

Abstract
Protein–protein interactions play critical roles in biology, and computational design of interactions could be useful in a range of applications. We describe in detail a general approach to de novo design of protein interactions based on computed, energetically optimized interaction hotspots, which was recently used to produce high-affinity binders of influenza hemagglutinin. We present several alternative approaches to identify and build the key hotspot interactions within both core secondary structural elements and variable loop regions and evaluate the method's performance in natural-interface recapitulation. We show that the method generates binding surfaces that are more conformationally restricted than previous design methods, reducing opportunities for off-target interactions.
Keywords: protein interactions, computational design, negative design, antibody engineering, conformational plasticity
Introduction
Protein–protein interactions play key roles in diverse cellular processes, including cell signaling, immune response, and host–pathogen recognition. The atomic structures of thousands of protein complexes have been solved by X-ray crystallography and nuclear magnetic resonance spectroscopy. Together with biophysical studies on the contribution of individual amino acid residues to binding, these experimental data provide a detailed view of the molecular basis for association of natural proteins.
Despite this wealth of information, until recently, general tools for computational design of novel protein binders were not available, suggesting deficiencies in our understanding of protein association. Indeed, most existing technologies for generating specific binding proteins have been confined to in vitro screening and selection of antibodies and specialized protein scaffolds such as ankyrins and fibronectin domains.1–5 Though such methods have resulted in promising therapeutics and diagnostics, they do not allow targeting of a specific region of interest on a target protein surface. They also rely on a handful of protein scaffolds from which new binders are evolved (e.g., the immunoglobulin fold), and an optimal topology for binding to a target surface might be inaccessible to any of the scaffolds in a given experiment. By contrast, computational design of protein binders allows the testing and refining of our understanding of molecular recognition and can take advantage of many different scaffolds.
Structures of biologically relevant protein–protein interfaces (in contrast to crystal lattice interactions) reveal that 1600 ± 400 Å2 of the previously solvent-accessible surface area is buried upon complexation.6 Shape complementarity (Sc)7 between the two surfaces is exquisite with clusters of core interactions providing a disproportionate amount of the binding energy (examples are shown in Fig. 1). These clusters, originally termed interaction hot-spots by Clackson and Wells, have been subsequently characterized in many protein complexes (reviewed in Ref. 11).12 Upon single-site mutation of hotspot residues to alanine, binding affinity typically decreases by several orders of magnitude.13 The interactions mediated by these hotspot residues are energetically very favorable, involving hydrogen bonds, tight van der Waals packing, and favorable electrostatics, and are more evolutionarily conserved on average than the remainder of the interface.14–16 Co-crystal structures show that several different proteins that interact with a single target surface often use analogous key residues in spite of large differences in the binders' folds. In a phenomenon termed structural mimicry,17 bacterial effectors of host proteins position the same hotspot residues in spatially overlapping locations with those of the host effector proteins (Fig. 1d), though sharing no secondary structural elements. Such evolutionary convergence on the same placement of key residues may indicate that these hotspot residues represent an optimal solution to the challenge of binding to the target surface.
Fig. 1.
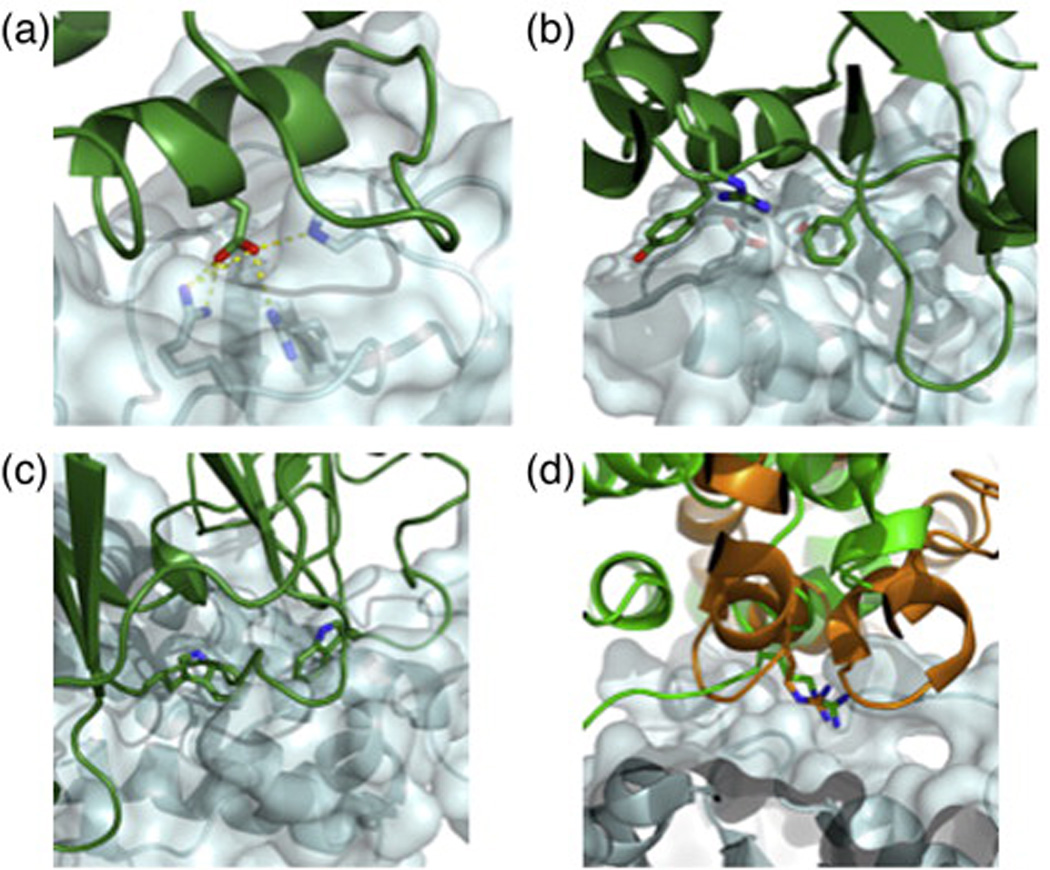
Examples of naturally occurring hotspots. (a) Bacterial ribonuclease barnase (pale cyan) and its inhibitor barstar (green). Hydrogen bonds between a hotspot aspartate on barstar and three basic side chains are marked as yellow dashed lines. (b) Bacterial colicin endonuclease E9 (green) and immunity protein Im9 (pale cyan). E9 hotspot residue Phe86 and Im9 hotspot residue Tyr54 are shown in sticks. The hotspot is supported by peripheral interactions, such as a salt bridge involving Arg54 (E9) and Glu30 (Im9). (c) Human growth hormone receptor (green) and human growth hormone (pale cyan). Two Trp hotspot residues are shown in sticks. (d) Structural mimicry shows key residues that are conserved between proteins that interact with a similar surface. Human Cdc42 GTPase-activating protein8 (green) and Salmonella SptP9 (gold) share a key arginine residue at the core of the interface, through which they interact with a GTPase (pale cyan). Sticks show key residues, including hotspot residues. All molecular representations were generated using PyMOL.10
Computational motif-grafting approaches have been used to design protein binders of DNA and other proteins. Liu et al. identified a set of three key residues in human erythropoietin (Epo) interacting with erythropoietin receptor (EpoR) and grafted these residues on an unrelated protein to obtain a high-affinity binder to EpoR.18 Havranek and Baker used restraints on backbone positions to guide backbone sampling to conformations that are compatible with positioning residue motifs for binding DNA sequences.19 Although powerful, a limitation of grafting methods is that they are restricted to the interactions previously observed in crystal structures. Such restrictions result in a limited repertoire of viable backbones and do not take full advantage of the plasticity of hotspots implied by the above-mentioned examples of structural mimicry.17
Side chains that make important contributions to binding are conformationally restricted by dense interaction networks involving surrounding side chains and backbone atoms on their host monomer, thereby disfavoring nonnative binding modes.20 Recently, a computational method for two-sided design centering on the incorporation of high-affinity residue interactions at the interface of two naturally noninteracting proteins yielded a de novo high-affinity interacting pair.21 However, a co-crystal structure of a close variant of the computational design showed significant rearrangements at the interface compared with that of the model, such that the experimentally determined interface utilized the same surfaces but reoriented by 180°. Taken together, these observations suggest that, to achieve specific molecular recognition, design methodology should incorporate elements of negative design, where off-target states are penalized.22 However, explicit modeling of all alternative states during design is computationally intractable for even modestly sized protein systems.23, 24 We reasoned that the clustering of hotspot residues, often observed in natural interfaces, may lead to dense interaction networks disfavoring alternative states as any rearrangement of the network would likely result in the elimination of favorable interactions and introduction of steric overlaps, conformational strain, or energetically unfavorable voids. The strategy of forming dense interaction networks is computationally affordable, as it does not require explicitly modeling alternative states.
Based on this reasoning, we recently developed a computational method to generate high-affinity binders of influenza hemagglutinin by constructing a diverse hotspot conception comprising thousands of potential amino acid residue combinations and incorporating these interactions on diverse scaffolds.25 This method opened the way, in principle, to the design of proteins binding any desired protein target. Here, we generalize this method to a range of design scenarios and show that it produces interfaces which recapitulate some key properties of native interfaces.
Results
A flowchart describing a generalization of our method is shown in Fig. 2. Our strategy centers on forming high-affinity interactions at the core of the interface. The first step (1A in Fig. 2) involves the construction of an interaction hotspot region by single-residue docking with RosettaDock26 using rigid-body sampling and side-chain repacking. We require that hotspot residues form dense interactions, interacting favorably with both one another and the target surface, as seen in natural interfaces (Fig. 1). This step can be used to precompute interactions with an arbitrarily large number of surfaces on the target protein. Hotspot residues can be of any type but, as in natural interfaces, are primarily larger amino acids such as aromatics. A specific hotspot region typically contains multiple types of amino acids to best complement the physicochemical properties of the binding surface. For each hotspot residue, all rotamers compatible with the computed binding mode are used—each results in an alternative position for the backbone of the scaffold position that will ultimately support it (step 2A). In a parallel step, which is completely independent from the first step, we use coarse-grained docking of the two protein partners to compute high-shape-complementary configurations of the designed scaffold protein and the target surface (1–2B). Next (step 3), the results from the first two steps are combined: in the vicinity of each of the coarse-grained binding modes found in the second step, a search is carried out for rigid-body orientations that support as many of the hotspot residue backbones as possible. In step 4, the hotspot-placement step, the hotspot residues are explicitly incorporated on the scaffold protein, followed by structural and energetic filters (step 5). The remainder of the residues in the scaffold that are at the protein–protein interface are then designed (step 6) and refined, and several filters including ones that are tailored specifically for the particular target (such as specific hydrogen-bonding patterns at the interface) are used to screen the resultant designs (step 7). RosettaScripts27 implementations for each of the cases described here are available as Supplemental Data.
Fig. 2.
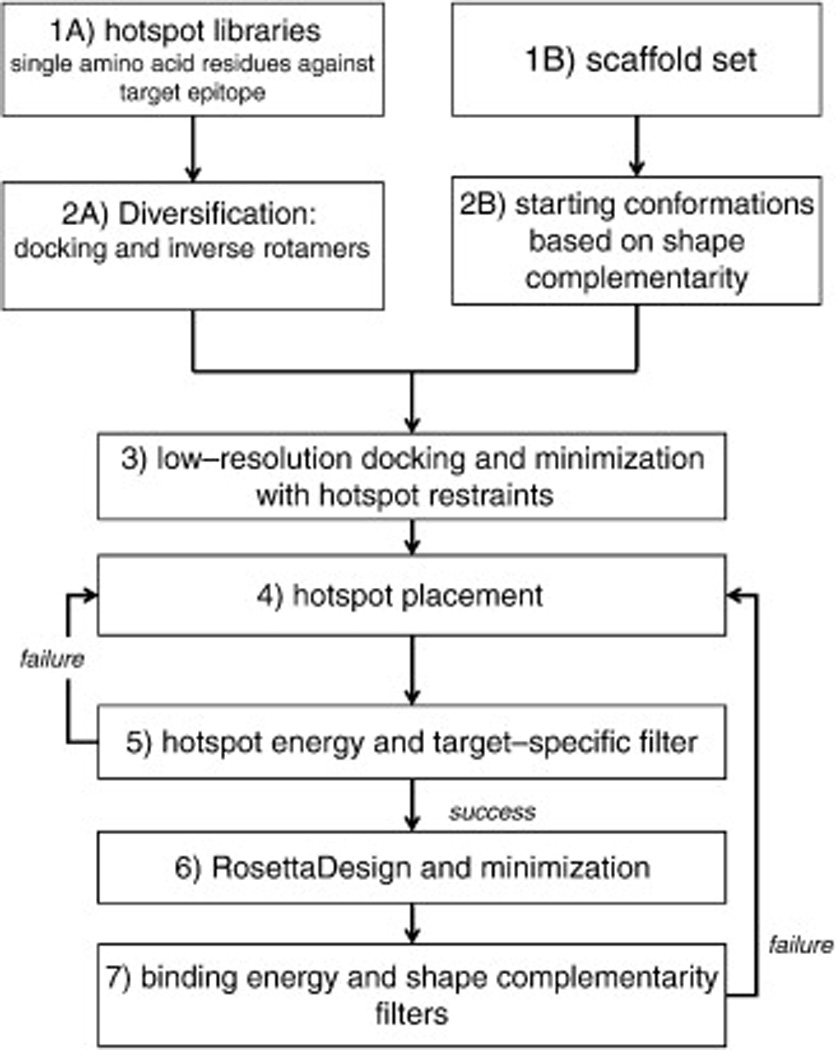
Overview of the hotspot-centric design strategy. Labels (1A, 1B, etc.) are used in Results to refer to individual steps in the design process. The first two steps, 1 and 2, are divided into two independent design paths, signified by labels A and B.
Hotspot interactions are dominated by short-range contacts, such as van der Waals packing, hydrogen bonding, and the burial of hydrophobic surfaces, which are generally well modeled by the Rosetta all-atom energy. Thus, the use of hotspot-based constraints is unlikely to improve design simply by compensating for errors in the energy function. Instead, by designing the hotspot residues so they interact favorably both with the target surface and one another, we increase the chances that the binding surface would adopt the designed conformation even in the target's absence, precluding other conformational states of the designed surface that would be incompatible with binding the target.20
Three strategies for generating hotspot residue libraries (steps 1A and 2A)
De novo design of high-affinity interactions
To identify high-affinity interactions in an unbiased way, we exhaustively dock all amino acid residues (except Cys, Gly, and Pro) against the target's surface. Each residue is treated as a small-molecule ligand, and RosettaDock26 is used to sample the rigid-body interactions between the side chain and the target protein, similar to the method of Ben-Shimon and Eisenstein.28 As the docked residue's backbone will not be fully accessible for interaction in the context of a designed protein, the force field used in docking the residue only considers the steric repulsion terms related to backbone atoms. The target protein can be held fixed or allowed to repack to identify potential binding sites not observed in the crystal structure, producing more diverse hotspots.
We find that the lowest-energy interactions identified in fixed target docking calculations form clusters of chemically similar residues with respect to the target surface, for example, of acidic residues interacting with basic patches on the target and vice versa (Fig. 3a). These interactions sometimes recapitulate natural hotspot interactions, as observed in the docking of Glu residues in a manner very similar to that of the barstar's Asp39 hotspot residue (Figs. 1a and 3b). However, by focusing on single-residue docking, this strategy cannot, by itself, generate clusters of interacting hotspot residues as observed in many natural interfaces (Fig. 1a–c). To generate such clusters of favorably interacting residues, one could conduct a second round of residue docking against a protein surface comprising the target site and previously identified hotspot residues and select residues that form high-affinity interactions with both the target surface and the previously identified hotspot residues.
Fig. 3.
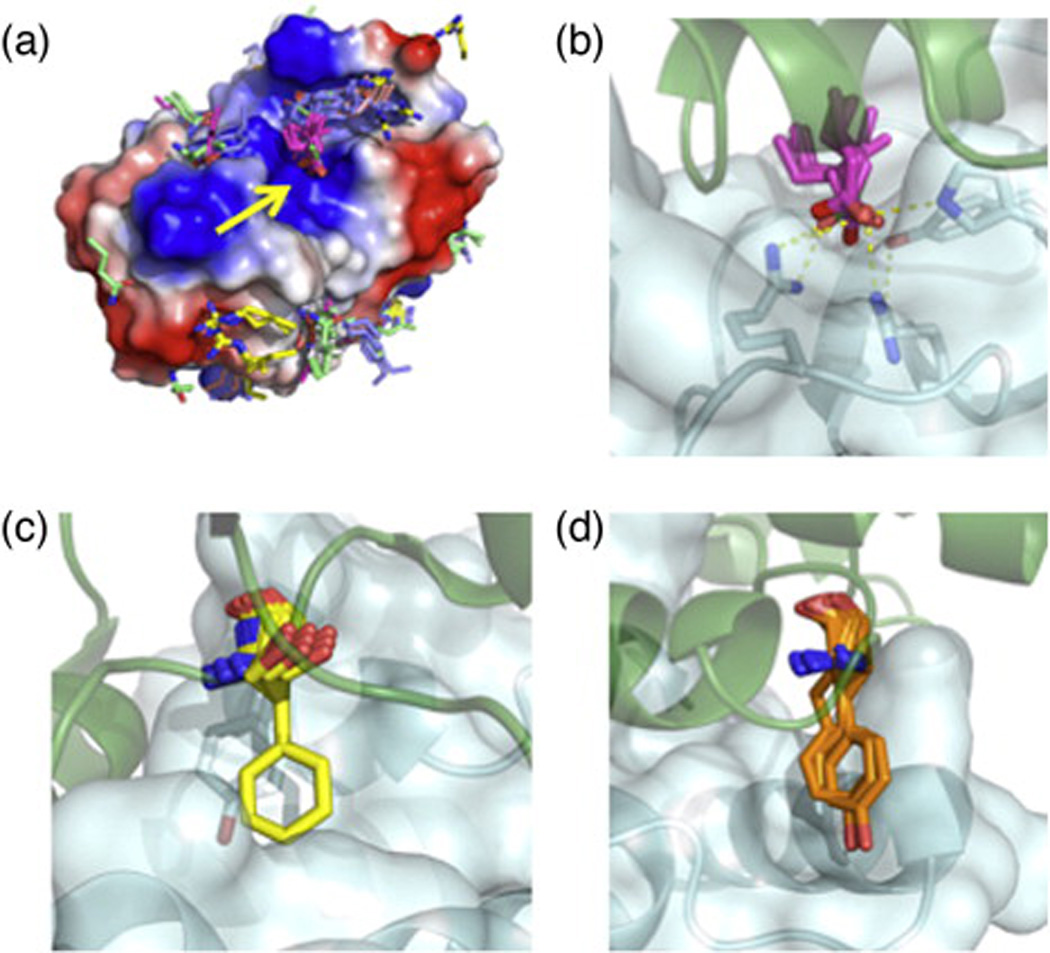
Construction of hotspot residue libraries. (a) A de novo hotspot library of the barnase surface highlights the barstar hotspot-binding site (yellow arrow). Ten thousand trajectories of all natural amino acids except Gly, Cys, and Pro were evaluated, and the best 1% by calculated binding energy of each residue identity was used to build a hotspot library. The barnase surface is colored by electrostatic charge using vacuum electrostatics in PyMOL. (b) De novo identified hotspot interactions of Glu (pink sticks) closely recapitulate the interaction of barstar Asp39 (green sticks) with barnase. (c) Inverse rotamers of the colicin E9 hotspot Phe86 (yellow sticks) against the surface of Im9. Im9 hotspot residue Tyr54 is shown in pale-cyan sticks. Highly optimal π-stacking interactions between E9 Phe86 and Im9 Tyr54 restrict the rigid-body orientation of the Phe hotspot residue. (d) E9 hotspot Tyr83 has more conformational freedom against the Im9 surface than the Phe86 of (c), allowing for rigid-body docking of the Tyr residue (orange). For each docked conformation, inverse rotamers were built as in (c) but, for clarity, are not shown in this figure.
Diversified native hotspots
Co-crystal structures of natural protein complexes reveal only one or, in favorable cases, a handful of different bound structures involving any target site, thus limiting the usefulness of crystal structures for design of novel binders. However, the hotspot interactions observed in natural binders can be diversified to generate a hypothetical hotspot region comprising thousands of combinations of residue interactions with the target surface. We developed two strategies for doing so: inverse rotamers starting from a fixed side-chain functional group and rigid-body docking.
Inverse rotamers
This approach is suitable for interactions in which the hotspot residue functional group makes very favorable and geometrically constrained interactions with the target surface. Since the functional group interactions are very well defined in this case, diversification is restricted to non-clashing residue conformations that position the functional group in the appropriate location for interaction with the target. This diversification procedure was used to generate a diverse set of interactions with the immunity protein (Im) 9 surface based on the Phe86 hotspot residue from the colicin endonuclease (E) 929 (Fig. 3c). The identities of the hotspot residues can be further diversified by choosing chemically similar side chains that are compatible with interacting with the target surface (e.g., the other aromatics in addition to Phe).
Rigid-body docking
In this approach, a hotspot residue observed in the co-crystal structure is docked against the target surface, isolating high-affinity interactions. This procedure is better suited to hotspot residues where the geometric constraint is more lax and where a larger diversity of backbone positions is desired, for example, the E9-Tyr83-based hotspot position against the Im9 surface (Fig. 3d). While docking the hotspot residue, previously isolated hotspot residues can be positioned in an optimal location ensuring that the docked side chain interacts favorably with other hotspot residues, as well as with the target surface forming native-like densely interacting clusters of hotspot residues.
Docking in a hotspot-restraint-based force field
Pairs of target and scaffold proteins are first docked using PatchDock30 to identify shape-complementary configurations without any hotspot-based restraints (steps 1B and 2B). In this study, we restrict ourselves to the top 100 PatchDock orientations, but typically, PatchDock produces several thousand configurations for each scaffold protein with respect to the target site, providing many alternative binding modes. These preliminary configurations are coarse-grained, and many steric overlaps are observed at the interface, which are relieved by subsequent rounds of sequence design and all-atom refinement.
The PatchDock orientations are then refined using low-resolution RosettaDock to generate rigid-body orientations compatible with the hotspot residues identified as described in the previous section.26 Docking refinement is limited to rigid-body orientations in the vicinity of the PatchDock-computed configuration to retain high shape complementarity. The force field that is normally used by RosettaDock is augmented with strong hotspot-based restraints to enrich for configurations of the scaffold that would be compatible with harboring as many hotspot residues as possible [Fig. 4a and Eqs. (1) and (2)], and configurations that do not satisfy at least one restraint are triaged, thereby pruning design trajectories that are unlikely to produce hotspot-containing interfaces.
Fig. 4.
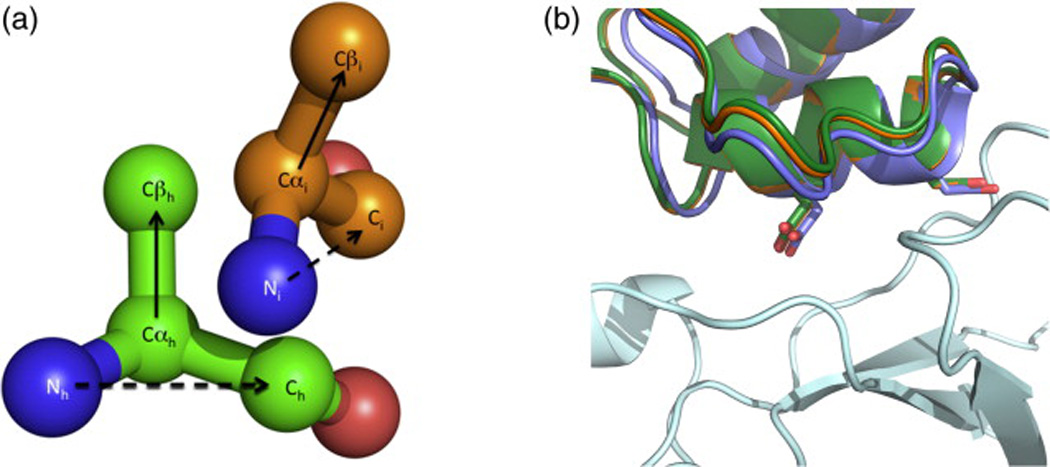
Hotspot-based restraints. (a) Hotspot-based restraints are derived according to Eq. (2) from hotspot residues (green) and applied to scaffold positions (gold). (b) The rigid-body conformation of native barstar (green) was locally docked (purple) and then minimized (orange) against its target surface on barnase (pale cyan) in the presence of strong hotspot-based restraints. The hotspot aspartates are shown as sticks in all three models. The distances between the Cα atoms of the hotspot residues in the native state compared to the redocked state are 1 Å, and after minimization, the distances are below 0.2 Å.
Following Monte-Carlo-based low-resolution docking, side chains on the scaffold protein within 10 Å of the target surface are reduced to their Cβ atom (excepting Pro, Gly, and disulfide-linked cysteines, which are not designed), and the side-chain conformations and rigid-body configuration of the two proteins are minimized, again with strong hotspot restraints (Fig. 4b and step 3). Even though the minimized configuration is often extremely close to the native, with the Cα atoms of the hotspot residues under 1 Å from those of the natural complex, we found that sequence design following this docking minimization step usually does not produce a native-like hotspot-containing region, highlighting the ruggedness of the energy landscape around the native configuration. We therefore follow docking with an explicit hotspot-placement step.
Hotspot residue placement on redesigned scaffolds
Following docking refinement, the scaffold protein is within close distance to a configuration that may be compatible with harboring the precomputed hotspot residues. We next iterate over scaffold positions within 4 Å of the hotspot residues within each hotspot residue library and place them on the scaffold (step 4). We implemented three methods of incorporating the hotspot residues, different combinations of which could be used in different design scenarios.
Scaffold placement superimposes the scaffold protein such that one of the scaffold residues matches one of the precomputed hotspot residues precisely. The advantage of this placement strategy is that it results in a designed scaffold that reproduces exactly the interaction with the target protein that was computed for the disembodied amino acid residue in the hotspot residue library. This strategy is optimal for interactions where a precise geometric relationship between the hotspot residue and the target surface is required. An example of such a scenario is given by Phe86 in the colicin E9, which is conformationally highly restricted (Fig. 3c). Following scaffold placement, the rigid body of the scaffold protein and the side-chain degrees of freedom of the designed hotspot residue are minimized to reduce strain. This step can result in eliminating the critical contacts between the hotspot residue and the target, for instance, the aromatic interactions between the Phe hotspot residue and Im9's Tyr54 (Fig. 3c), and is followed by energetic and structural filters to ensure that the hotspot residues retain their close-to-optimal positions (step 5). Scaffold placement cannot be invoked more than once without deforming the scaffold protein's main chain, and thus, subsequent placements require other strategies, two of which are outlined below.
Hotspot residue placement iterates over each hotspot residue in a library and minimizes the configuration of the scaffold protein matching a randomly chosen scaffold position within 4 Å of the idealized hotspot residue with a single hotspot residue subject to the restraints of Eqs. (1) and (2). If other hotspot residues were previously placed, we take advantage of the dihedral degrees of freedom of these side chains by freeing them during minimization, subject to the constraint that the hotspot residue's functional group maintains its position with respect to the target. For colicin E9–Im9 design (Fig. 3c and d), we minimized the dihedral degrees of freedom of the first placed aromatic hotspot residue (corresponding to E9's Phe86) starting from the side chain's Cγ going back to the scaffold's backbone. Following minimization, the second hotspot residue (corresponding to E9's Tyr83) is explicitly modeled on the selected scaffold position, and the configuration of the complex is further minimized in an all-atom force field.
If more than two hotspot residues are to be placed or if many different conformations need to be scanned, such as when backbone conformational changes are modeled, the iterative grafting protocols described above become computationally very demanding. We therefore implemented a third variant called simultaneous placement. In this protocol, multiple scaffold positions are simultaneously coupled to multiple-site hotspot residue libraries. The choice of which amino acid to design at each position is determined by the fit between the orientation of the backbone position and the hotspot residue as well as the distance between their Cβ atoms [Eq. (1) and further details in Methods]. These scaffold residues are then redesigned to identities of the hotspot residues that were coupled to them. This strategy is computationally efficient and was used in the flexible backbone design of antibody loops (see below).
Following each hotspot-placement step, the resulting configurations are filtered based on the all-atom energy to ensure that the hotspot residue is energetically favorable in the context of the scaffold protein (step 5). In some cases, we impose additional target-dependent structural filters, as different hotspot placements imply different filtering requirements, such as contacts with the target surface and the formation of hydrogen bonds with specific chemical groups on the target. These filtering steps prune the majority of modeling trajectories that are unlikely to result in productive binding modes prior to the computationally intensive steps of full design and refinement of the interface. The rigid-body orientation and the hotspot side chains are then minimized. Following the design of the core hotspot region on the scaffold protein, the surface of the scaffold protein is designed using RosettaDesign31 for increased interaction energy (step 6). Scaffold hotspot residues are not allowed to repack during this stage so as to maintain the clustering of the hotspot originally envisioned in the hotspot libraries.
At the end of the design process, the complexes are filtered (step 7) for computed binding energy32 [below −15 Rosetta energy units (R.e.u.), roughly −7.5 kcal/mol33], buried surface area (above 1200 Å2), and shape complementarity (Sc7 above 0.66). Additional target-specific structural filters, such as hotspot residues satisfying particular hydrogen bonds, were also used (see RosettaScripts in Supplemental Data). In this report, only automatic filters were used, but prior to experimental characterization, the designs are generally visually inspected for fine tuning and selection of candidates for testing;25 the effect (productive or counterproductive) of this final step based on human intuition requires further investigation.
A recapitulation benchmark for protein-interface design
Standard native complex recapitulation tests are not sufficient to test the hotspot-based method since if the natural hotspot residues are included in the hotspot libraries, the natural binding mode would very likely be recapitulated. Indeed, the more restrictive the hotspot conception, the higher the likelihood that the protocol would single out the natural complex from alternatives, so that high preference for the native binder and binding mode is a trivial outcome of our method. However, in the context of de novo design, we are interested in generating proteins that incorporate native binder-like features in scaffolds other than the natural binding partner. In the following, we use an interface design recapitulation benchmark to demonstrate that an appropriately diverse set of hotspots generates native-like interfaces in both natural and proteins that are not the natural partners of the target protein.
We assembled a diverse set of naturally occurring protein complexes (Table 1). All of these interactions are non-obligatory and span a wide range of dissociation constants, from the femtomolar (barnase–barstar38 and colicin immunity39) to the mid-nanomolar (Fc–protein A).40 Some are electrostatically steered (barnase–barstar and colicin), and others comprise mainly hydrophobic interactions at the interface (hemagglutinin–CR6261). The benchmark spans several functional classes, including pathogen–host recognition (Fc–protein A), antibody inhibitors of protein function (hemagglutinin–CR6261) and enzymes (lysozyme–antibody), and signaling (hGH–hGHR). Two complexes, barnase–barstar and colicin E9–Im9, are used twice each, once with one chain as the target and the other as scaffold and vice versa.
Table 1.
Protein-interface design benchmark
| Protein Data Bank entrya |
Interactionb | Hotspot diversificationc |
Hotspot residues |
RMSD (Å) | Sequence ID (%)d | Ranke | Alternative scaffoldsf |
|---|---|---|---|---|---|---|---|
| 1l6x | Fc–protein A | Native | Asn/Gln, Phe/Trp | 0.7 | 20 | 1/2 | 2 |
| Combined | Asn/Gln, Phe/Trp | 1.1 | 36 | 1/3 | 7 | ||
| 1sq2 | Lysozyme–antibody | Native | Tyr, Arg | 3.7 | 33 | 1/1 | 0 |
| Combined | Tyr, Arg | 3.94 | 10 | 1/1 | 0 | ||
| 1emv | Colicin E9–Im9 | Native | Tyr, Tyr | 0.8 | 39 | 1/2 | 1 |
| Combined | Tyr, Tyr | 0.8 | 33 | 1/1 | 0 | ||
| 1emv | Colicin Im9–E9 | Native | Phe, Tyr | 0.7 | 31 | 1/2 | 0 |
| Combined | Phe, Tyr | 0.5 | 35 | 1/1 | 0 | ||
| 1brs | Barnase–barstar | Native | His, Arg | 0.4 | 35 | 1/4 | 2 |
| Combined | His, Arg | 0.4 | 35 | 1/5 | 4 | ||
| 1brs | Barstar–barnase | Native | Asp, Asp | 0.6 | 20 | 1/1 | 0 |
| Combined | Asp, Asp | 0.8 | 20 | 1/1 | 1 | ||
| 3gbn | Hemagglutinin–antibody | Native | Ile, Phe | 3.2 | 10 | 2/2 | 0 |
| Combined | Ile, Phe | 4.5 | 20 | 1/1 | 0 | ||
| 1a22 | hGHR–hGH | Native | Trp, Trp | 0.9 | 23 | 1/1 | 0 |
| Combined | Trp, Trp | 0.9 | 37 | 1/1 | 0 |
Each scaffold monomer was designed to bind each target.
Target and scaffold are on the left-hand side and right-hand side, respectively.
Native: hotspot residue library contains diversified hotspot residues observed in natives; combined: hotspot residue library contains diversified hotspot residues observed in natives as well as de novo generated hotspot residues.
Computed as a percentage of the total number of residues on the scaffold within a 10-Å shell around the target.
The ranking in terms of computed binding energy32 of the close-to-native redesign (left) out of the total number of binding modes identified. Binding modes are defined as configurations that cluster within 4 Å.
The total number of scaffolds other than the native binder that produced designs that passed all of the reported filters.
Each protein scaffold was repacked in the absence of the target to eliminate memory of the binding configuration. It was then redocked using PatchDock.30 We find that, in the constrained setup mentioned here, a close-to-native configuration [< 4-Å root-mean-square deviation (RMSD) over all backbone atoms] is often identified among the highest-ranking 100 PatchDock solutions (but not in the case of hGH and its receptor, see below). The 100 best-scoring PatchDock configurations were subjected to the hotspot-centric design protocols provided as Supplemental Information using two different sets of hotspot residue libraries: (1) diversified from the co-crystal structure (see Table 1 entries labeled Native) and (2) the hotspot residues in (1) augmented with de novo computed hotspot residues (combined in Table 1).
The majority of scaffolds designed with the hotspot-centric method find configurations that are very close (<1.0 Å) to the configuration observed in the natural complexes (Table 1). Even trajectories that start from a PatchDock generated configuration that is as remote as 24 Å from the native end in a configuration that is very similar to that of the experimentally determined one (hGH and its receptor in Table 1), indicating that, in some cases, restraint-driven docking and hotspot placement have a very large radius of convergence. Sequence recovery rates in hotspot-guided simulations are largely in line with those reported for an enzyme design benchmark.41 These rates are approximately 30%, in keeping with the findings that amino acid identities outside of the hotspot region are less evolutionarily conserved14,15,42 and considerably more insensitive to mutation than those at the hotspot,13,43–46 though improved recapitulation rates may be obtained by using an energy function expressly optimized for protein interfaces.47
As a concrete example of the characteristically high recapitulation of the natural interaction using this method, we demonstrate the results of subjecting the natural complex between the bacterial ribonuclease, barnase, and its protein inhibitor barstar38 to our design protocol (Fig. 5). The side chains of the hotspot residues are recaptured with high accuracy, and other interacting residues are also largely recapitulated. Despite starting from 100 nonredundant rigid-body configurations for the pair, no other configuration of the barnase–barstar system could incorporate the desired hotspot and satisfy the specified energetic and structural filters, illustrating the protocol's specificity and restrictiveness. Similar results were obtained for all interfaces in the natural-interface recapitulation set (Table 1).
Fig. 5.
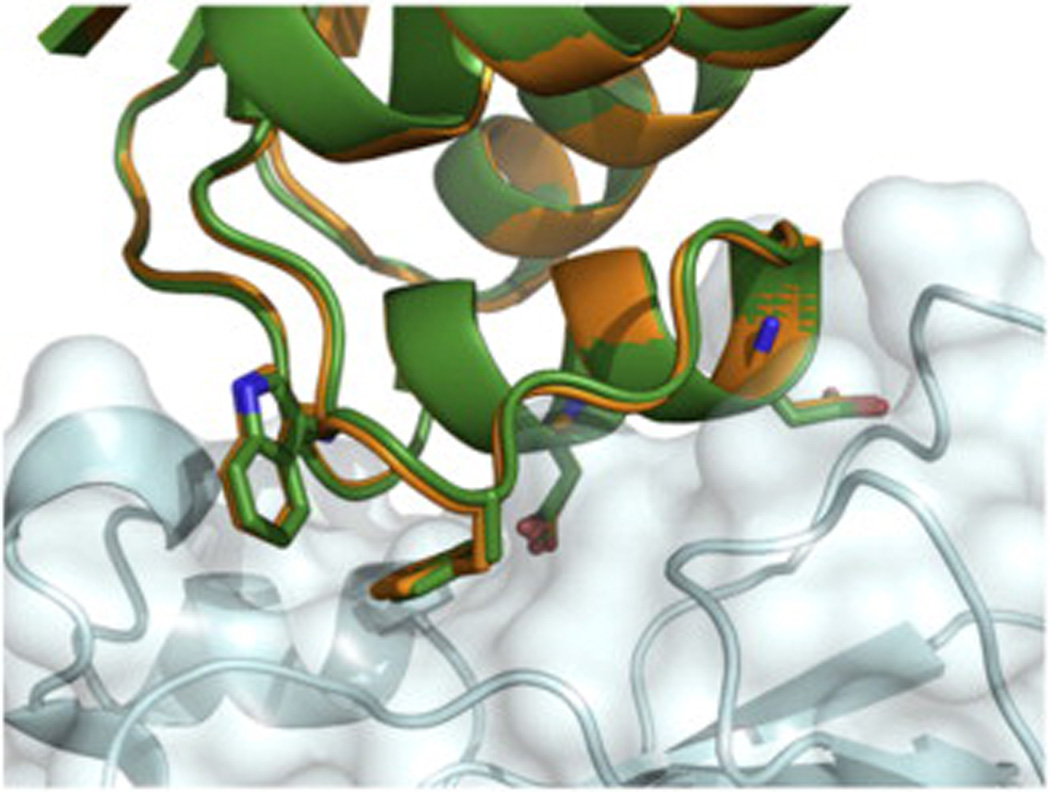
Redesigned barnase–barstar interaction closely recapitulates the native complex. The native interface is shown in green, and the redesign is shown in gold. Barnase is represented in pale cyan. Conserved residues at the interface are shown as sticks.
Redesign of nonnative scaffold proteins to incorporate hotspot-like interactions
To test the method's ability to design proteins that are not the natural partners of the target protein as binders, we conducted a cross-design experiment, where each target protein in the natural-interface recapitulation test was coupled to all nonnative binders in the set, and the same procedure as in the natural-interface recapitulation test was used. In this experiment, the constraints implied by the hotspot strategy almost completely preclude alternative scaffolds (Table 1, under “Alternative scaffolds”). By contrast, the Fc target, when designed with a combined set of diversified native and de novo hotspot residues, produces designs from all seven nonnative binders in the cross-docking experiment. The high permissibility of Fc observed here is likely due to the fact that the two hotspot residues we selected from protein A34 occupy positions that are separated by a single helical turn—a structural feature that is quite common on exposed protein surfaces. The lower permissibility of the majority of the scaffolds can be overcome by using many more scaffold proteins for design than the seven used here. Indeed, in the design of influenza hemagglutinin binders using a total of 865 nonredundant scaffolds, multiple plausible designs were obtained despite the conformationally restrictive nature of the target site.25
Figure 6 shows two examples in which scaffold proteins other than barstar were subjected to this protocol to generate designed barnase binders. A redesigned protein A shows a solution where a helix that harbors the hotspot Asp residues in a way that highly resembles the interaction between barnase and barstar was identified (Fig. 6a). By contrast, a redesigned colicin E9 incorporates the hotspot residues derived from barstar on a very different backbone (Fig. 6b), with the hotspot residues nevertheless matching those seen in barstar very closely. This solution is reminiscent of structural mimicry (Fig. 1d). In both redesigned scaffolds, the procedure finds opportunities to form favorable interactions across the interface that are different from those observed in the barnase–barstar complex.
Fig. 6.
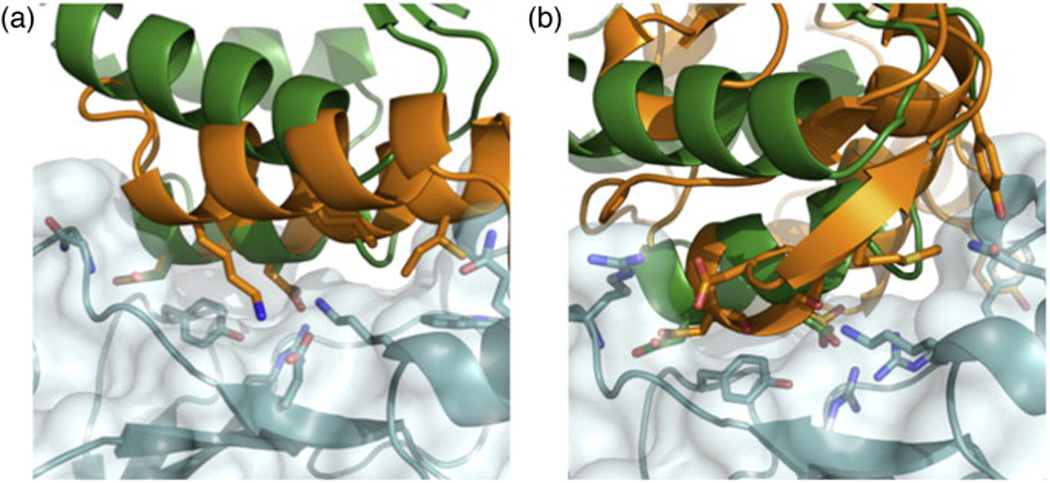
Redesigned proteins that mimic the hotspot barnase–barstar interaction and form additional interactions across the interface. Barnase is shown in pale cyan, and the native barstar is shown in green. The redesigned scaffold protein is in gold. (a) Redesigned protein A. (b) Redesigned colicin E9. The hotspot aspartates and the additional interactions between the designed proteins and barnase are shown in sticks.
Hotspot residue placement on loops generates antibody-like interactions
So far, we have discussed only scenarios where the modeled scaffold protein's backbone was minimally perturbed. By contrast, interactions involving antibodies clearly demonstrate the potential utility of sequence changes in loops. Our approach is easily adapted to handle a scenario where an initial interaction has been structurally characterized and where further interactions involving a loop segment are desired (e.g., for added affinity or specificity). In such a case, the rigid-body orientation between the two partners can be kept fixed, and the hotspot residues placed on completely remodeled loops of variable length. We developed a protocol (see Supplemental Material) in which the rigid-body orientation of the scaffold with respect to the target protein was held fixed, and residues were either inserted or deleted from a specified loop. The loop was then modeled from scratch using kinematic loop closure as implemented in the Rosetta software suite48 in the presence of strong hotspot-based restraints [Eqs. (1) and (2)] and minimized. A similar approach has been used to computationally reengineer enzyme specificity.49 Other loop-modeling strategies can be used in place of kinematic loop closure.
As an example of the potential usefulness of this strategy, we remodeled a loop on an antibody that interacts with lysozyme via Tyr and Arg residues. As in the approach described above, the Tyr and Arg hotspot residues were first docked against the lysozyme surface in the vicinity of the antibody hotspot residues and were diversified to include all energetically compatible rotamers for each residue. The hotspot-restraint-guided loop protocol was used to remodel the backbone by inserting one residue, two residues, or three residues or by deleting one residue followed by simultaneous placement of Tyr and Arg hotspot residues. (The protocol is available in Supplemental Data.) Only one solution comprising a two-residue insertion compared to the original antibody's sequence was identified (Fig. 7). In this solution, the functional groups of the hotspot tyrosine and arginine residues closely aligned with those of the original antibody's even though their backbones were very different: whereas the original pair of residues are sequential, in the design, they are separated by an insertion.
Fig. 7.
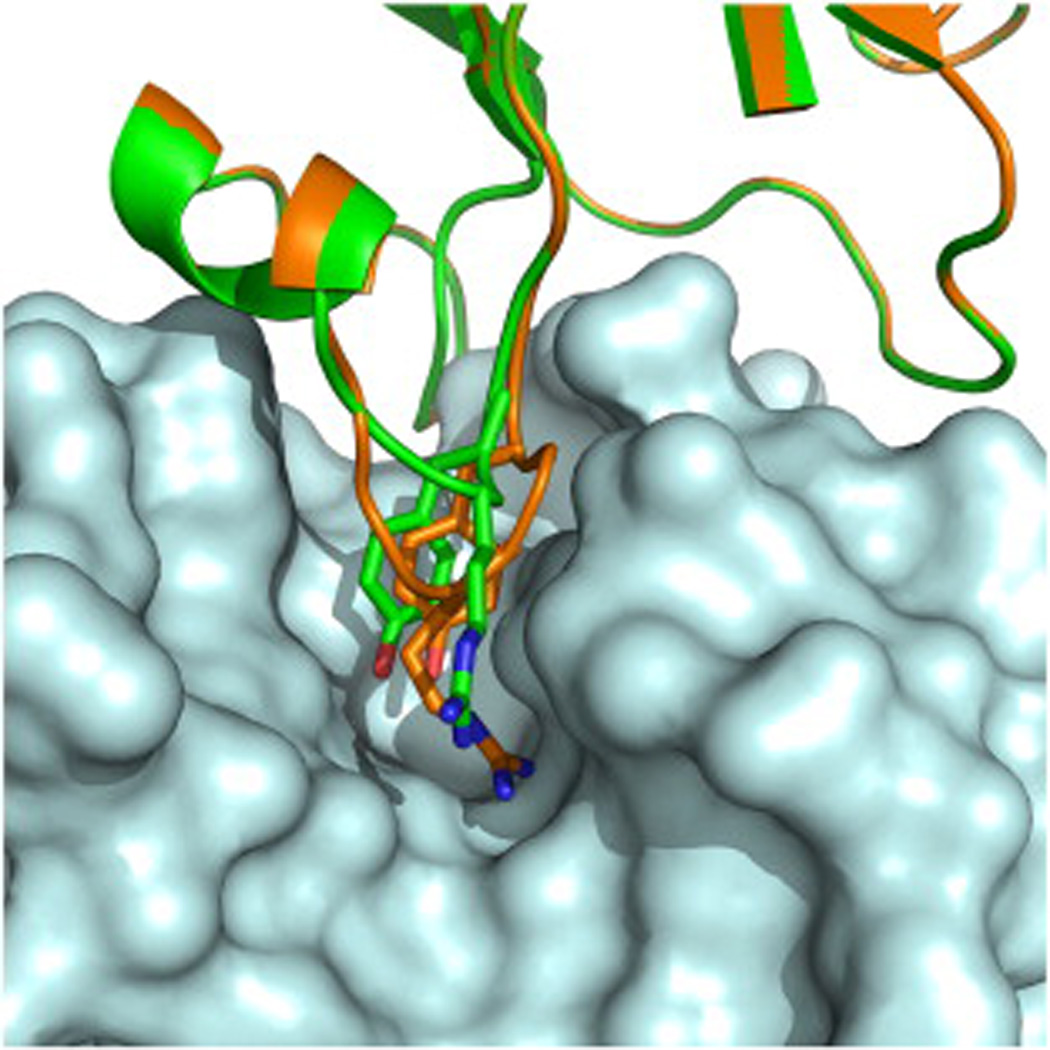
Complete redesign of antibody-like loop interactions with a target surface. Lysozyme (pale cyan) interacts with an antibody (green). Hotspot residues on the antibody are shown in sticks. Following complete redesign of a loop, a two-residue insertion was identified in the antibody loop that encompasses the two hotspot residues, which places the functional groups of the hotspot residues on the redesigned antibody (gold) in close proximity to their natural counterparts.
Hotspot-centric design yields native-like side-chain conformational probabilities
We observed previously that residues that make substantial contributions to binding affinity in natural interfaces are conformationally restricted in the unbound state, disfavoring conformations that would be incompatible with binding.20 Residues that were computed to contribute substantially to binding energy were found to form dense interaction networks with their host monomers in natural protein interfaces. By contrast, proteins designed using a “traditional design strategy”, where binders were selected based mostly on computed binding energy, showed lower side-chain conformational probabilities in the unbound state than key side chains in natives. The designed binders' key side chains made more atomic interactions across the interface than natives but fewer stabilizing interactions with their host monomer. In fact, the contribution to binding energy from these designed residues was computed to be higher on average than those in natural hotspots. This finding suggested that hotspot residue stabilization in the unbound state is a negative design aspect disfavoring alternative conformations of natural hotspots that would be incompatible with the natural binding mode.
To test whether hotspot-centric design yields higher-probability conformations for interface side chains, we used a recently published benchmark comprising 87 protein complexes that were de novo designed using the hotspot-centric strategy (Fleishman et al., in press) and contrasted the Boltzmann conformational probability distributions in the unbound state with those observed in the docking benchmark comprising 120 natural protein complexes50, 51 (Fig. 8). The hotspot-centric de novo designed interfaces were computed and selected without considering the interface side chains' Boltz-mann conformational probabilities. We additionally compared these two distributions to the conformational side-chain probabilities obtained by a more traditional dock and design strategy, where the chief criterion for design selection was computed binding affinity (data taken from Ref. 20).
Fig. 8.
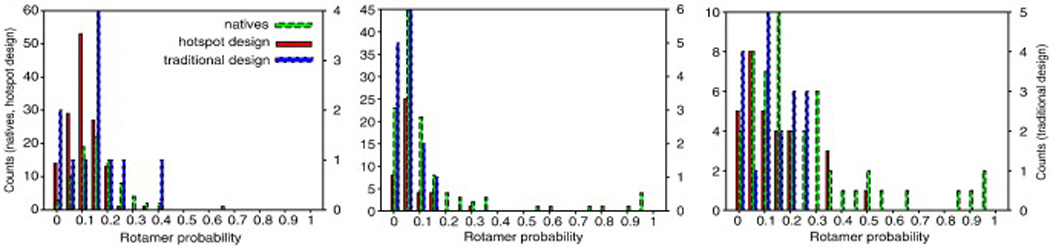
Comparison of side-chain conformational probabilities in natural and designed complexes. The side-chain conformational probabilities in the unbound state [Eq. (3)] were computed using the method in Ref. 20. Hotspot-centric design yields complexes with a comparable proportion of low-probability conformations (≤ 0.05 probability) to native complexes, whereas the “traditional designs” selected on the basis of binding affinity show proportionately more low-probability side-chain conformations. Both design strategies have fewer high-probability conformations (>0.50) than do natural interfaces, potentially explaining the low success rate in the protein binder design. The energetic consequences of differences in the intermediate probability range (0.1–0.5) are less significant than in the extremes of the probability. The native set contains the docking benchmark complexes (120 proteins);52 the hotspot designed set is taken from the protein-interface design benchmark (87 proteins) (Fleishman et al., in press), and the traditional designs are taken from Ref. 20.
Encouragingly, the proportion of very low probability conformations (<0.05 probability) in the hotspot-centric designs and natural sets is comparable and considerably less than that in designs selected solely based on binding energy. While there are differences in the distribution in the intermediate probability range (0.1–0.5), the energetic consequences of probability differences in this range are probably within the error of the method. A few side chains in natural interfaces have very high probability conformations (>0.50 probability), whereas none of the designs do. This difference may in part underlie the low success rate in protein-interface design25 (Fleishman et al., in press) and suggests that explicit modeling of side-chain conformational probabilities may be helpful in future methodological developments. The very high conformational probabilities in natural hotspots were found previously to be largely due to backbone and Cβ atoms in the host monomer eliminating a significant fraction of alternative side-chain conformations for residues making interactions across the interface. Hence, designing very high probability side-chain conformations may require explicit backbone remodeling strategies such as the one described above for loop remodeling (in most of the calculations described here, the backbone degrees of freedom determining the positioning of backbone and Cβ atoms were not varied to avoid introducing further sensitivity to errors in the energy function or insufficient sampling). Nevertheless, the reduced proportion of very low probability conformations is an encouraging sign that the hotspot-centric design strategy implicitly incorporates elements of negative design, disfavoring alternative side-chain conformations in the unbound state.
Discussion
We have described in detail a method to design protein binders of, in principle, any desired protein surface. The method centers on the observation that cores of natural protein interfaces have energetically highly optimized and spatially clustered residue interactions. Our approach can target surfaces for which crystal structures of natural binders are not known by using hotspot interactions found in de novo residue docking calculations. The key elements in the binder design procedure are to start with a configuration of the scaffold protein that shows high shape complementarity toward the target surface and then to sculpt it in a way that favors the incorporation of the precomputed hotspot residues. The resultant redesigned scaffolds incorporate the hotspot and form additional interactions with the target.
Molecular recognition demands very high precision. To achieve this precision, binders must favor the bound state as well as disfavor the many alternatives that are incompatible with binding the target and those that would associate with myriad off-target molecules. Thus, binders must employ elements of positive53 and negative22 design. Our method produces designs with side-chain conformational probabilities in the unbound state that are comparable to those of native complexes, thus disfavoring alternative, nonproductive conformations. This result is significant as it shows that the clustering of side chains on designed protein surfaces implicitly introduces elements of negative design, which are likely to be crucial to preclude off-target binding modes as recently observed.21 As explicit negative design is too computationally demanding for all but very limited design problems,23,24 similar approaches could make headway in still intractable biopolymer design problems such as flexible loop redesign and ligand binding.
Conclusions
Advances in gene synthesis and computing54 are yielding a continuing decrease in the cost and effort needed to assemble new genes and an increase in capabilities of sampling conformations and sequences in silico. New protein structures are added to public databases at a high rate, not least through the various Structural Genomics Initiatives.55 The hotspot-centric approach provides a means to leverage these technological developments for identifying many different native-like solutions to binding of a target surface. These will hopefully lead to efficient design of novel, high-affinity inhibitors, diagnostics, and laboratory molecular probes that target specific surfaces on proteins.
Methods
The design strategy follows that introduced in Ref. 25. To effectively generate hotspot-containing de novo binders of target epitopes, we divided the computational design procedure into three steps (Fig. 2): (1) hotspot-like interactions comprising two or more disembodied residues interacting with the target epitope were precomputed. The residues were clustered into separate libraries, in each of which the residues constitute a spatially distinct interaction site. (2) Hotspot residue libraries then bias the conformational sampling of scaffold proteins, and attempts are made to incorporate hotspot residues from the libraries in each scaffold. (3) RosettaDesign31 was used to design interfacial residues on the scaffold protein outside the hotspot with the aim of enhancing affinity.
De novo hotspots
To explore the effects of diversification of hotspots on protein design, we developed a de novo search that identifies hotspot-like interactions with the target epitope. The results of the search can be used if it is necessary to identify many more potential hotspot-like interactions with the target site than existing crystal structures provide. Our approach is to treat the “disembodied” amino acid of a potential hotspot residue as a ligand, searching for favorable interactions by exhaustively docking the side chain against the target protein. Resulting conformations are filtered on the basis of binding energy, and a small percentage is used as candidates in a hotspot residue library. Thus, a “cloud” of residues interacting with the target protein is narrowed down to a small number of very favorable interactions. Ben-Shimon and Eisenstein28 have recently described a similar strategy for mapping residue interactions with a protein target, using a pre-calculated surface representation to identify pockets that might accommodate a hotspot.
The search for hotspot interactions is carried out using residue identities typical of a hotspot (e.g., Trp, Tyr, and Arg).11 However, specific knowledge on a target may indicate the use of other residue identities. For recapitulation of a native interaction, the identity of the experimentally determined hotspot residue was used, together with conservative substitutions. The force field used for a hotspot search up-weights the contribution of hydrogen bonds and ignores the environmental dependence of the hydrogen-bonding energy, attempting to approximate the burial of the hotspot upon its incorporation on a scaffold protein. Hotspot searches can also be constrained to a particular epitope or allowed to explore the entire target surface, depending on the amount of prior knowledge available. Here, the hotspot search was constrained to within 25 Å of the center of the native interface. Of 10,000 search trajectories with computed binding energy of less than −1.0 Rosetta energy units (R.e.u.), only the top 1% per residue identity was used to construct hotspot libraries (Fig. 3a).
Native-complex-based hotspot libraries
A diverse hotspot residue library based on a known protein–protein interaction
Hotspot residues in a natural complex can be identified through experimental13 or computational alanine-scanning mutagenesis.56,57 These hotspot residues are then excised from the protein binder and diversified by two means: (1) they are docked against the target epitope using RosettaDock26 to generate a diversity of hotspot residues (Fig. 3d). (2) Starting froman atomat the root of the functional group (the Cγ of aromatic residues, the Cδ of glutamine, etc.), we construct inverse rotamers. A “fan” of residue backbones that maintain the position of the amino acid's functional group vis-à-vis the target protein is thus generated (Fig. 3c).
For each target, multiple libraries of spatially distinct hotspot residues are used. For example, one library may contain Tyr and Trp side chains that interact favorably with the target, while a second library may contain hydrophobic amino acids that engage in favorable interactions with the target protein. As clustering of hotspot residues is often observed in natural interfaces and could be a source of the reduced plasticity of binding sites in natural proteins,20 docking of hotspot residues could be performed in the presence of other hotspot residues to ensure favorable interaction energies between them.
Low-resolution docking of scaffold proteins against the target epitope
To obtain high-shape-complementary configurations of the scaffold protein with respect to the target epitope, we employ the PatchDock feature-matching algorithm.30 Constraints are used to prune conformations of each scaffold protein that do not interact with the target epitope. The surviving conformations are clustered at 4-Å RMSD. PatchDock was run with default parameters.
Backbone restraints guide low-resolution docking of scaffold proteins to configurations that favor hotspot residue placement
The hotspot residue libraries are used to identify configurations of the scaffold protein with respect to the target that may accommodate the placement of these hotspot residues. Each hotspot residue computed in the library implies an approximate location for a position on the scaffold protein and an orientation for the Cα−Cβ and the C–N vectors. Using Fig. 4a as a guide, we formulate scoring restraints to bias conformational sampling to configurations that would favor the placement of the hotspot residues. For each hotspot residue h and each scaffold position i, we formulate scoring restraints to bias conformational sampling to configurations that would favor the placement of the hotspot residues:
| (1) |
where ΔGh is the computed binding energy for hotspot residue h, is always negative, and was chosen to be −3 in all design trajectories; β, α, C, and N are the coordinates of the Cβ, Cα, C, and N atoms; k (the spring constant) is arbitrarily set to 0.5; min is the minimum function ensuring that the restraint is negative or zero; the quantities within the square brackets are the dot products of the relevant vectors; and is a normalization constant.
This form of the restraint function reaches a minimum when the distance between the Cβ of the hotspot residue and a position on the scaffold is 0 and when the Cα–Cβ and C–N vectors are matched. Thus, a given restraint is best satisfied when a potential grafting position on the scaffold is perfectly aligned with a precomputed hotspot residue. If the orientation of either of the two vectors of position i with respect to hotspot h is more than 90°, then is set to 0.
A library of n hotspot residues thus implies n restraints. Each residue i is then assigned the smallest of these n restraints:
| (2) |
Eq. (2) then assigns the minimal restraint to each amino acid position on the scaffold, so that each scaffold position is affected only by the most appropriate hotspot restraint at any given time during conformational search.
Since only the locations of the Cβ and the backbone atoms are required in evaluating , the restraints can be computed efficiently during low-resolution Monte-Carlo-based docking of the scaffold protein with respect to the target. Importantly, the restraints can be used during minimization as Eq. (1) is readily differentiable.
Hotspot residue placement
Following the identification of a configuration of the two partners that may be favorable for placing the hotspot residues contained in the library on the scaffold protein, we explicitly model hotspot residues onto the scaffold protein. We developed three methods for hotspot residue placement for use in different contexts. For all methods, we start with the configuration of the scaffold protein that was obtained from hotspot-residue-guided docking and minimization and with one of the hotspot residue libraries. With the exception of Gly, Pro, and disulfide-linked cysteines, interfacial residues on the scaffold protein within 10 Å from the target protein are reduced to alanine to increase the chances of accommodating the hotspot residues.
Placement of the scaffold onto an idealized hotspot residue
The residues within the hotspot residue libraries define configurations that are optimal for realizing the hotspot interaction. Here, for a given interfacial scaffold position, we iterate over each of the nearby hotspot residues in the library and rotate and translate the scaffold protein so as to align it perfectly with the rotamer of the hotspot residue. Scaffold positions, for which the Cβ atoms are farther than 4.0 Å from the relevant hotspot residue or whose C–N or Cα–Cβ vectors are misaligned with the hotspot residues by more than 60°, are triaged to avoid compromising the initial high-shape-complementarity configuration of the two partners (see Fig. 4a for an illustration of these vectors). We then minimize the rigid-body orientation and the side-chain degrees of freedom of the placed hotspot residue in a reduced force field that only considers the punitive energy terms for van der Waals clashes and rotameric energies. If the energy of the placed hotspot residue is higher than 1.0 R.e.u., we discard this placement.
Placement of a hotspot residue onto a scaffold position
Here, for each interfacial scaffold position, we minimize the configuration of the scaffold protein with respect to the target in the context of a single restraint [Eq. (1)] derived from the hotspot residue. All other parameters and cutoffs are as in the previous section.
Simultaneous placement of multiple hotspot residues
For each hotspot residue library, we identify a position on the scaffold protein that produces themost favorable restraint score as defined by Eq. (1) compared to the remainder of the hotspot residue libraries. Each such scaffold position is then coupled to the appropriate hotspot residue library. If not all hotspot residue libraries are matched to different scaffold positions, the configuration of the scaffold with respect to the target is discarded. Upon success, we simultaneously redesign the identities of the relevant scaffold positions to those amino acid identities contained in their matched hotspot residue libraries. Since only a handful of positions are designed in this scheme and since the identities of the designed residues are limited based on the relevant hotspot residue library, adding off-rotameric conformations into the design step is computationally affordable.
Redesign of residues outside of the hotspot
Following the successful placement of residues from all hotspot residue libraries, a shell of scaffold positions that are at most 10 Å from the target protein is redesigned using RosettaDesign,31 while the target protein side chains are allowed to repack. Gly, Pro, and disulfide-linked cysteines are left as in the wild-type sequence. Minimization of backbone, rigid-body, and side-chain degrees of freedom at the interface is interspersed between various design schemes that ramp up and down the van der Waals clash and rotameric strain penalties. These minimization and design steps are useful in obtaining higher sequence diversity in design. The last design step uses a force field with high weights on the punitive energy terms, such as steric clashes and rotameric strain to ensure that the designed residues do not assume high-energy conformations.
During these design simulations, the side chains of the grafted hotspot residues are biased toward the coordinates of the idealized hotspot residues as present in the hotspot residue library (similar to the implementation in Ref. 19). This bias is implemented as harmonic coordinate restraints on, typically, three atoms that define the functional group of the side chain, in effect pulling the placed hotspot residue's functional group toward its idealized position with respect to the target protein. For example, these atoms would be the three carbon atoms at the root of an aromatic ring or the three polar atoms in the side chains of Gln, Asn, Glu, and Asp residues. To ensure that the placed residues are stable in their position on the scaffold, we gradually remove all restraints during the simulation, and carry out the last packing and minimization step in the absence of restraints.
Each resulting model is automatically filtered according to computed binding energy32 and shape complementarity.7 In this study, we do not carry out any additional manual filtering.
Binding-energy calculations
In keeping with Ref. 32, the binding energy is defined as the difference between the total system energy in the bound and unbound states. In each state, interface residues are allowed to repack. For numerical stability, binding-energy calculations were repeated three times, and the average was taken.
Boltzmann conformational probabilities of interface side chains
For comparison with published results, we used the same method, parameters, and reference set of natural complexes as in Ref. 20. For each complex, the method first separated the partners, and for each residue that makes an appreciable contribution to binding (binding energy increases by more than 1.5 R.e.u. upon mutation to alanine), it iterated over all of its rotameric states as defined in the Dunbrack library of backbone-dependent rotamers,58 excluding rotamers that are predicted to form steric clashes with protein main chain or Cβ atoms. For each rotamer placement, all residues within a 6-Å shell were repacked and minimized. The energy E of each such state was then evaluated using the Rosetta all-atom energy function,59 which is dominated by van der Waals, hydrogen bonding, and solvation terms. The probability of the conformation of residue i, pi, was then computed assuming a Boltzmann distribution:
| (3) |
where s is the rotameric state, kB is the Boltzmann constant, and T is the absolute temperature. kBT was set to 0.8 R.e.u. in all simulations. Ei is the energy of the unbound state.
Shape complementarity
Shape complementarity was computed using the CCP4 v6.02 sc program.60
Source code availability
The methods have been implemented within the Rosetta macromolecular modeling software suite and are available through the Rosetta Commons agreement†. All of the methods have been implemented through Rosetta-Scripts, and all scripts are available as Supplemental Data.
Supplementary Material
Acknowledgements
We thank Ingemar Andre for many insights and Rickard Hedman for help with computations of lysozyme and its associated antibody. We thank Dina Schneidman-Duhovny and Haim Wolfson for suggestions and for providing source code for manipulating PatchDock results. Computations were carried out on computational resources generously provided by participants in Rosetta@Home and the Argonne Leadership Computing Facility. We thank Darwin O. V. Alonso and Keith E. Leidig for their support and maintenance of the computational infrastructure in the Baker laboratory. S.J.F. was supported by a long-term fellowship from the Human Frontier Science Program. J.E.C. was supported by the Jane Coffin Childs Memorial Fund. E.-M.S. was supported by a career development award from the Northwest Regional Center of Excellence National Institutes of Health/National Institute of Allergy and Infectious Diseases AI057141. This research was supported by the Protein Design Processes grant from the Defense Advanced Research Program Agency, the Defense Threat Reduction Agency, the National Institutes of Health Yeast Resource Center, and the Howard Hughes Medical Institute.
Footnotes
Supplementary Data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.jmb.2011.09.001
References
- 1.Koide A, Koide S. Monobodies: antibody mimics based on the scaffold of the fibronectin type III domain. Methods Mol. Biol. 2007;352:95–109. doi: 10.1385/1-59745-187-8:95. [DOI] [PubMed] [Google Scholar]
- 2.Binz HK, Amstutz P, Kohl A, Stumpp MT, Briand C, Forrer P, et al. High-affinity binders selected from designed ankyrin repeat protein libraries. Nat. Biotechnol. 2004;22:575–582. doi: 10.1038/nbt962. [DOI] [PubMed] [Google Scholar]
- 3.Binz HK, Amstutz P, Pluckthun A. Engineering novel binding proteins from nonimmunoglobulin domains. Nat. Biotechnol. 2005;23:1257–1268. doi: 10.1038/nbt1127. [DOI] [PubMed] [Google Scholar]
- 4.Binz HK, Pluckthun A. Engineered proteins as specific binding reagents. Curr. Opin. Biotechnol. 2005;16:459–469. doi: 10.1016/j.copbio.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 5.Binz HK, Stumpp MT, Forrer P, Amstutz P, Pluckthun A. Designing repeat proteins: well-expressed, soluble and stable proteins from combinatorial libraries of consensus ankyrin repeat proteins. J. Mol. Biol. 2003;332:489–503. doi: 10.1016/s0022-2836(03)00896-9. [DOI] [PubMed] [Google Scholar]
- 6.Lo Conte L, Chothia C, Janin J. The atomic structure of protein–protein recognition sites. J. Mol. Biol. 1999;285:2177–2198. doi: 10.1006/jmbi.1998.2439. [DOI] [PubMed] [Google Scholar]
- 7.Lawrence MC, Colman PM. Shape complementarity at protein/protein interfaces. J. Mol. Biol. 1993;234:946–950. doi: 10.1006/jmbi.1993.1648. [DOI] [PubMed] [Google Scholar]
- 8.Nassar N, Hoffman GR, Manor D, Clardy JC, Cerione RA. Structures of Cdc42 bound to the active and catalytically compromised forms of Cdc42GAP. Nat. Struct. Biol. 1998;5:1047–1052. doi: 10.1038/4156. [DOI] [PubMed] [Google Scholar]
- 9.Stebbins CE, Galan JE. Modulation of host signaling by a bacterial mimic: structure of the Salmonella effector SptP bound to Rac1. Mol. Cell. 2000;6:1449–1460. doi: 10.1016/s1097-2765(00)00141-6. [DOI] [PubMed] [Google Scholar]
- 10.DeLano WL. The PyMOL Molecular Graphics Systems. Palo Alto, CA: DeLano Scientific; 2002. [Google Scholar]
- 11.Bogan AA, Thorn KS. Anatomy of hot spots in protein interfaces. J. Mol. Biol. 1998;280:1–9. doi: 10.1006/jmbi.1998.1843. [DOI] [PubMed] [Google Scholar]
- 12.Clackson T, Wells JA. A hot spot of binding energy in a hormone–receptor interface. Science. 1995;267:383–386. doi: 10.1126/science.7529940. [DOI] [PubMed] [Google Scholar]
- 13.Clackson T, Ultsch MH, Wells JA, de Vos AM. Structural and functional analysis of the 1:1 growth hormone:receptor complex reveals the molecular basis for receptor affinity. J. Mol. Biol. 1998;277:1111–1128. doi: 10.1006/jmbi.1998.1669. [DOI] [PubMed] [Google Scholar]
- 14.Ofran Y, Rost B. Protein–protein interaction hotspots carved into sequences. PLoS Comput. Biol. 2007;3:e119. doi: 10.1371/journal.pcbi.0030119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu Z, Ma B, Wolfson H, Nussinov R. Conservation of polar residues as hot spots at protein interfaces. Proteins. 2000;39:331–342. [PubMed] [Google Scholar]
- 16.Ma B, Elkayam T, Wolfson H, Nussinov R. Protein–protein interactions: structurally conserved residues distinguish between binding sites and exposed protein surfaces. Proc. Natl Acad. Sci. USA. 2003;100:5772–5777. doi: 10.1073/pnas.1030237100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stebbins CE, Galan JE. Structural mimicry in bacterial virulence. Nature. 2001;412:701–705. doi: 10.1038/35089000. [DOI] [PubMed] [Google Scholar]
- 18.Liu S, Zhu X, Liang H, Cao A, Chang Z, Lai L. Nonnatural protein–protein interaction-pair design by key residues grafting. Proc. Natl Acad. Sci. USA. 2007;104:5330–5335. doi: 10.1073/pnas.0606198104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Havranek JJ, Baker D. Motif-directed flexible backbone design of functional interactions. Protein Sci. 2009;18:1293–1305. doi: 10.1002/pro.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fleishman SJ, Khare SD, Koga N, Baker D. Restricted sidechain plasticity in the structures of native proteins and complexes. Protein Sci. 2011;20:753–757. doi: 10.1002/pro.604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karanicolas J, Corn JE, Chen I, Joachimiak LA, Dym O, Peck SH, et al. A de novo protein binding pair by computational design and directed evolution. Mol. Cell. 2011;42:250–260. doi: 10.1016/j.molcel.2011.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Richardson JS, Richardson DC, Tweedy NB, Gernert KM, Quinn TP, Hecht MH, et al. Looking at proteins: representations, folding, packing, and design. Biophysical Society National Lecture, 1992. Biophys. J. 1992;63:1185–1209. [PMC free article] [PubMed] [Google Scholar]
- 23.Havranek JJ, Harbury PB. Automated design of specificity in molecular recognition. Nat. Struct. Biol. 2003;10:45–52. doi: 10.1038/nsb877. [DOI] [PubMed] [Google Scholar]
- 24.Grigoryan G, Reinke AW, Keating AE. Design of protein-interaction specificity gives selective bZIP-binding peptides. Nature. 2009;458:859–864. doi: 10.1038/nature07885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fleishman SJ, Whitehead TA, Ekiert DC, Dreyfus C, Corn JE, Strauch EM, et al. Computational design of proteins targeting the conserved stem region of influenza hemagglutinin. Science. 2011;332:816–821. doi: 10.1126/science.1202617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gray JJ, Moughon S, Wang C, Schueler-Furman O, Kuhlman B, Rohl CA, Baker D. Protein–protein docking with simultaneous optimization of rigid-body displacement and side-chain conformations. J. Mol. Biol. 2003;331:281–299. doi: 10.1016/s0022-2836(03)00670-3. [DOI] [PubMed] [Google Scholar]
- 27.Fleishman SJ, Leaver-Fay A, Corn JE, Strauch EM, Khare SD, Koga N, et al. Rosetta-Scripts: a scripting language interface to the Rosetta macromolecular modeling suite. PLoS ONE. 2011 doi: 10.1371/journal.pone.0020161. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ben-Shimon A, Eisenstein M. Computational mapping of anchoring spots on protein surfaces. J. Mol. Biol. 2010;402:259–277. doi: 10.1016/j.jmb.2010.07.021. [DOI] [PubMed] [Google Scholar]
- 29.Kuhlmann UC, Pommer AJ, Moore GR, James R, Kleanthous C. Specificity in protein–protein interactions: the structural basis for dual recognition in endonuclease colicin-immunity protein complexes. J. Mol. Biol. 2000;301:1163–1178. doi: 10.1006/jmbi.2000.3945. [DOI] [PubMed] [Google Scholar]
- 30.Schneidman-Duhovny D, Inbar Y, Nussinov R, Wolfson HJ. PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res. 2005;33:W363–W367. doi: 10.1093/nar/gki481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kuhlman B, Dantas G, Ireton GC, Varani G, Stoddard BL, Baker D. Design of a novel globular protein fold with atomic-level accuracy. Science. 2003;302:1364–1368. doi: 10.1126/science.1089427. [DOI] [PubMed] [Google Scholar]
- 32.Kortemme T, Baker D. A simple physical model for binding energy hot spots in protein–protein complexes. Proc. Natl Acad. Sci. USA. 2002;99:14116–14121. doi: 10.1073/pnas.202485799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kellogg EH, Leaver-Fay A, Baker D. Role of conformational sampling in computing mutation-induced changes in protein structure and stability. Proteins. 2011;79:830–838. doi: 10.1002/prot.22921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Idusogie EE, Presta LG, Gazzano-Santoro H, Totpal K, Wong PY, Ultsch M, et al. Mapping of the C1q binding site on rituxan, a chimeric antibody with a human IgG1 Fc. J. Immunol. 2000;164:4178–4184. doi: 10.4049/jimmunol.164.8.4178. [DOI] [PubMed] [Google Scholar]
- 35.Stanfield RL, Dooley H, Flajnik MF, Wilson IA. Crystal structure of a shark single-domain antibody V region in complex with lysozyme. Science. 2004;305:1770–1773. doi: 10.1126/science.1101148. [DOI] [PubMed] [Google Scholar]
- 36.Buckle AM, Schreiber G, Fersht AR. Protein–protein recognition: crystal structural analysis of a barnase–barstar complex at 2.0-Å resolution. Biochemistry. 1994;33:8878–8889. doi: 10.1021/bi00196a004. [DOI] [PubMed] [Google Scholar]
- 37.Ekiert DC, Bhabha G, Elsliger MA, Friesen RH, Jongeneelen M, Throsby M, et al. Antibody recognition of a highly conserved influenza virus epitope. Science. 2009;324:246–251. doi: 10.1126/science.1171491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schreiber G, Fersht AR. Rapid, electro-statically assisted association of proteins. Nat. Struct. Biol. 1996;3:427–431. doi: 10.1038/nsb0596-427. [DOI] [PubMed] [Google Scholar]
- 39.Wallis R, Moore GR, James R, Kleanthous C. Protein–protein interactions in colicin E9 DNase–immunity protein complexes. 1. Diffusion-controlled association and femtomolar binding for the cognate complex. Biochemistry. 1995;34:13743–13750. doi: 10.1021/bi00042a004. [DOI] [PubMed] [Google Scholar]
- 40.Braisted AC, Wells JA. Minimizing a binding domain from protein A. Proc. Natl Acad. Sci. USA. 1996;93:5688–5692. doi: 10.1073/pnas.93.12.5688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zanghellini A, Jiang L, Wollacott AM, Cheng G, Meiler J, Althoff EA, et al. New algorithms and an in silico benchmark for computational enzyme design. Protein Sci. 2006;15:2785–2794. doi: 10.1110/ps.062353106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guharoy M, Chakrabarti P. Conservation and relative importance of residues across protein–protein interfaces. Proc. Natl Acad. Sci. USA. 2005;102:15447–15452. doi: 10.1073/pnas.0505425102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pal G, Kouadio JL, Artis DR, Kossiakoff AA, Sidhu SS. Comprehensive and quantitative mapping of energy landscapes for protein–protein interactions by rapid combinatorial scanning. J. Biol. Chem. 2006;281:22378–22385. doi: 10.1074/jbc.M603826200. [DOI] [PubMed] [Google Scholar]
- 44.Humphris EL, Kortemme T. Prediction of protein–protein interface sequence diversity using flexible backbone computational protein design. Structure. 2008;16:1777–1788. doi: 10.1016/j.str.2008.09.012. [DOI] [PubMed] [Google Scholar]
- 45.Jin L, Wells JA. Dissecting the energetics of an antibody–antigen interface by alanine shaving and molecular grafting. Protein Sci. 1994;3:2351–2357. doi: 10.1002/pro.5560031219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Castro MJ, Anderson S. Alanine point-mutations in the reactive region of bovine pancreatic trypsin inhibitor: effects on the kinetics and thermodynamics of binding to β-trypsin and α-chymotrypsin. Biochemistry. 1996;35:11435–11446. doi: 10.1021/bi960515w. [DOI] [PubMed] [Google Scholar]
- 47.Sharabi O, Dekel A, Shifman JM. Triathlon for energy functions: who is the winner for design of protein–protein interactions? Proteins. 2011;79:1487–1498. doi: 10.1002/prot.22977. [DOI] [PubMed] [Google Scholar]
- 48.Mandell DJ, Coutsias EA, Kortemme T. Sub-angstrom accuracy in protein loop reconstruction by robotics-inspired conformational sampling. Nat. Methods. 2009;6:551–552. doi: 10.1038/nmeth0809-551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Murphy PM, Bolduc JM, Gallaher JL, Stoddard BL, Baker D. Alteration of enzyme specificity by computational loop remodeling and design. Proc. Natl Acad. Sci. USA. 2009;106:9215–9220. doi: 10.1073/pnas.0811070106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pierce B, Weng Z. ZRANK: reranking protein docking predictions with an optimized energy function. Proteins. 2007;67:1078–1086. doi: 10.1002/prot.21373. [DOI] [PubMed] [Google Scholar]
- 51.Chen R, Li L, Weng Z. ZDOCK: an initial-stage protein-docking algorithm. Proteins. 2003;52:80–87. doi: 10.1002/prot.10389. [DOI] [PubMed] [Google Scholar]
- 52.Hwang H, Pierce B, Mintseris J, Janin J, Weng Z. Protein–protein docking benchmark version 3.0. Proteins. 2008;73:705–709. doi: 10.1002/prot.22106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bowie JU, Luthy R, Eisenberg D. A method to identify protein sequences that fold into a known three-dimensional structure. Science. 1991;253:164–170. doi: 10.1126/science.1853201. [DOI] [PubMed] [Google Scholar]
- 54.Moore GE. Lithography and the future of Moore's law. Proc. SPIE. 1995;2438 [Google Scholar]
- 55.Joachimiak A. High-throughput crystallography for structural genomics. Curr. Opin. Struct. Biol. 2009;19:573–584. doi: 10.1016/j.sbi.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kruger DM, Gohlke H. DrugScorePPI webserver: fast and accurate in silico alanine scanning for scoring protein–protein interactions. Nucleic Acids Res. 2010;38:W480–W486. doi: 10.1093/nar/gkq471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kortemme T, Kim DE, Baker D. Computational alanine scanning of protein–protein interfaces. Sci. STKE. 2004;2004:pl2. doi: 10.1126/stke.2192004pl2. [DOI] [PubMed] [Google Scholar]
- 58.Dunbrack RL, Jr, Karplus M. Conformational analysis of the backbone-dependent rotamer preferences of protein sidechains. Nat. Struct. Biol. 1994;1:334–340. doi: 10.1038/nsb0594-334. [DOI] [PubMed] [Google Scholar]
- 59.Das R, Baker D. Macromolecular modeling with Rosetta. Annu. Rev. Biochem. 2008;77:363–382. doi: 10.1146/annurev.biochem.77.062906.171838. [DOI] [PubMed] [Google Scholar]
- 60.Collaborative Computational Project, Number 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.