

Abstract
Background
The AID/APOBECs are deaminases that act on cytosines in a diverse set of pathways and some of them have been linked to the onset of genetic alterations in cancer. Among them, APOBEC1 is the only family member to physiologically target RNA, as the catalytic subunit in the Apolipoprotein B mRNA editing complex. APOBEC1 has been linked to cancer development in mice but its oncogenic mechanisms are not yet well understood.
Results
We analyze whether expression of APOBEC1 induces a mutator phenotype in vertebrate cells, likely through direct targeting of genomic DNA. We show its ability to increase the inactivation of a stably inserted reporter gene in a chicken cell line that lacks any other AID/APOBEC proteins, and to increase the number of imatinib-resistant clones in a human cellular model for chronic myeloid leukemia through induction of mutations in the BCR-ABL1 fusion gene. Moreover, we find the presence of an AID/APOBEC mutational signature in esophageal adenocarcinomas, a type of tumor where APOBEC1 is expressed, that mimics the one preferred by APOBEC1 in vitro.
Conclusions
Our findings suggest that the ability of APOBEC1 to trigger genetic alterations represents a major layer in its oncogenic potential. Such APOBEC1-induced mutator phenotypes could play a role in the onset of esophageal adenocarcinomas. APOBEC1 could be involved in cancer promotion at the very early stages of carcinogenesis, as it is highly expressed in Barrett's esophagus, a condition often associated with esophageal adenocarcinoma.
Electronic supplementary material
The online version of this article (doi:10.1186/s13059-014-0417-z) contains supplementary material, which is available to authorized users.
Background
APOBEC1 (Apolipoprotein B mRNA editing enzyme, catalytic polypeptide 1) is part of the RNA editing complex that physiologically deaminates C6666 to U in the transcript of human Apolipoprotein B, a major component in lipid transport [1-3]. APOBEC1 exerts this function in the small intestine in humans, and in the liver in rodents.
APOBEC1 was the first identified member of the AID/APOBEC protein family, a group of cytosine deaminases that target nucleic acids in a diverse set of pathways to induce C > U changes [4,5]. Most of these proteins are DNA mutators: AID (Activation Induced Deaminase) is essential for all secondary antibody diversification processes [6,7], and the APOBEC3s act in a defense pathway against retroviruses and mobile elements [8,9].
The only well-characterized target for APOBEC1 is the mRNA for Apolipoprotein B; however, additional target mRNAs have been identified [10-12], and it has been also suggested that APOBEC1 regulates mRNA stability through its ability to bind RNA [13,14]. On the other hand, APOBEC1 can also target DNA in bacteria and in vitro [15-17]. Based on this activity, other roles have been suggested, from controlling DNA methylation [18,19], to being part of a restriction pathway against retroviruses and mobile elements, similar to the APOBEC3s [20-25].
APOBEC1 expression has been linked to cancer: transgenic mice and rabbits constitutively expressing APOBEC1 in the liver develop hepatocellular carcinoma [26], and APOBEC1 deficiency in cancer-prone APCmin mice reduces the number of polyps and tumors in the gastrointestinal tract [27]. The oncogenic potential of APOBEC1 has been attributed mostly to its ability to target RNA [3]. However, we hypothesize that the oncogenic role of APOBEC1 is related to its ability to target DNA. This hypothesis is corroborated by the evidence that aberrant activity of other AID/APOBECs underlies the onset of genetic alterations in human cancer [28-35].
In direct support of this notion, here we show in two cellular models that expression of APOBEC1 induces a mutator phenotype. In addition, we show the presence of the mutational signature of the AID/APOBECs in human esophageal adenocarcinomas, a type of tumor in which APOBEC1 is highly expressed.
Results and discussion
Rat APOBEC1 induces a mutator phenotype in a chicken cell line
In order to investigate the ability of rat APOBEC1 - the best characterized among the mammalian homologues - to induce a mutator phenotype, we used DT40 cells, a chicken lymphoma B cell line that has been extensively employed to study the mutational activity of AID [36], the only catalytically active member of the AID/APOBEC family present in the chicken genome [17,37]. A derivative DT40 clone in which AID has been knocked out (sIgM+ ψV− AID−/−) [38] has enabled us to assay the effects of APOBEC1 in the absence of other DNA mutators. We first inserted a single-copy enhanced green fluorescent protein (EGFP) reporter gene in the DT40 genome, so that inactivation of the EGFP could be easily detected through fluorescence-activated cell sorting (FACS) analysis, in a similar way to that used to study AID-dependent mutations [36]. We then transfected independent DT40GFP clones with constructs expressing either rat APOBEC1 or a control plasmid, and we followed the expression of the EGFP reporter gene in clones selected to stably express APOBEC1 (Figure S1A in Additional file 1). Starting from 2 weeks after transfection we observed the rise of a distinct EGFP(−) population in the clones expressing APOBEC1 (Figure 1A,B). At 4 weeks after transfection, the median percentage of the EGFP(−) cells in the APOBEC1-expressing clones was four times larger than in the control clones. Indeed, sorted EGFP(−) cells remained negative after expansion. It seemed unlikely that this finding could result from an effect of APOBEC1 on RNA, since this would likely alter the overall level of EGFP RNA rather than abolish its expression in individual cells. To confirm this we sorted the GFP(−) population from APOBEC1- and control-transfected cells and sequenced the EGFP coding region to detect the presence of mutations. Approximately a quarter of the sequences bore mutations. The mutation pattern observed in the samples expressing APOBEC1 was different from the mutational background in the controls (Figure 1C): C/G base pairs were preferentially mutated in the APOBEC1 samples, and the changes were mainly transversions, a pattern reminiscent of the one observed after AID-dependent deamination in DT40 cells [38].
Figure 1.
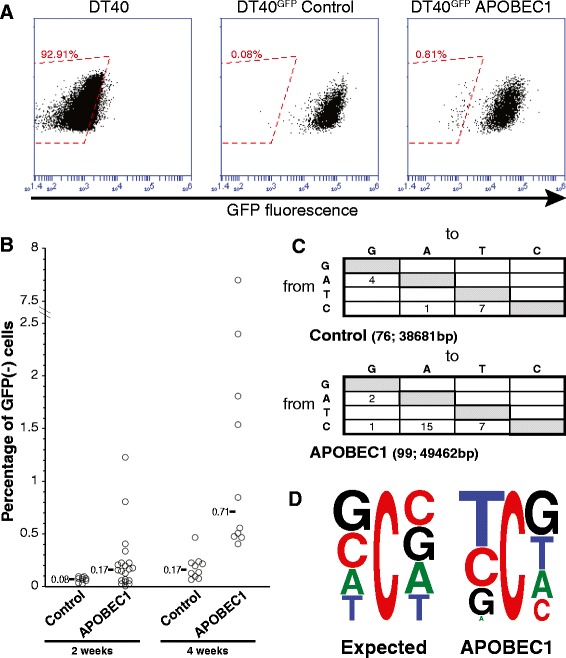
APOBEC1 induces mutations in DT40 cells. (A) The inactivation of the EGFP transgene in DT40 cells was assayed by monitoring the size of a distinct EGFP(−) population in transfectants expressing either APOBEC1 or a control plasmid (Figure S1A in Additional file 1). Flow cytometric analysis of representative DT40GFP clones stably transfected with APOBEC1 or with the control plasmid. The red boxed area indicates the EGFP(−) population considered for the analysis. (B) Plot depicting the inactivation of the EGFP in independent transfectants, with the median for each construct indicated. The measurements were taken at the indicated times (P < 0.05 for each time-point). The sorted EGFP(−) cells did not regain fluorescence after expansion. (C) Mutation pattern obtained after sequence analysis of independent EGFP fragments from sorted EGFP(−) APOBEC1- and vector-transfected DT40 cells. The number of sequences analyzed and the total number of bases is indicated in parentheses (P = 0.012 by Fisher’s exact test). Only non-clonal mutations were considered, which is likely to underestimate the number of mutations in the case of preferred mutational hotspots. (D) Local sequence context for the cytosine residues present on both strands of the analyzed GFP fragments (expected) and for the mutated cytosines (observed) in the APOBEC1-expressing clones.
Each member of the AID/APOBEC family preferentially targets cytosines within a specific sequence context: this preference can be quite strict (for example, CCC for APOBEC3G, TC for APOBEC3B) or more relaxed (for example, WRC for AID, YC for APOBEC1). Indeed, the sequence context of the mutations at cytosine residues observed in the EGFP reflects that observed in vitro and in bacteria for APOBEC1 [15,16,39], especially with regard to the preference for a thymine directly upstream to the mutated C and the avoidance of adenines (Figure 1D; Additional file 2).
Human APOBEC1 is able to mutate DNA in bacteria
Human APOBEC1 - in contrast to other mammalian homologues - exerts only a minor effect on retroviruses and mobile elements [20,22,24,40]. This has cast doubts on its ability to target DNA. We therefore cloned human APOBEC1 in a bacterial expression vector and used this in the rifampicin-resistance reversion assay, a test widely used to study AID/APOBEC-mediated induction of a mutator phenotype. The outcome of the experiments reveals that human APOBEC1 is effective as a DNA mutator (Figure 2A). Sequence analysis of the rpoB gene from rifampicin-resistant clones shows that the mutations induced by human APOBEC1 closely resemble those induced by the rat homologue (Figure 2B).
Figure 2.
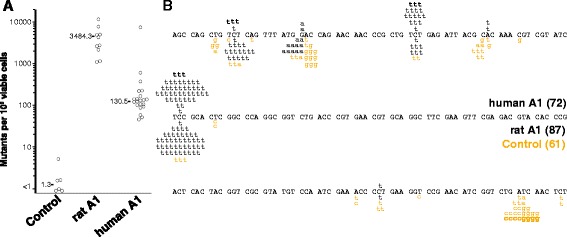
Human APOBEC1 induces a mutator phenotype in bacteria. (A) Frequency of rifampicin-resistant clones in cultures of Escherichia coli transformed with APOBEC1 expression constructs (rat or human) or empty vector. Each point represents the mutation frequency of an independent overnight culture. The median mutation frequency is indicated. (B) Distribution of independent rpoB point mutations identified in rifampicin-resistant clones from bacteria transformed with human APOBEC1 (black changes above the sequence), rat APOBEC1 (black changes below the sequence), or empty plasmid (brown changes below the sequence). The number of independent colonies is indicated in parentheses.
APOBEC1 induces a mutator phenotype in human K562 cells
Having confirmed that both rat and human APOBEC1 could effectively target DNA, we tested whether the APOBEC1-induced mutator phenotype could affect drug-resistance dynamics in a cancer cellular model. To this end, we used human K562 cells, a model for chronic myeloid leukemia (CML) bearing a BCR-ABL1 chromosomal translocation. The BCR-ABL1 fusion gene provides support for the cellular growth, and treatment with imatinib, a tyrosine kinase inhibitor, results in cell death due to blockade of the BCR-ABL1 kinase domain. Mutations in BCR-ABL1 can produce imatinib-resistant clones: it has been shown that exogenous overexpression of AID induces mutations that can confer resistance to the drug by disrupting the interaction of BCR-ABL1 with the drug [41,42]. AID/APOBECs do not seem to be expressed in these cells, either treated or not with imatinib. After obtaining stably transfected clones, we cultured K562 cells expressing APOBEC1, AID as a positive control, or a control plasmid (Figure S1B in Additional file 1) in the presence of imatinib to select clones resistant to the drug. In line with our expectations, a higher number of colonies was present in the samples expressing AID or the APOBEC1, both rat or human, compared with controls, with APOBEC1-expressing clones outnumbering those expressing AID (Figure 3). After confirming that the selected clones were able to grow in imatinib-supplemented medium, we amplified the region of the BCR-ABL1 gene encoding the imatinib binding site to evaluate the presence of mutations. Sequence analysis of individual clones confirmed the presence of mutations in the ABL1 tyrosine kinase domain, mostly located at residues near the catalytic pocket (Table 1). Taking into account the number of observed mutations and the fact that the selected phenotype depends on mutations at specific sites of the fusion gene (as opposed to loss-of-function mutations in the EGFP), the type and the sequence context of the mutations observed are not indicative of the AID/APOBEC1 mutational preferences. This is in line with previous studies in which A/T mutations represented 33 to 50% of all unique BCR-ABL1 mutations induced by AID [41,42].
Figure 3.
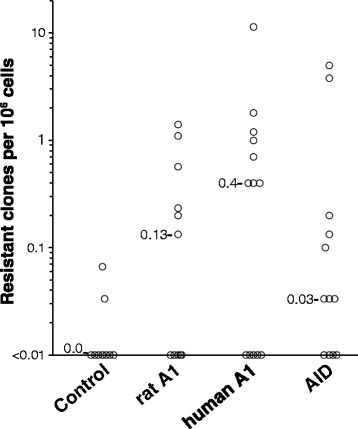
APOBEC1 induces a mutator phenotype in a human chronic myeloid leukemia cell line. Frequency of imatinib-resistant clones arising from 107 K562 cells expressing either mammalian APOBEC1s, AID, or a control plasmid seeded in the presence of 1 μM imatinib (control/APOBEC1, P < 0.05). Imatinib resistance was confirmed by growing the clones in imatinib-supplemented medium.
Table 1.
List of the non-clonal mutations identified in the BCR-ABL1 fusion gene from imatinib-resistant K562 clones
| Samples | Position | AA change | Codon change | Sequence context |
|---|---|---|---|---|
| 1,503 | E495G | GAA > GGA | ||
| Control | 1,472 | R491Q | CGG > CAG | GCCGG |
| 1,503 | Silent | TTT > TTC | ||
| 633 | G100C | GGC > TGC | GCCTA | |
| AID | 395 | Silent | AAA > AAG | |
| 568 | G190D | GGC > GAC | TGCCA | |
| 633 | T212A | ACG > GCG | ||
| 987 | G155D | GGC > GAC | GGCCA | |
| 607 | L203M | CTG > ATG | CCCTG | |
| Rat APOBEC1 | 613 | E205K | GAG > AAG | CTCGG |
| 764 | K255T | AAG > ACG | ||
| 987 | Silent | GGG > GGT | TTCCC | |
| 1,245 | silent | AGC > AGT | AGCCG | |
| 758 | R253H | CGC > CAC | TGCGC | |
| 841* | silent | CTG > TTG | AGCTG | |
| 697 | H233D | CAT > GAT | TTCAT | |
| Human APOBEC1 | 699 | H233D | CAT > GAC | |
| 1,149* | Silent | GCC > GCA | GCCAT | |
| 1,245* | F415L | TTT > TTG |
The region analyzed (encompassing exon 13 of BCR and exon 9 of ABL1) includes the imatinib-binding region of the fusion gene. The asterisk indicates mutations found in the same clone. The local sequence context for the mutations at cytosines is shown. Compared to the AID-induced mutations found in previous reports (mutations in approximately 30% of the sequences) [41,42], we found approximately one mutation in each of the clones analyzed. This is explained by the different procedures we used to select resistant clones: whereas the other studies focused on competing bulk populations of AID-transfected GFP(+) cells and control GFP(−) cells, we analyzed individual clones arising from the same number of cells plated in the presence of imatinib.
APOBEC1 expression in esophageal adenocarcinomas
Thus, our findings show that APOBEC1 can induce somatic mutations in vertebrate cells. Such an APOBEC1-induced mutator phenotype could play a role in the onset of cancer previously observed in mice. On the other hand, there is no evidence so far linking APOBEC1 to any human cancer.
We therefore searched the Gene Expression Omnibus (GEO) repository [43] for specific types of cancer in which APOBEC1 was overexpressed. We found that, in striking contrast to normal esophageal mucosa, the expression levels of APOBEC1 in esophageal adenocarcinomas (EACs) are comparable to those observed in the small intestine, where APOBEC1 is expressed at its highest levels [44-46] (Figure 4A; Additional file 3). Indeed, in individual matched pairs of normal esophageal mucosa and EAC, APOBEC1 is consistently overexpressed in EACs (Figure S3B in Additional file 3). In comparison, other AID/APOBECs such as AID and APOBEC2 are not expressed. An increased expression of APOBEC3s in EACs cannot be excluded: in the dataset from Kim et al. [44] there is a statistically significant increase of APOBEC3A (Additional file 3), which is lost when comparing normal/EAC pairs (data not shown); on the other hand, in the dataset from Kimchi et al. [45], it is APOBEC3B that is increased in EACs, albeit not at the levels of APOBEC1 (data not shown).
Figure 4.
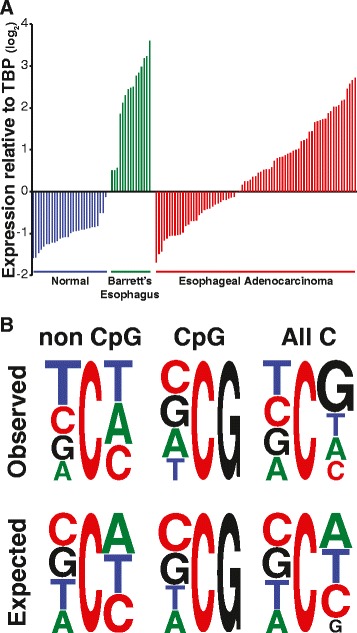
Upregulation of APOBEC1 and mutational context in esophageal adenocarcinomas. (A) Expression levels of APOBEC1 in a cohort of samples from normal esophagus, Barrett’s esophagus and EACs. The data were extracted from a recent study analyzing the transcriptome of EAC using microarrays [44], normalized on TATA-binding protein (TBP) expression levels and shown as median-centered Log2 values. Differences between the various sets of data are statistically significant (P < 10−4 by one-way ANOVA coupled with Tukey’s test). Details of APOBEC1 expression patterns from this and other studies are shown in Additional file 3. (B) Sequence context for the mutated cytosine residues in EAC exomes from a recent study [47]. The trinucleotide sequence context is shown for mutations at Cs (All C), at CpG and at non-CpG sites as a weblogo [48]. The expected sequence context for random mutations at cytosines in the human exomes is shown (P < 0.0001 for the first position at non-CpG sites by Chi-square analysis).
The assessment of the expression levels of the APOBEC3s in the datasets from Kimchi et al. [45] and Stairs et al. [46] is confused by the U133A Affymetrics platform used (due to cross-hybridizing probes, as discussed in Burns et al. [30]). On the other hand, the data from Kim et al. [44] could be informative of the expression levels of several APOBEC3s (see discussion in Additional file 4).
Interestingly, a marked increase of APOBEC1 is also found in Barrett’s esophagus, a condition closely related to adenocarcinomas, both epidemiologically and genetically, as the same hallmark DNA/genomic alterations are seen in both [49-52]. In contrast, this is not the case for the other AID/APOBECs (Figure S3A in Additional file 3). The overexpression of APOBEC1 is in line with the metaplasia that is characteristic of Barrett’s esophagus, whereby the normal esophageal epithelium is replaced by cells with intestinal features.
AID/APOBEC mutational signature in esophageal adenocarcinomas
In human tumors derived from mature B cells there are characteristic chromosomal translocations and other mutations that can be regarded as a veritable AID mutational signature [28,53]. Other AID/APOBECs have been shown to induce DNA damage and somatic mutations [30,54,55], and they are involved in kataegis, clusters of mutations observed in some cancer genomes [29,32,35,56,57]. An AID/APOBEC mutational signature can be observed in genes highly mutated in cancer [39] and in cancer genomes and exomes [31,33-35,58]. Based on this, we have looked at the data from a recent study on EACs [47] in which at least two mutational signatures had been observed: A to C transversions in the context of the ApA dinucleotide, and C to T transitions at CpG sites. Our re-analysis shows that mutations at non-CpG sites represent about 55% of all mutations at Cs, and the sequence context observed in these non-CpG mutations is similar to that ascribed to AID/APOBEC action (Figure 4B). Such sequence context also resembles that observed for APOBEC1 [15,16,39] as well that we found in APOBEC1-expressing DT40 cells (Figure 1D).
The mutational sequence context at position −1 in EACs is closer to that observed in tumor types in which APOBEC3B is expressed alongside other AID/APOBECs, including APOBEC1 [31]. Moreover, an analysis of the dinucleotides 5’ to a mutated C shows that the ratio between pyrimidine and purine residues at position −2 varies among the tumor types in which an AID/APOBEC mutational signature is present [30,31,33-35,58]. Indeed, the pyrimidine/purine ratio in EACs is the lowest among the different tumors, with the tumors with high expression of APOBEC3B displaying the highest ratio (Additional file 5).
There is no direct evidence of correlation between mutation signature and APOBEC1 expression because of the lack of expression data in Dulak et al. [47]. However, different studies show variable levels of APOBEC1 overexpression in EACs (Figure 4; Figure S3A-C in Additional file 3), providing indirect evidence of such correlation. It is noteworthy that APOBEC1 is markedly overexpressed in all samples from Barrett’s esophagus (Figure 4; Figure S3A,C,D in Additional file 3).
Indeed, a recent analysis of the somatic mutations in Barrett’s esophagus reports that 71% of all mutations are targeted to C/G base pairs, with a strong bias towards transition mutations (Ts/Tv ratio at CpH sites of 1.69) [52]. Such a trend is present also in EACs associated with Barrett’s esophagus compared with those that are not associated (Additional file 6; Ts/Tv of 1.22 versus 1.01). If we consider the marked overexpression of APOBEC1 in Barrett’s esophagus, it is suggestive that the AID/APOBEC mutational signature is more evident (albeit not significantly different) in EACs associated with Barrett’s esophagus compared with those that are not (Additional file 7).
Conclusions
DNA deaminases perform various functions within an organism. Unlike the APOBEC3 genes, which physiologically exhibit a broad expression pattern [59], the expression of AID and of APOBEC1 is limited to few tissues in which they exert their physiologic activity. Ectopic expression of AID (especially outside the B-cell lineage) results in mutations and cancer formation [60,61]. This suggests that tissues and cells that require these deaminases in the nucleus and hence near their own genome have developed protective regulatory mechanisms. This is seen in mature B cells, where AID expression is regulated at the transcriptional level as well as in terms of protein stability and localization. On the other hand, cells that do not physiologically express these deaminases could lack this layer of protection and be more susceptible to self-induced DNA damage (for example, [60,62]).
By analogy, we can presume that cells physiologically expressing APOBEC1 have devices to protect them from its aberrant activity, whereas those not normally expressing APOBEC1, such as the esophageal epithelium, may have no such protection. We have shown here that APOBEC1 can induce mutations in genomic DNA. Together with previous findings linking APOBEC1 presence/absence with the induction of tumors in mice [26,27], our experimental results and our observations in EACs indicate that APOBEC1 could be involved in the onset of cancer by targeting genomic DNA directly.
We do not know yet at which stage in EAC oncogenesis AID/APOBECs exert their mutagenic potential; however, since Barrett’s esophagus can be regarded as a precursor of EAC, and since genomic alterations of EAC are already seen in Barrett’s esophagus [49-52], we surmise that AID/APOBECs play a role at an early stage, even though further studies will be needed to assess the specific contribution of APOBEC1 compared with other AID/APOBECs. In addition, we must consider that, through RNA targeting, APOBEC1 can alter a number of cellular functions [11,13,14,27]. It is possible, therefore, that these changes, associated with APOBEC1 mutagenic activity, may also be boosting its oncogenic potential and drive both the onset and the progression of cancer.
Materials and methods
Plasmids and mutator assay in bacteria
The EGFP-expressing construct used for the DT40 experiments was built by subcloning on a pBluescript SK + backbone the beta-actin promoter (XhoI/NheI), the EGFP coding sequence (NheI/KpnI) and a Blasticidin-S resistance cassette (BamHI). Rat and human APOBEC1 were PCR-amplified (rat forward primer AAAGCTAGCATGAGTTCCGAGACAGGCCCTGTA, rat reverse primer AAATGTACAAGATCTCATTTCAACCCTGTGGC; human forward primer AAAGCTAGCATGACTTCTGAGAAAGGTCCT, human reverse primer AAATGTACAAGATCTCATCTCCAAGCCACAGAAGG) and cloned into the pAIDexpressPuro2 expression vector [63] under the control of the beta-actin promoter. Depending on the restriction enzymes used for cloning, the final constructs encoded either APOBEC1 or APOBEC1-IRES-EGFP (NheI-BsrGI or NheI-BglII). The construction of the AID-IRES-EGFP expression construct is detailed in [64]. An empty plasmid with a puromycin cassette [65] was used as control for the DT40 experiments. An analogous one expressing EGFP was used in the experiments on K562 cells. The rat APOBEC1 vector for bacterial expression was described in Harris et al. [15]. The human APOBEC1 coding sequence was cloned into pTrc99a (forward primer, TTTCCATGGCCATGACTTCTGAGAAAGGTCC; reverse primer, AAATGTACAAGATCTCATCTCCAAGCCACAGAAGG; NcoI/BglII). The rifampicin-resistance reversion assay used to test the induction of a mutator phenotype was performed as described in Petersen-Mahrt et al. [66] and Harris et al. [15], but with induction of the AID/APOBEC at 18°C for 24 hours in order to increase the viability of the bacteria and to obtain better resolution (Additional file 8).
Cells, transfections, and protein expression
sIgM+ ψV− AID−/− DT40 cells [38] were maintained in RPMI1640 9% fetal bovine serum (FBS), 1% chicken serum (Life Technologies, Carlsbad, CA, USA), 50 μM 2-mercaptoethanol at 37°C in 5% CO2, and transfected as previously described [63]. Cells were selected with 25 μg/ml blasticidin or 0.25 μg/ml puromycin, depending on the plasmid used. DT40GFP cells were prepared by selecting independent clones stably transfected with the EGFP construct. Southern blot analysis using an EGFP fragment as probe were performed to select for DT40GFP clones bearing a single EGFP copy.
K562 cells were maintained in DMEM supplemented with 10% FBS at 37°C in 5% CO2; 106 cells and 5 μg of plasmid were used to electroporate K562 cells (250 V, 950 μF, Biorad GenePulserII Hercules, CA, USA). K562 cells were selected with 3 μg/ml puromycin.
EGFP fluorescence was assayed by flow cytometry on a BD Accuri C6 cytometer (Franklin Lakes, NJ, USA). Expression of APOBEC1 and AID was monitored by western blot. Cells were lysed (RIPA) and - after SDS-PAGE - the proteins were detected using either a primary goat anti-APOBEC1 antibody (1:5,000; Santa Cruz Biotechnology (Dallas, TX, USA), a monoclonal anti-AID antibody (hAnp52-1, 1:8,000) [64], or a beta-actin antibody (1:10,000; Sigma (St. Louis, MO, USA).
Assaying APOBEC1 activity in DT40 cells
After electroporation with the APOBEC1/control constructs, DT40GFP cells were plated in 96-well plates for selection. Independent DT40GFP clones were used. Single clones were picked after approximately 8 days and expanded. We then analyzed 105 cells by flow cytometry to assay the loss of EGFP fluorescence at 14 and 28 days after transfection. All experiments were repeated at least three times. For the mutation analysis, the GFP(−) population from independent clones was sorted using a BD FACSAria (Franklin Lakes, NJ, USA). After expansion the genomic DNA of the sorted population was prepared and the EGFP coding sequence was amplified by touch-down PCR using the KOD polymerase (CGTAAACGGCCACAAGTTCAG, ACTGGGTGCTCAGGTAGTGGT, 10 touchdown cycles plus 20 amplification cycles). The PCR fragments were cloned in pCR-Blunt II-TOPO according to the manufacturer’s instructions (Life Technologies, Carlsbad, CA, USA), and independent bacterial clones were sequenced (Additional file 9).
Assaying APOBEC1 activity in K562 cells
Our failure to amplify by RT-PCR the catalytically active AID/APOBECs in K562 cells in the presence/absence of imatinib suggests that they are not expressed. This is in line with available data in GEO datasets GSE26821 and GSE51083. We have produced independent clones of K562 cells stably expressing APOBEC1-IRES-EGFP, AID-IRES-EGFP, or the control vector. We seeded 107 cells from these clones in 96-well plates in medium supplemented with 1 μM of imatinib. Imatinib-resistant colonies grew after approximately 3 weeks, and were picked for expansion. After confirming the ability of these clones to grow in the presence of imatinib, total RNA was prepared and the mutations in the region encompassing exon 13 of BCR and exon 9 of ABL1 were analyzed as described in [42]. The number of independent imatinib-resistant clones that were analyzed is: rat APOBEC1, 8; human APOBEC1, 5; AID, 6; control, 6. There are between 5 and 11 copies of the BCR-ABL1 fusion gene in K562, with many of these copies inactivated by indels. In our sequencing we found evidence for at least five variants of the PCR-amplified BCR-ABL1 fragment, only one of which could translate a proper BCR-ABL1 fusion protein. Typically - after cloning in a plasmid - we sequenced between 12 and 24 bacterial colonies for each imatinib-resistant clone in order to discriminate the active BCR-ABL1 copy and obtain sufficient information on its mutational status (Additional files 10 and 11).
Expression of APOBEC1 in the esophagus and mutational analysis
Expression data were downloaded from GEO [43]. The data from Kimchi et al. [45], Kim et al. [44], and Stairs et al. [46] correspond to GEO dataset accession numbers GSE1420, GSE13898 and GSE13083, respectively. All data were normalized on TATA-binding protein expression levels and their value centered on the median of the samples as Log2 values. Details of the statistics used are provided in the figure legends.
Sequence context analysis
The sequence context of the mutations at cytosines in EACs was calculated using the mutation data from the exome analysis reported in Dulak et al. [47]. Only single-nucleotide changes were considered for analysis. Duplicate mutations were used only if originating from different tumor samples. For the analysis, we used the mutations at cytosines on both strands. Based on the genomic coordinates of the mutations, the local sequence context was extracted using a Perl script (Additional file 12). The expected mutational context was calculated based on the exonic sequences of the reference genome used for the exomes (build GRCh37). The trinucleotide representing the local context of mutations at C either from the DT40 experiments, from the EAC data, or from the control exome were then fed into the weblogo interface [48].
Acknowledgements
We thank the CRBA in Bologna for cell sorting support. We are grateful to Rosario Notaro, Lucio Luzzatto, Mike Fainzilber, Javier Di Noia and Svend Petersen-Mahrt for the long discussions and helpful comments on the manuscript. SGC is especially grateful to Michael Neuberger, who took an entire day off from his illness to chat about life, science, and unfinished manuscripts. I take this opportunity to give tribute to years of mentorship and friendship by this great scientist. This work was supported by an institutional grant from the Istituto Toscano Tumori, and by a grant of the Italian Ministry of Health (GR-2008-1141464).
Abbreviations
- AID
Activation Induced Deaminase
- APOBEC1
Apolipoprotein B mRNA editing enzyme, catalytic polypeptide 1
- bp
base pair
- EAC
esophageal adenocarcinoma
- EGFP
enhanced green fluorescent protein
- FBS
fetal bovine serum
- GEO
Gene Expression Omnibus
- PCR
polymerase chain reaction
- TBP
TATA-binding protein
Additional files
Expression of the transduced proteins in stably transfected cells. (A) Western blot analysis of representative DT40 clones stably expressing APOBEC1. (B,C) Western blot analysis of representative K562 clones stably expressing rat APOBEC1, human APOBEC1, or AID as indicated. The blots were stripped and rehybridized with beta-actin antibody as control for loading.
Mutational sequence context in DT40 cells. Local sequence context for all residues present on both strands of the analyzed EGFP fragments (expected), for the mutated residues in the APOBEC1-expressing clones (APOBEC1) and in the controls (Control).
Expression levels of APOBEC1 in esophageal tissues. All data were normalized on TATA-binding protein (TBP) expression levels and shown as median-centered Log2 values. (A) Expression levels of the AID/APOBECs in the study by Kim et al. [44], comparing normal esophagus samples to esophageal adenocarcinoma (EAC) and Barrett’s esophagus samples. Each circle represents either a normal (blue), tumor (red), or Barrett’s (green) sample. The median is indicated as a black bar. Only differences in the expression levels of APOBEC1 (Normal/Tumor/Barrett’s samples) and APOBEC3A (Normal/Tumor samples) reach statistical significance (P < 0.0001 by one-way ANOVA coupled with Tukey’s test). (B) Levels of APOBEC1 in samples from matched normal/EAC pairs [44]. While an increased expression was present also with regards to APOBEC3A, only in the case of APOBEC1 did the differences reached statistical significance (P = 0.0002 by Wilcoxon signed rank test). (C) Levels of APOBEC1 in matched samples from normal esophagus, EACs, and Barrett’s esophagus in [45] (P = 0.0155 by repeated measures ANOVA coupled with Tukey’s test, with differences between tumors and Barrett’s samples not statistically significant). (D) Comparison of APOBEC1 expression levels in matched samples from normal/Barrett’s esophagus pairs and in small intestine [46] (P = 0.015 by Wilcoxon signed rank test).
Possible cross-hybridization of the APOBEC3 probes in the Illumina GPL6102 chip. The data from Kim et al. [44] originate from the Illumina GPL6102 beadchip. This platform uses 50-mer probes that allow for a more specific analysis compared with the Affymetrix ones: some probes (A3A, A3C, A3D, A3F) could still cross-hybridize with other genes (albeit with non-transcribed/intronic portions of other APOBEC3s), and others (A3B, A3G, A3H) should not. The cross-hybridization potential of the individual probes (matching bases/length of the probe) is shown. The location of the probe on the gene if outside of the coding region is shown in parentheses (intron, in; untranscribed region in the 3’ of the gene, downstream).
Mutational sequence context at position −2 in tumors expressing AID/APOBEC genes. The table shows the pyrimidine/purine ratio at position −2 in cancers for which the AID/APOBEC mutational signature has been associated with AID/APOBEC expression [30,31,33,58]. The expression of the various AID/APOBECs has been taken from Lin et al. [58] for the ESCC, and from Burns et al. [30,31] and Roberts et al. [33] for the other tumor types. The expression in EACs has been extrapolated from Kim et al. [44]. An asterisk indicates whenever the expression has been established through microarray analysis (as opposed to real-time PCR). The parentheses indicate the possible cross-reactivity of a microarray probe with other AID/APOBECs (Additional file 4).
Transition/transversion ratio in mutations at CpH sites in EAC associated or not with Barrett’s esophagus [47]. The number of tumors and the average in each subset are indicated in the legend and beside the box, respectively (P = 0.0192 by two-tailed t-test with Welch’s correction). The extra points represent the values external to the 5th to 95th percentile interval.
Sequence context for the mutated cytosine residues in EAC associated or not with Barrett’s esophagus [47]. The trinucleotide sequence context is shown for both CpG and non-CpG mutations as a weblogo [48].
File containing the chromatograms of the sequences obtained from the Rifampicin assay.
File containing the chromatograms of the sequences obtained from the DT40 experiments.
File containing the chromatograms of the sequences obtained from the K562 experiments.
File containing the chromatograms of the sequences obtained from the K562 experiments.
File containing the Perl scripts used in the mutation analysis.
Footnotes
Giulia Saraconi, and Francesco Severi are equal contributors.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
GS designed and performed the experiments, and drafted the manuscript. FS designed and performed the experiments and helped to draft the manuscript. CS prepared the cells with the EGFP reporter gene. GM performed the bioinformatic analysis. SGC conceived the study, participated in its design, performed the bioinformatic analysis and wrote the manuscript. All authors have read and approved the manuscript.
Contributor Information
Giulia Saraconi, Email: giulia.saraconi@ittumori.it.
Francesco Severi, Email: francesco.severi@ittumori.it.
Cesare Sala, Email: cesare.sala@ittumori.it.
Giorgio Mattiuz, Email: giorgio.mattiuz@ittumori.it.
Silvestro G Conticello, Email: silvo.conticello@ittumori.it.
References
- 1.Navaratnam N, Morrison JR, Bhattacharya S, Patel D, Funahashi T, Giannoni F, Teng BB, Davidson NO, Scott J. The p27 catalytic subunit of the apolipoprotein B mRNA editing enzyme is a cytidine deaminase. J Biol Chem. 1993;268:20709–20712. [PubMed] [Google Scholar]
- 2.Teng B, Burant CF, Davidson NO. Molecular cloning of an apolipoprotein B messenger RNA editing protein. Science. 1993;260:1816–1819. doi: 10.1126/science.8511591. [DOI] [PubMed] [Google Scholar]
- 3.Blanc V, Davidson NO. APOBEC-1-mediated RNA editing. Wiley Interdiscip Rev Syst Biol Med. 2010;2:594–602. doi: 10.1002/wsbm.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Conticello SG. The AID/APOBEC family of nucleic acid mutators. Genome Biol. 2008;9:229. doi: 10.1186/gb-2008-9-6-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith HC, Bennett RP, Kizilyer A, McDougall WM, Prohaska KM. Functions and regulation of the APOBEC family of proteins. Semin Cell Dev Biol. 2012;23:258–268. doi: 10.1016/j.semcdb.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102:553–563. doi: 10.1016/S0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- 7.Di Noia JM, Neuberger MS. Molecular mechanisms of antibody somatic hypermutation. Annu Rev Biochem. 2007;76:1–22. doi: 10.1146/annurev.biochem.76.061705.090740. [DOI] [PubMed] [Google Scholar]
- 8.Sheehy AM, Gaddis NC, Choi JD, Malim MH. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature. 2002;418:646–650. doi: 10.1038/nature00939. [DOI] [PubMed] [Google Scholar]
- 9.Refsland EW, Harris RS. The APOBEC3 family of retroelement restriction factors. Curr Top Microbiol Immunol. 2013;371:1–27. doi: 10.1007/978-3-642-37765-5_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Skuse GR, Cappione AJ, Sowden M, Metheny LJ, Smith HC. The neurofibromatosis type I messenger RNA undergoes base-modification RNA editing. Nucleic Acids Res. 1996;24:478–485. doi: 10.1093/nar/24.3.478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yamanaka S, Poksay KS, Arnold KS, Innerarity TL. A novel translational repressor mRNA is edited extensively in livers containing tumors caused by the transgene expression of the apoB mRNA-editing enzyme. Genes Dev. 1997;11:321–333. doi: 10.1101/gad.11.3.321. [DOI] [PubMed] [Google Scholar]
- 12.Rosenberg BR, Hamilton CE, Mwangi MM, Dewell S, Papavasiliou FN. Transcriptome-wide sequencing reveals numerous APOBEC1 mRNA-editing targets in transcript 3' UTRs. Nat Struct Mol Biol. 2011;18:230–236. doi: 10.1038/nsmb.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anant S, Davidson NO. An AU-rich sequence element (UUUN[A/U]U) downstream of the edited C in apolipoprotein B mRNA is a high-affinity binding site for Apobec-1: binding of Apobec-1 to this motif in the 3' untranslated region of c-myc increases mRNA stability. Mol Cell Biol. 2000;20:1982–1992. doi: 10.1128/MCB.20.6.1982-1992.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anant S, Murmu N, Houchen CW, Mukhopadhyay D, Riehl TE, Young SG, Morrison AR, Stenson WF, Davidson NO. Apobec-1 protects intestine from radiation injury through posttranscriptional regulation of cyclooxygenase-2 expression. Gastroenterology. 2004;127:1139–1149. doi: 10.1053/j.gastro.2004.06.022. [DOI] [PubMed] [Google Scholar]
- 15.Harris RS, Petersen-Mahrt SK, Neuberger MS. RNA editing enzyme APOBEC1 and some of its homologs can act as DNA mutators. Mol Cell. 2002;10:1247–1253. doi: 10.1016/S1097-2765(02)00742-6. [DOI] [PubMed] [Google Scholar]
- 16.Petersen-Mahrt SK, Neuberger MS. In vitro deamination of cytosine to uracil in single-stranded DNA by apolipoprotein B editing complex catalytic subunit 1 (APOBEC1) J Biol Chem. 2003;278:19583–19586. doi: 10.1074/jbc.C300114200. [DOI] [PubMed] [Google Scholar]
- 17.Severi F, Chicca A, Conticello SG. Analysis of reptilian APOBEC1 suggests that RNA editing may not be its ancestral function. Mol Biol Evol. 2011;28:1125–1129. doi: 10.1093/molbev/msq338. [DOI] [PubMed] [Google Scholar]
- 18.Morgan HD, Dean W, Coker HA, Reik W, Petersen-Mahrt SK. Activation-induced cytidine deaminase deaminates 5-methylcytosine in DNA and is expressed in pluripotent tissues: implications for epigenetic reprogramming. J Biol Chem. 2004;279:52353–52360. doi: 10.1074/jbc.M407695200. [DOI] [PubMed] [Google Scholar]
- 19.Guo JU, Su Y, Zhong C, Ming GL, Song H. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell. 2011;145:423–434. doi: 10.1016/j.cell.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bishop KN, Holmes RK, Sheehy AM, Malim MH. APOBEC-mediated editing of viral RNA. Science. 2004;305:645. doi: 10.1126/science.1100658. [DOI] [PubMed] [Google Scholar]
- 21.Bishop KN, Holmes RK, Sheehy AM, Davidson NO, Cho SJ, Malim MH. Cytidine deamination of retroviral DNA by diverse APOBEC proteins. Curr Biol. 2004;14:1392–1396. doi: 10.1016/j.cub.2004.06.057. [DOI] [PubMed] [Google Scholar]
- 22.Ikeda T, Ohsugi T, Kimura T, Matsushita S, Maeda Y, Harada S, Koito A. The antiretroviral potency of APOBEC1 deaminase from small animal species. Nucleic Acids Res. 2008;36:6859–6871. doi: 10.1093/nar/gkn802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Petit V, Guétard D, Renard M, Keriel A, Sitbon M, Wain-Hobson S, Vartanian JP. Murine APOBEC1 is a powerful mutator of retroviral and cellular RNA in vitro and in vivo. J Mol Biol. 2009;385:65–78. doi: 10.1016/j.jmb.2008.10.043. [DOI] [PubMed] [Google Scholar]
- 24.Ikeda T, Abd El Galil KH, Tokunaga K, Maeda K, Sata T, Sakaguchi N, Heidmann T, Koito A. Intrinsic restriction activity by apolipoprotein B mRNA editing enzyme APOBEC1 against the mobility of autonomous retrotransposons. Nucleic Acids Res. 2011;39:5538–5554. doi: 10.1093/nar/gkr124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gee P, Ando Y, Kitayama H, Yamamoto SP, Kanemura Y, Ebina H, Kawaguchi Y, Koyanagi Y. APOBEC1-mediated editing and attenuation of herpes simplex virus 1 DNA indicate that neurons have an antiviral role during herpes simplex encephalitis. J Virol. 2011;85:9726–9736. doi: 10.1128/JVI.05288-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamanaka S, Balestra ME, Ferrell LD, Fan J, Arnold KS, Taylor S, Taylor JM, Innerarity TL. Apolipoprotein B mRNA-editing protein induces hepatocellular carcinoma and dysplasia in transgenic animals. Proc Natl Acad Sci U S A. 1995;92:8483–8487. doi: 10.1073/pnas.92.18.8483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blanc V, Henderson JO, Newberry RD, Xie Y, Cho SJ, Newberry EP, Kennedy S, Rubin DC, Wang HL, Luo J, Davidson NO. Deletion of the AU-rich RNA binding protein Apobec-1 reduces intestinal tumor burden in Apc(min) mice. Cancer Res. 2007;67:8565–8573. doi: 10.1158/0008-5472.CAN-07-1593. [DOI] [PubMed] [Google Scholar]
- 28.Robbiani DF, Nussenzweig MC. Chromosome translocation, B cell lymphoma, and activation-induced cytidine deaminase. Annu Rev Pathol. 2013;8:79–103. doi: 10.1146/annurev-pathol-020712-164004. [DOI] [PubMed] [Google Scholar]
- 29.Nik-Zainal S, Alexandrov LB, Wedge DC, Van Loo P, Greenman CD, Raine K, Jones D, Hinton J, Marshall J, Stebbings LA, Menzies A, Martin S, Leung K, Chen L, Leroy C, Ramakrishna M, Rance R, Lau KW, Mudie LJ, Varela I, McBride DJ, Bignell GR, Cooke SL, Shlien A, Gamble J, Whitmore I, Maddison M, Tarpey PS, Davies HR, Papaemmanuil E, et al. Mutational processes molding the genomes of 21 breast cancers. Cell. 2012;149:979–993. doi: 10.1016/j.cell.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burns MB, Lackey L, Carpenter MA, Rathore A, Land AM, Leonard B, Refsland EW, Kotandeniya D, Tretyakova N, Nikas JB, Yee D, Temiz NA, Donohue DE, McDougle RM, Brown WL, Law EK, Harris RS. APOBEC3B is an enzymatic source of mutation in breast cancer. Nature. 2013;494:366–370. doi: 10.1038/nature11881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burns MB, Temiz NA, Harris RS. Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat Genet. 2013;45:977–983. doi: 10.1038/ng.2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Taylor BJ, Nik-Zainal S, Wu YL, Stebbings LA, Raine K, Campbell PJ, Rada C, Stratton MR, Neuberger MS. DNA deaminases induce break-associated mutation showers with implication of APOBEC3B and 3A in breast cancer kataegis. Elife. 2013;2:e00534. doi: 10.7554/eLife.00534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roberts SA, Lawrence MS, Klimczak LJ, Grimm SA, Fargo D, Stojanov P, Kiezun A, Kryukov GV, Carter SL, Saksena G, Harris S, Shah RR, Resnick MA, Getz G, Gordenin DA. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat Genet. 2013;45:970–976. doi: 10.1038/ng.2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alexandrov LB, Nik-Zainal S, Wedge DC, Campbell PJ, Stratton MR. Deciphering signatures of mutational processes operative in human cancer. Cell Rep. 2013;3:246–259. doi: 10.1016/j.celrep.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale AL, Boyault S, Burkhardt B, Butler AP, Caldas C, Davies HR, Desmedt C, Eils R, Eyfjörd JE, Foekens JA, Greaves M, Hosoda F, Hutter B, Ilicic T, Imbeaud S, Imielinsk M, Jäger N, Jones DT, Jones D, Knappskog S, Kool M, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arakawa H, Buerstedde JM. Activation-induced cytidine deaminase-mediated hypermutation in the DT40 cell line. Philos Trans R Soc Lond B Biol Sci. 2009;364:639–644. doi: 10.1098/rstb.2008.0202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Conticello SG, Thomas CJ, Petersen-Mahrt SK, Neuberger MS. Evolution of the AID/APOBEC family of polynucleotide (deoxy)cytidine deaminases. Mol Biol Evol. 2005;22:367–377. doi: 10.1093/molbev/msi026. [DOI] [PubMed] [Google Scholar]
- 38.Arakawa H, Saribasak H, Buerstedde JM. Activation-induced cytidine deaminase initiates immunoglobulin gene conversion and hypermutation by a common intermediate. PLoS Biol. 2004;2:E179. doi: 10.1371/journal.pbio.0020179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Beale RC, Petersen-Mahrt SK, Watt IN, Harris RS, Rada C, Neuberger MS. Comparison of the differential context-dependence of DNA deamination by APOBEC enzymes: correlation with mutation spectra in vivo. J Mol Biol. 2004;337:585–596. doi: 10.1016/j.jmb.2004.01.046. [DOI] [PubMed] [Google Scholar]
- 40.Suspène R, Aynaud MM, Guétard D, Henry M, Eckhoff G, Marchio A, Pineau P, Dejean A, Vartanian JP, Wain-Hobson S. Somatic hypermutation of human mitochondrial and nuclear DNA by APOBEC3 cytidine deaminases, a pathway for DNA catabolism. Proc Natl Acad Sci U S A. 2011;108:4858–4863. doi: 10.1073/pnas.1009687108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Klemm L, Duy C, Iacobucci I, Kuchen S, von Levetzow G, Feldhahn N, Henke N, Li Z, Hoffmann TK, Kim YM, Hofmann WK, Jumaa H, Groffen J, Heisterkamp N, Martinelli G, Lieber MR, Casellas R, Müschen M. The B cell mutator AID promotes B lymphoid blast crisis and drug resistance in chronic myeloid leukemia. Cancer Cell. 2009;16:232–245. doi: 10.1016/j.ccr.2009.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Orthwein A, Patenaude AM, Affar EB, Lamarre A, Young JC, Di Noia JM. Regulation of activation-induced deaminase stability and antibody gene diversification by Hsp90. J Exp Med. 2010;207:2751–2765. doi: 10.1084/jem.20101321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim SM, Park YY, Park ES, Cho JY, Izzo JG, Zhang D, Kim SB, Lee JH, Bhutani MS, Swisher SG, Wu X, Coombes KR, Maru D, Wang KK, Buttar NS, Ajani JA, Lee JS. Prognostic biomarkers for esophageal adenocarcinoma identified by analysis of tumor transcriptome. PLoS One. 2010;5:e15074. doi: 10.1371/journal.pone.0015074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kimchi ET, Posner MC, Park JO, Darga TE, Kocherginsky M, Karrison T, Hart J, Smith KD, Mezhir JJ, Weichselbaum RR, Khodarev NN. Progression of Barrett's metaplasia to adenocarcinoma is associated with the suppression of the transcriptional programs of epidermal differentiation. Cancer Res. 2005;65:3146–3154. doi: 10.1158/0008-5472.CAN-04-2490. [DOI] [PubMed] [Google Scholar]
- 46.Stairs DB, Nakagawa H, Klein-Szanto A, Mitchell SD, Silberg DG, Tobias JW, Lynch JP, Rustgi AK. Cdx1 and c-Myc foster the initiation of transdifferentiation of the normal esophageal squamous epithelium toward Barrett's esophagus. PLoS One. 2008;3:e3534. doi: 10.1371/journal.pone.0003534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dulak AM, Stojanov P, Peng S, Lawrence MS, Fox C, Stewart C, Bandla S, Imamura Y, Schumacher SE, Shefler E, McKenna A, Carter SL, Cibulskis K, Sivachenko A, Saksena G, Voet D, Ramos AH, Auclair D, Thompson K, Sougnez C, Onofrio RC, Guiducci C, Beroukhim R, Zhou Z, Lin L, Lin J, Reddy R, Chang A, Landrenau R, Pennathur A, et al. Exome and whole-genome sequencing of esophageal adenocarcinoma identifies recurrent driver events and mutational complexity. Nat Genet. 2013;45:478–486. doi: 10.1038/ng.2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maley CC, Galipeau PC, Li X, Sanchez CA, Paulson TG, Blount PL, Reid BJ. The combination of genetic instability and clonal expansion predicts progression to esophageal adenocarcinoma. Cancer Res. 2004;64:7629–7633. doi: 10.1158/0008-5472.CAN-04-1738. [DOI] [PubMed] [Google Scholar]
- 50.Spechler SJ, Sharma P, Souza RF, Inadomi JM, Shaheen NJ. American Gastroenterological Association: American Gastroenterological Association technical review on the management of Barrett's esophagus. Gastroenterology. 2011;140:e18–e52. doi: 10.1053/j.gastro.2011.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ellsworth E, Jackson SA, Thakkar SJ, Smith DM, Finkelstein S. Correlation of the presence and extent of loss of heterozygosity mutations with histological classifications of Barrett's esophagus. BMC Gastroenterol. 2012;12:181. doi: 10.1186/1471-230X-12-181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Streppel MM, Lata S, DelaBastide M, Montgomery EA, Wang JS, Canto MI, Macgregor-Das AM, Pai S, Morsink FH, Offerhaus GJ, Antoniou E, Maitra A, McCombie WR. Next-generation sequencing of endoscopic biopsies identifies ARID1A as a tumor-suppressor gene in Barrett's esophagus. Oncogene. 2014;33:347–357. doi: 10.1038/onc.2012.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Küppers R, Dalla-Favera R. Mechanisms of chromosomal translocations in B cell lymphomas. Oncogene. 2001;20:5580–5594. doi: 10.1038/sj.onc.1204640. [DOI] [PubMed] [Google Scholar]
- 54.Landry S, Narvaiza I, Linfesty DC, Weitzman MD. APOBEC3A can activate the DNA damage response and cause cell-cycle arrest. EMBO Rep. 2011;12:444–450. doi: 10.1038/embor.2011.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shinohara M, Io K, Shindo K, Matsui M, Sakamoto T, Tada K, Kobayashi M, Kadowaki N, Takaori-Kondo A. APOBEC3B can impair genomic stability by inducing base substitutions in genomic DNA in human cells. Sci Rep. 2012;2:806. doi: 10.1038/srep00806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Roberts SA, Sterling J, Thompson C, Harris S, Mav D, Shah R, Klimczak LJ, Kryukov GV, Malc E, Mieczkowski PA, Resnick MA, Gordenin DA. Clustered mutations in yeast and in human cancers can arise from damaged long single-strand DNA regions. Mol Cell. 2012;46:424–435. doi: 10.1016/j.molcel.2012.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lada AG, Dhar A, Boissy RJ, Hirano M, Rubel AA, Rogozin IB, Pavlov YI. AID/APOBEC cytosine deaminase induces genome-wide kataegis. Biol Direct. 2012;7:47. doi: 10.1186/1745-6150-7-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lin DC, Hao JJ, Nagata Y, Xu L, Shang L, Meng X, Sato Y, Okuno Y, Varela AM, Ding LW, Garg M, Liu LZ, Yang H, Yin D, Shi ZZ, Jiang YY, Gu WY, Gong T, Zhang Y, Xu X, Kalid O, Shacham S, Ogawa S, Wang MR, Koeffler HP. Genomic and molecular characterization of esophageal squamous cell carcinoma. Nat Genet. 2014;46:467–473. doi: 10.1038/ng.2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Refsland EW, Stenglein MD, Shindo K, Albin JS, Brown WL, Harris RS. Quantitative profiling of the full APOBEC3 mRNA repertoire in lymphocytes and tissues: implications for HIV-1 restriction. Nucleic Acids Res. 2010;38:4274–4284. doi: 10.1093/nar/gkq174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Okazaki IM, Hiai H, Kakazu N, Yamada S, Muramatsu M, Kinoshita K, Honjo T. Constitutive expression of AID leads to tumorigenesis. J Exp Med. 2003;197:1173–1181. doi: 10.1084/jem.20030275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Muto T, Okazaki IM, Yamada S, Tanaka Y, Kinoshita K, Muramatsu M, Nagaoka H, Honjo T. Negative regulation of activation-induced cytidine deaminase in B cells. Proc Natl Acad Sci U S A. 2006;103:2752–2757. doi: 10.1073/pnas.0510970103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shen HM, Bozek G, Pinkert CA, McBride K, Wang L, Kenter A, Storb U. Expression of AID transgene is regulated in activated B cells but not in resting B cells and kidney. Mol Immunol. 2008;45:1883–1892. doi: 10.1016/j.molimm.2007.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Arakawa H, Hauschild J, Buerstedde JM. Requirement of the activation-induced deaminase (AID) gene for immunoglobulin gene conversion. Science. 2002;295:1301–1306. doi: 10.1126/science.1067308. [DOI] [PubMed] [Google Scholar]
- 64.Conticello SG, Ganesh K, Xue K, Lu M, Rada C, Neuberger MS. Interaction between antibody-diversification enzyme AID and spliceosome-associated factor CTNNBL1. Mol Cell. 2008;31:474–484. doi: 10.1016/j.molcel.2008.07.009. [DOI] [PubMed] [Google Scholar]
- 65.Arakawa H, Lodygin D, Buerstedde JM. Mutant loxP vectors for selectable marker recycle and conditional knock-outs. BMC Biotechnol. 2001;1:7. doi: 10.1186/1472-6750-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Petersen-Mahrt SK, Harris RS, Neuberger MS. AID mutates E. coli suggesting a DNA deamination mechanism for antibody diversification. Nature. 2002;418:99–103. doi: 10.1038/nature00862. [DOI] [PubMed] [Google Scholar]