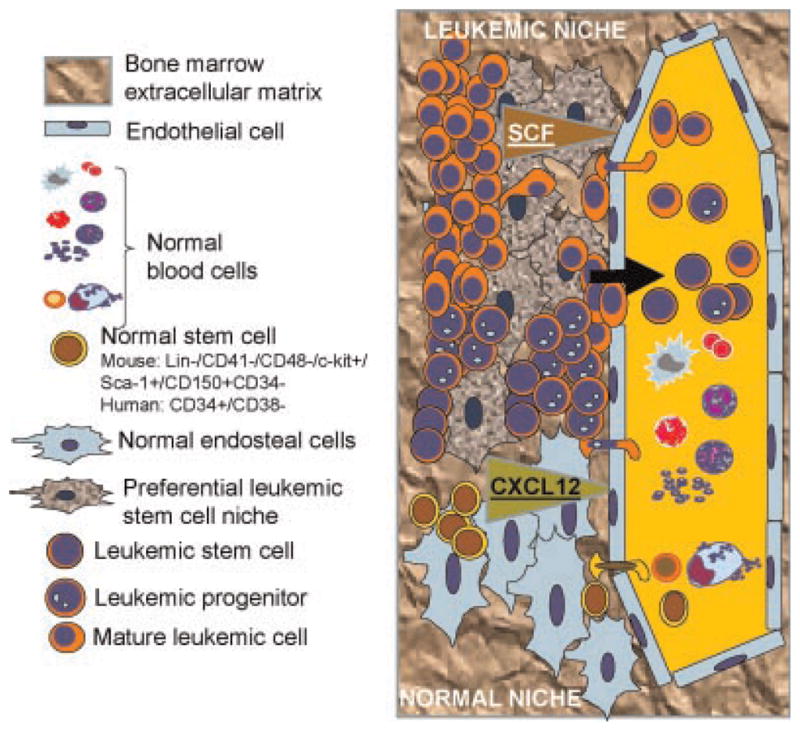
Migration within the leukemic stem cell (LSC) niches of the bone marrow endosteal space in relationship with their different components. The normal bone marrow stem cell niche comprises of mesenchymal cells and vascular cells within an acellular bone mineral matrix (fibronectin, osteopontin, collagen, hemonectin, and calcium). The niche cells comprise endothelial cells, CXCL12-expressing adventital reticular (CAR) cells, osteoprogenitors, osteoblast precursors, and mesenchymal stem cells. The LSC and their niche interact by expressing cytokines (e.g., SCF) and adhesion molecules like hialuronid acid to bind c-kit and CD44 receptors in leukemic stem cells. The LSC may impair the function of the normal HSC–niche, infiltrate and disrupt the normal pattern of proliferation and differentiation as shown later in Figure 2, leading to formation of a proposed alternative niche, which would allow increased migration of leukemic cells out of the bone marrow while retaining a significant fraction of the LSC population.
Implications of the CSC Hypothesis for Cancer Therapy
While the stochastic model explains chemoresistance as the selection of pre-existing clones with drug-resistance conferring mutations, the CSC model postulates that the tumor cell heterogeneity arises because the tumor is organized as a cellular hierarchy sustained by a CSC at the highest level, where the CSC is a quiescent, primary chemotherapy-resistant cell, and where all the relevant mutations in cancer maintenance and progression accumulate.
The frustrations associated with developing therapy approaches based on the stochastic, random organization of cancer have allowed the rapid (and perhaps not-so-critically discussed) emergence of the CSC model as a therapeutic alternative. The CSC model has favored the conceptual development of targeted therapies towards molecular pathways relevant in CSC maintenance and progression, independent of their ability to induce cell death of non-CSC. Targeted therapies on the CSC compartment could provide cancer curability through, (a) elimination of primary tumor chemoresistance inducing apoptosis or proliferation blockade of CSC; (b) differentiation of CSC into non-tumorigenic cells, in association with conventional cytotoxic therapies; or (c) interference in the interaction of stem cells with their nurturing microenvironment. The overall expectation is that combinatorial treatments involving both cytotoxic and targeted therapies would ablate the bulk tumor burden, including the CSC component. A schema of all the cancer properties attributed to the CSC and potential areas of therapeutic intervention is depicted in Figure 2.
Fig. 2.

Fates of cancer stem cells relevant for therapeutic intervention. A: Normal stem cell population is homeostatically controlled by intrinsic and extrinsic genetic programs that allow cells to maintain an equilibrium between self-renewal, differentiation, senescence/apoptosis, and migration out of the bone marrow. B: Cancer stem cells, however, display increased proliferation and self-renewal, decreased apoptosis/senescence and increased migration of tumor-initiating cells ready for invasiveness of distant tissues. Cancer-induced increased expansion of the transient amplifying cells may be responsible of mutation accumulation in the context of chromosomal instability, with the ability to create new populations of cancer stem cells. Green areas denote areas of opportunity for cancer therapy. Arrows’ weight depict the relative weight of that specific fate.
Induction of chemoresistance through selection of more aggressive CSC has been documented in vitro and in vivo. Hermann et al. (2007) showed that CD133+ pancreatic cancer cells showed a preferential resistance to the drug gemcitabine in vitro and in vivo when compared with CD133− cells from the same tumor. In colon cancer, CD133− cells treated with oxaliplatin and/or 5-FU showed a selective, high sensitivity to both in vitro and in vivo drug treatment (Dalerba et al., 2007; O’Brien et al., 2007), while CD133+ cells showed primary chemoresistance through an autocrine IL-4 loop which could be interfered in vivo by administration of anti-IL-4 neutralizing antibodies. Colon CSC (CD44+/ESA+) (Dalerba et al., 2007) also seem to express high levels of aldehyde dehydrogenase, which has been shown to mediate chemoresistance to cyclophosphamide but not to other unrelated drugs.
Several clinical studies are in progress to correlate CSC content in tumors and clinical outcomes. In one, lapatinib, a human epidermal growth factor receptor 2 (EGFR/HER2) pathway inhibitor, has been shown to specifically target the CD44+/CD24− breast cancer cell population in the setting of a neoadjuvant clinical trial for locally advanced breast cancer patients (Al-Hajj et al., 2003). CD44+/CD24−, mammosphere-forming ability and xenografting-cell content in residual breast cancer specimens increased after conventional chemotherapy with docetaxel or a combination of cyclophosphamide and doxorubicin, while remaining steady in the residual cancer of patients treated with the combination of lapatinib and conventional chemotherapy. Finally, radioresistance has also been shown to be associated with enriched CSC content in brain tumors. Bao et al. (2006a) found that glioma CD133+ cells were enriched in vitro and in vivo after irradiation through DNA damage response activation, which could be interfered with using a small molecule inhibitor of Chk1/2 kinases.
Blocking of differentiation is a common hallmark in cancer. Differentiation-inducing therapies in cancer have been proven successful since all-trans retinoic acid (ATRA) was incorporated into the first-line therapy for APL. The combination of ATRA and conventional chemotherapy has revolutionized the treatment of this disease. Recently, in a syngeneic murine model, an APL-initiating cell population (CD34+, c-kit+, FcγRIII/II+, Gr1bright) has been shown to represent a promyelocytic phenotype, generate leukemia upon transplantation, and exhibit down-regulation of C/EBPα (CCAAT/enhancer-binding protein alpha) by a methylation-dependent mechanism (Guibal et al., 2009). In solid tumors, transient exposure of glioblastoma cells to bone morphogenetic protein-4 (BMP-4) in vitro or in vivo resulted in no cell death, and reduction of proliferation and clonogenic ability, while displaying increased expression of markers of neural differentiation (Piccirillo et al., 2006). Interestingly, glioblastoma CSC can defend themselves from the differentiation action of BMPs by enhancer of zeste homolog 2 (EZH2)-dependent epigenetic silencing of BMP receptor 1B (BMPR1B) (Lee et al., 2008). As many tumor-initiating cells may depend on a particular niche to maintain their identity and fate (Colmone et al., 2008; Wei et al., 2008), targeting the CSC–microenvironment in specific ways could also be a strategy to indirectly inhibit or differentiate tumor-initiating cells. There is increasing interest in the possibility of exploiting the putative CSC–niche for drug targeting. CSCs may dictate expansion of the normal niche as they proliferate. For instance, CD133+ colon cancer cells depend on IL-4 signaling in a paracrine manner, possibly tumor microenvironment-dependent mechanism, in order to protect themselves from apoptosis (Todaro et al., 2007). A recent study has shown that blocking IL-4 signaling can significantly enhance the sensitivity of CD133+ cells against chemotherapy (Todaro et al., 2007). In addition, treatment of CD133+ cells with bevacizumab, a VEGF-neutralizing monoclonal antibody, markedly inhibited their ability to initiate tumors in vivo and depleted both blood vessels and self-renewing CD133+ cells from tumor xenografts (Bao et al., 2006b; Calabrese et al., 2007). Therefore, anti-angiogenic therapy in conjunction with cytotoxic chemotherapy could be effective in targeting tissue-specific CSCs.
Summary and Future Trends
Undoubtedly, proper characterization and refinement of the tools used for the identification, isolation, and propagation of CSCs may lead to a better understanding of how these cells initiate and sustain tumor growth. Many of the models of CSC propagation published are based on “too-large” populations of putative CSC to believe that all of them share the required property of an individual ability to initiate tumors in vivo. Analysis of the role of accessory (extrinsic) cells and pathways of CSC activation may be the next wave of work. The generation of mouse models that develop heterogeneous tumors and more closely resemble the human malignancies and xenograft tumors from patients, would provide powerful tools for evaluating the therapeutic sensitivity of CSCs. Finally, a low level of dogmatism and high level of skepticism towards each new CSC model are necessary. As an example, for half-a-century, we have been building a hierarchical system in hematopoiesis based on the presence of a unique, multipotential, self-renewing stem cell, able to generate all the different progenies of hematopoietic cells, and responsive to an internal program and external cues. Very recently, such a model was challenged and possible alternative explanations for the heterogeneity of differentiation-biased HSC were proposed (Challen et al., 2010). This suggests that heterogeneity of stem cells in highly refined competitive repopulation assays could be eventually translated into CSC models. Furthermore, although many cancers are initiated by CSC, however, during the terminal acute and metastatic phase of the disease, the tumor burden may not be completely explained or rather tackled only at the CSC level. According to the “Two-hit” model of cancer development, as elegantly proposed by Gary Gilliland, cancer progression is not only defined by the initial hit at the CSC level, rather accumulation of additional secondary changes significantly contribute towards the malignant transformation (Gilliland and Griffin, 2002; Kelly et al., 2002). Therefore, although targeting only at the CSC compartment could be effective at the initial phase of cancer, it may not be so at the subsequent malignant phase of the disease.
Developing effective cancer treatment(s) by focusing therapy on the relatively more malignant and quiescent cells could be a direct result of the application of CSC models to tumor growth. In order to achieve this goal it is important to determine which cancers follow a CSC model and which do not, and to address technical issues related to tumorigenesis assays. Independent of whether the CSC model may be relevant to all tumors or not, the hunt for novel approaches to target tumor cells that are refractory to current conventional therapies continues.
Acknowledgments
Contract grant sponsor: NIH;
Contract grant number: HL087159.
Contract grant sponsor: Department of Defense;
Contract grant number: CM064050.
Contract grant sponsor: National Blood Foundation.
Contract grant sponsor: Alex Lemonade Stand Foundation.
A.S. is a recipient of The Institute of Cancer Research, UK/Lady Tata Postdoctoral Fellowship in Leukemia. J.A.C. acknowledges funding from NIH (HL087159), Department of Defense (CM064050), National Blood Foundation, and Alex Lemonade Stand Foundation. We thank Margaret O’Leary for editorial assistance.
Literature Cited
- Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006a;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- Bao S, Wu Q, Sathornsumetee S, Hao Y, Li Z, Hjelmeland AB, Shi Q, McLendon RE, Bigner DD, Rich JN. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res. 2006b;66:7843–7848. doi: 10.1158/0008-5472.CAN-06-1010. [DOI] [PubMed] [Google Scholar]
- Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730–737. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, Hamner B, Oh EY, Gaber MW, Finklestein D, Allen M, Frank A, Bayazitov IT, Zakharenko SS, Gajjar A, Davidoff A, Gilbertson RJ. A perivascular niche for brain tumor stem cells. Cancer Cell. 2007;11:69–82. doi: 10.1016/j.ccr.2006.11.020. [DOI] [PubMed] [Google Scholar]
- Calabretta B, Perrotti D. The biology of CML blast crisis. Blood. 2004;103:4010–4022. doi: 10.1182/blood-2003-12-4111. [DOI] [PubMed] [Google Scholar]
- Cancelas JA, Lee AW, Prabhakar R, Stringer KF, Zheng Y, Williams DA. Rac GTPases differentially integrate signals regulating hematopoietic stem cell localization. Nat Med. 2005;11:886–891. doi: 10.1038/nm1274. [DOI] [PubMed] [Google Scholar]
- Castor A, Nilsson L, Astrand-Grundstrom I, Buitenhuis M, Ramirez C, Anderson K, Strombeck B, Garwicz S, Bekassy AN, Schmiegelow K, Lausen B, Hokland P, Lehmann S, Juliusson G, Johansson B, Jacobsen SE. Distinct patterns of hematopoietic stem cell involvement in acute lymphoblastic leukemia. Nat Med. 2005;11:630–637. doi: 10.1038/nm1253. [DOI] [PubMed] [Google Scholar]
- Challen GA, Boles NC, Chambers SM, Goodell MA. Distinct hematopoietic stem cell subtypes are differentially regulated by TGF-beta1. Cell Stem Cell. 2010;6:265–278. doi: 10.1016/j.stem.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005;65:10946–10951. doi: 10.1158/0008-5472.CAN-05-2018. [DOI] [PubMed] [Google Scholar]
- Colmone A, Amorim M, Pontier AL, Wang S, Jablonski E, Sipkins DA. Leukemic cells create bone marrow niches that disrupt the behavior of normal hematopoietic progenitor cells. Science. 2008;322:1861–1865. doi: 10.1126/science.1164390. [DOI] [PubMed] [Google Scholar]
- Dalerba P, Dylla SJ, Park IK, Liu R, Wang X, Cho RW, Hoey T, Gurney A, Huang EH, Simeone DM, Shelton AA, Parmiani G, Castelli C, Clarke MF. Phenotypic characterization of human colorectal cancer stem cells. Proc Natl Acad Sci USA. 2007;104:10158–10163. doi: 10.1073/pnas.0703478104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick JE. Stem cell concepts renew cancer research. Blood. 2008;112:4793–4807. doi: 10.1182/blood-2008-08-077941. [DOI] [PubMed] [Google Scholar]
- Eramo A, Lotti F, Sette G, Pilozzi E, Biffoni M, Di Virgilio A, Conticello C, Ruco L, Peschle C, De Maria R. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2008;15:504–514. doi: 10.1038/sj.cdd.4402283. [DOI] [PubMed] [Google Scholar]
- Gilliland DG, Griffin JD. The roles of FLT3 in hematopoiesis and leukemia. Blood. 2002;100:1532–1542. doi: 10.1182/blood-2002-02-0492. [DOI] [PubMed] [Google Scholar]
- Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, Jacquemier J, Viens P, Kleer CG, Liu S, Schott A, Hayes D, Birnbaum D, Wicha MS, Dontu G. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1:555–567. doi: 10.1016/j.stem.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodell MA, Brose K, Paradis G, Conner AS, Mulligan RC. Isolation and functional properties of murine hematopoietic stem cells that are replicating in vivo. J Exp Med. 1996;183:1797–1806. doi: 10.1084/jem.183.4.1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodell MA, Rosenzweig M, Kim H, Marks DF, DeMaria M, Paradis G, Grupp SA, Sieff CA, Mulligan RC, Johnson RP. Dye efflux studies suggest that hematopoietic stem cells expressing low or undetectable levels of CD34 antigen exist in multiple species. Nat Med. 1997;3:1337–1345. doi: 10.1038/nm1297-1337. [DOI] [PubMed] [Google Scholar]
- Gu Y, Filippi MD, Cancelas JA, Siefring JE, Williams EP, Jasti AC, Harris CE, Lee AW, Prabhakar R, Atkinson SJ, Kwiatkowski DJ, Williams DA. Hematopoietic cell regulation by Rac1 and Rac2 guanosine triphosphatases. Science. 2003;302:445–449. doi: 10.1126/science.1088485. [DOI] [PubMed] [Google Scholar]
- Guibal FC, Alberich-Jorda M, Hirai H, Ebralidze A, Levantini E, Di Ruscio A, Zhang P, Santana-Lemos BA, Neuberg D, Wagers AJ, Rego EM, Tenen DG. Identification of a myeloid committed progenitor as the cancer-initiating cell in acute promyelocytic leukemia. Blood. 2009;114:5415–5425. doi: 10.1182/blood-2008-10-182071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna J, Saha K, Pando B, van Zon J, Lengner CJ, Creyghton MP, van Oudenaarden A, Jaenisch R. Direct cell reprogramming is a stochastic process amenable to acceleration. Nature. 2009;462:595–601. doi: 10.1038/nature08592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann PC, Huber SL, Herrler T, Aicher A, Ellwart JW, Guba M, Bruns CJ, Heeschen C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell. 2007;1:313–323. doi: 10.1016/j.stem.2007.06.002. [DOI] [PubMed] [Google Scholar]
- Hess DA, Meyerrose TE, Wirthlin L, Craft TP, Herrbrich PE, Creer MH, Nolta JA. Functional characterization of highly purified human hematopoietic repopulating cells isolated according to aldehyde dehydrogenase activity. Blood. 2004;104:1648–1655. doi: 10.1182/blood-2004-02-0448. [DOI] [PubMed] [Google Scholar]
- Hiratsuka S, Nakamura K, Iwai S, Murakami M, Itoh T, Kijima H, Shipley JM, Senior RM, Shibuya M. MMP9 induction by vascular endothelial growth factor receptor-1 is involved in lung-specific metastasis. Cancer Cell. 2002;2:289–300. doi: 10.1016/s1535-6108(02)00153-8. [DOI] [PubMed] [Google Scholar]
- Hope KJ, Jin L, Dick JE. Acute myeloid leukemia originates from a hierarchy of leukemic stem cell classes that differ in self-renewal capacity. Nat Immunol. 2004;5:738–743. doi: 10.1038/ni1080. [DOI] [PubMed] [Google Scholar]
- Hosokawa K, Arai F, Yoshihara H, Nakamura Y, Gomei Y, Iwasaki H, Miyamoto K, Shima H, Ito K, Suda T. Function of oxidative stress in the regulation of hematopoietic stem cell-niche interaction. Biochem Biophys Res Commun. 2007;363:578–583. doi: 10.1016/j.bbrc.2007.09.014. [DOI] [PubMed] [Google Scholar]
- Ito K, Hirao A, Arai F, Takubo K, Matsuoka S, Miyamoto K, Ohmura M, Naka K, Hosokawa K, Ikeda Y, Suda T. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat Med. 2006;12:446–451. doi: 10.1038/nm1388. [DOI] [PubMed] [Google Scholar]
- Jamieson CH, Ailles LE, Dylla SJ, Muijtjens M, Jones C, Zehnder JL, Gotlib J, Li K, Manz MG, Keating A, Sawyers CL, Weissman IL. Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N Engl J Med. 2004;351:657–667. doi: 10.1056/NEJMoa040258. [DOI] [PubMed] [Google Scholar]
- Jin L, Hope KJ, Zhai Q, Smadja-Joffe F, Dick JE. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat Med. 2006;12:1167–1174. doi: 10.1038/nm1483. [DOI] [PubMed] [Google Scholar]
- Jin L, Lee EM, Ramshaw HS, Busfield SJ, Peoppl AG, Wilkinson L, Guthridge MA, Thomas D, Barry EF, Boyd A, Gearing DP, Vairo G, Lopez AF, Dick JE, Lock RB. Monoclonal antibody-mediated targeting of CD123, IL-3 receptor alpha chain, eliminates human acute myeloid leukemic stem cells. Cell Stem Cell. 2009;5:31–42. doi: 10.1016/j.stem.2009.04.018. [DOI] [PubMed] [Google Scholar]
- Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, MacDonald DD, Jin DK, Shido K, Kerns SA, Zhu Z, Hicklin D, Wu Y, Port JL, Altorki N, Port ER, Ruggero D, Shmelkov SV, Jensen KK, Rafii S, Lyden D. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature. 2005;438:820–827. doi: 10.1038/nature04186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly LM, Liu Q, Kutok JL, Williams IR, Boulton CL, Gilliland DG. FLT3 internal tandem duplication mutations associated with human acute myeloid leukemias induce myeloproliferative disease in a murine bone marrow transplant model. Blood. 2002;99:310–318. doi: 10.1182/blood.v99.1.310. [DOI] [PubMed] [Google Scholar]
- Kelly PN, Dakic A, Adams JM, Nutt SL, Strasser A. Tumor growth need not be driven by rare cancer stem cells. Science. 2007;317:337. doi: 10.1126/science.1142596. [DOI] [PubMed] [Google Scholar]
- Kim CF, Jackson EL, Woolfenden AE, Lawrence S, Babar I, Vogel S, Crowley D, Bronson RT, Jacks T. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell. 2005;121:823–835. doi: 10.1016/j.cell.2005.03.032. [DOI] [PubMed] [Google Scholar]
- Krause DS, Lazarides K, von Andrian UH, Van Etten RA. Requirement for CD44 in homing and engraftment of BCR-ABL-expressing leukemic stem cells. Nat Med. 2006;12:1175–1180. doi: 10.1038/nm1489. [DOI] [PubMed] [Google Scholar]
- Lee J, Son MJ, Woolard K, Donin NM, Li A, Cheng CH, Kotliarova S, Kotliarov Y, Walling J, Ahn S, Kim M, Totonchy M, Cusack T, Ene C, Ma H, Su Q, Zenklusen JC, Zhang W, Maric D, Fine HA. Epigenetic-mediated dysfunction of the bone morphogenetic protein pathway inhibits differentiation of glioblastoma-initiating cells. Cancer Cell. 2008;13:69–80. doi: 10.1016/j.ccr.2007.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessard J, Sauvageau G. Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature. 2003;423:255–260. doi: 10.1038/nature01572. [DOI] [PubMed] [Google Scholar]
- Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, Adsay V, Wicha M, Clarke MF, Simeone DM. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67:1030–1037. doi: 10.1158/0008-5472.CAN-06-2030. [DOI] [PubMed] [Google Scholar]
- Meissner A, Jaenisch R. Generation of nuclear transfer-derived pluripotent ES cells from cloned Cdx2-deficient blastocysts. Nature. 2006;439:212–215. doi: 10.1038/nature04257. [DOI] [PubMed] [Google Scholar]
- Morton JP, Mongeau ME, Klimstra DS, Morris JP, Lee YC, Kawaguchi Y, Wright CV, Hebrok M, Lewis BC. Sonic hedgehog acts at multiple stages during pancreatic tumorigenesis. Proc Natl Acad Sci USA. 2007;104:5103–5108. doi: 10.1073/pnas.0701158104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notta F, Doulatov S, Dick JE. Engraftment of human hematopoietic stem cells is more efficient in female NOD/SCID/IL-2Rgcnull recipients. Blood. 2010 Mar 5; doi: 10.1182/blood-2009-10-249326. (E pub) [DOI] [PubMed] [Google Scholar]
- O’Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106–110. doi: 10.1038/nature05372. [DOI] [PubMed] [Google Scholar]
- Piccirillo SG, Reynolds BA, Zanetti N, Lamorte G, Binda E, Broggi G, Brem H, Olivi A, Dimeco F, Vescovi AL. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature. 2006;444:761–765. doi: 10.1038/nature05349. [DOI] [PubMed] [Google Scholar]
- Prince ME, Sivanandan R, Kaczorowski A, Wolf GT, Kaplan MJ, Dalerba P, Weissman IL, Clarke MF, Ailles LE. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc Natl Acad Sci USA. 2007;104:973–978. doi: 10.1073/pnas.0610117104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana E, Shackleton M, Sabel MS, Fullen DR, Johnson TM, Morrison SJ. Efficient tumour formation by single human melanoma cells. Nature. 2008;456:593–598. doi: 10.1038/nature07567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raaijmakers MH, Mukherjee S, Guo S, Zhang S, Kobayashi T, Schoonmaker JA, Ebert BL, Al-Shahrour F, Hasserjian RP, Scadden EO, Aung Z, Matza M, Merkenschlager M, Lin C, Rommens JM, Scadden DT. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature. 2010;464:852–857. doi: 10.1038/nature08851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen JM, Jordan CT. The increasing complexity of the cancer stem cell paradigm. Science. 2009;324:1670–1673. doi: 10.1126/science.1171837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schatton T, Murphy GF, Frank NY, Yamaura K, Waaga-Gasser AM, Gasser M, Zhan Q, Jordan S, Duncan LM, Weishaupt C, Fuhlbrigge RC, Kupper TS, Sayegh MH, Frank MH. Identification of cells initiating human melanomas. Nature. 2008;451:345–349. doi: 10.1038/nature06489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta A, Banerjee S. Pleiotropic p27(Kip1), BCR-ABL and leukemic stem cell: The trio in concert. Leukemia. 2007;21:2559–2561. doi: 10.1038/sj.leu.2404842. [DOI] [PubMed] [Google Scholar]
- Sengupta A, Banerjee D, Chandra S, Banerji SK, Ghosh R, Roy R, Banerjee S. Deregulation and cross talk among Sonic hedgehog, Wnt, Hox and Notch signaling in chronic myeloid leukemia progression. Leukemia. 2007;21:949–955. doi: 10.1038/sj.leu.2404657. [DOI] [PubMed] [Google Scholar]
- Sengupta A, Arnett J, Dunn S, Williams DA, Cancelas JA. Rac2 GTPase deficiency depletes BCR-ABL+ leukemic stem cells and progenitors in vivo. Blood. 2010 Apr 20; doi: 10.1182/blood-2009-10-247437. (E pub) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- Somervaille TC, Matheny CJ, Spencer GJ, Iwasaki M, Rinn JL, Witten DM, Chang HY, Shurtleff SA, Downing JR, Cleary ML. Hierarchical maintenance of MLL myeloid leukemia stem cells employs a transcriptional program shared with embryonic rather than adult stem cells. Cell Stem Cell. 2009;4:129–140. doi: 10.1016/j.stem.2008.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taussig DC, Miraki-Moud F, Anjos-Afonso F, Pearce DJ, Allen K, Ridler C, Lillington D, Oakervee H, Cavenagh J, Agrawal SG, Lister TA, Gribben JG, Bonnet D. Anti-CD38 antibody-mediated clearance of human repopulating cells masks the heterogeneity of leukemia-initiating cells. Blood. 2008;112:568–575. doi: 10.1182/blood-2007-10-118331. [DOI] [PubMed] [Google Scholar]
- Thomas EK, Cancelas JA, Chae HD, Cox AD, Keller PJ, Perrotti D, Neviani P, Druker BJ, Setchell KD, Zheng Y, Harris CE, Williams DA. Rac guanosine triphosphatases represent integrating molecular therapeutic targets for BCR-ABL-induced myeloproliferative disease. Cancer Cell. 2007;12:467–478. doi: 10.1016/j.ccr.2007.10.015. [DOI] [PubMed] [Google Scholar]
- Todaro M, Alea MP, Di Stefano AB, Cammareri P, Vermeulen L, Iovino F, Tripodo C, Russo A, Gulotta G, Medema JP, Stassi G. Colon cancer stem cells dictate tumor growth and resist cell death by production of interleukin-4. Cell Stem Cell. 2007;1:389–402. doi: 10.1016/j.stem.2007.08.001. [DOI] [PubMed] [Google Scholar]
- Uchida N, Buck DW, He D, Reitsma MJ, Masek M, Phan TV, Tsukamoto AS, Gage FH, Weissman IL. Direct isolation of human central nervous system stem cells. Proc Natl Acad Sci USA. 2000;97:14720–14725. doi: 10.1073/pnas.97.26.14720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: Accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8:755–768. doi: 10.1038/nrc2499. [DOI] [PubMed] [Google Scholar]
- Walkley CR, Olsen GH, Dworkin S, Fabb SA, Swann J, McArthur GA, Westmoreland SV, Chambon P, Scadden DT, Purton LE. A microenvironment-induced myeloproliferative syndrome caused by retinoic acid receptor gamma deficiency. Cell. 2007;129:1097–1110. doi: 10.1016/j.cell.2007.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Krivtsov AV, Sinha AU, North TE, Goessling W, Feng Z, Zon LI, Armstrong SA. The Wnt/beta-catenin pathway is required for the development of leukemia stem cells in AML. Science. 2010;327:1650–1653. doi: 10.1126/science.1186624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J, Wunderlich M, Fox C, Alvarez S, Cigudosa JC, Wilhelm JS, Zheng Y, Cancelas JA, Gu Y, Jansen M, Dimartino JF, Mulloy JC. Microenvironment determines lineage fate in a human model of MLL-AF9 leukemia. Cancer Cell. 2008;13:483–495. doi: 10.1016/j.ccr.2008.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JP, Wu J, Johansson G, Rizvi TA, Miller SC, Geiger H, Malik P, Li W, Mukouyama YS, Cancelas JA, Ratner N. Nf1 mutation expands an EGFR-dependent peripheral nerve progenitor that confers neurofibroma tumorigenic potential. Cell Stem Cell. 2008;3:658–669. doi: 10.1016/j.stem.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Williams JP, Rizvi TA, Kordich JJ, Witte D, Meijer D, Stemmer-Rachamimov AO, Cancelas JA, Ratner N. Plexiform and dermal neurofibromas and pigmentation are caused by Nf1 loss in desert hedgehog-expressing cells. Cancer Cell. 2008;13:105–116. doi: 10.1016/j.ccr.2007.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang ZF, Ho DW, Ng MN, Lau CK, Yu WC, Ngai P, Chu PW, Lam CT, Poon RT, Fan ST. Significance of CD90+ cancer stem cells in human liver cancer. Cancer Cell. 2008;13:153–166. doi: 10.1016/j.ccr.2008.01.013. [DOI] [PubMed] [Google Scholar]
- Zhao C, Chen A, Jamieson CH, Fereshteh M, Abrahamsson A, Blum J, Kwon HY, Kim J, Chute JP, Rizzieri D, Munchhof M, VanArsdale T, Beachy PA, Reya T. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature. 2009;458:776–779. doi: 10.1038/nature07737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou BB, Zhang H, Damelin M, Geles KG, Grindley JC, Dirks PB. Tumour-initiating cells: Challenges and opportunities for anticancer drug discovery. Nat Rev Drug Discov. 2009;8:806–823. doi: 10.1038/nrd2137. [DOI] [PubMed] [Google Scholar]