

Abstract
To examine whether intestinal helminth infection may be a risk factor for enteric bacterial infection, a murine model was established using the intestinal helminth Heligomosomoides polygyrus and a murine pathogen Citrobacter rodentium, which causes infectious colitis. Using this model we recently have shown that coinfection with the Th2-inducing H. polygyrus and C. rodentium promotes bacterial-associated disease and colitis. In this study, we expand our previous observations and examine the hypothesis that dendritic cells (DC) stimulated by helminth infection may play an important role in the regulation of the intestinal immune response to concurrent C. rodentium infection as well as in the modulation of the bacterial pathogenesis. We show that H. polygyrus infection induces DC activation and IL-10 expression, and that adoptive transfer of parasite-primed DC significantly impairs host protection to C. rodentium infection, resulting in an enhanced bacterial infection and in the development of a more severe colonic injury. Furthermore, we demonstrate that adoptive transfer of parasite-primed IL-10-deficient DCs fails to result in the development of a significantly enhanced C. rodentium-mediated colitis. Similarly, when the DC IL-10 response was neutralized by anti-IL-10 mAb treatment in mice that received parasite-primed DC, no deleterious effect of the parasite-primed DC on the host intestinal response to C. rodentium was detected. Thus, our results provide evidence to indicate that the H. polygyrus-dependent modulation of the host response to concurrent C. rodentium infection involves IL-10-producing DCs.
Human helminth infections are estimated to infect 3 billion people worldwide (1, 2). The major importance of helminth infections includes not only the direct pathogenic effect of the worms, but also the modulatory role of the parasite on the host immune system, which may alter the response to other Ags and cause additional immunopathology. In many developing countries, exposure to helminth infections and simultaneous infection with other pathogens, such as enteric bacteria, is quite common. Recent evidence indicates that in the developed world, a complete absence of helminth infection may be a predisposing factor for the development of certain immune-mediated disorders (3). Thus, there is a growing interest in understanding how an existing helminth infection affects the capacity of the host to develop an appropriate immune response to other concomitant infections or Ags. A cross-regulatory suppression of Th1 responses by a helminth-driven strong Th2 response has been suggested as a contributing factor to the alteration of the host response to concurrent bacterial infections. Indeed, multiple studies (4–7) have revealed that helminth-induced Th2 responses can attenuate damaging Th1-driven inflammatory responses in the host.
Recently, we established a murine model to begin to examine the mechanism underlying the immune regulation that occurs when an intestinal helminth infection is coupled with a bacterial insult (8). The framework of this model is based on using two murine infectious agents that induce distinct Th responses: 1) the parasitic helminth Heligomosomoides polygyrus, which provokes a strong Th2 response; and 2) Citrobacter rodentium, which selectively stimulates a Th1-type immune response (9, 10). C. rodentium is a murine bacterial pathogen that shares important functional and structural similarities with enteropathogenic Escherichia coli (EPEC)3 isolates (11). C. rodentium produces attaching and effacing lesions in the colon indistinguishable from those caused by clinical EPEC strains, and the gene coding for the outer membrane protein responsible for intimate attachment, intimin, is functionally homologous in C. rodentium and clinical EPEC strains. Infection of mice with C. rodentium causes crypt hyperplasia, loss of goblet cells, and mucosal infiltration with macrophages, lymphocytes, and neutrophils. Thus, this organism is ideally suited to study the pathogenesis of EPEC and to explore host-enteric bacterial pathogen interactions. Our initial observations suggested that mice coinfected with C. rodentium and the intestinal nematode, H. polygyrus, displayed a marked increase in morbidity and mortality compared with mice that were infected with C. rodentium alone (8). The enhanced susceptibility and severe intestinal injury in coinfected mice were found to be associated with helminth-induced Th2 responses and correlated with a dysregulated proinflammatory Th1 response that resulted in an increase in bacterial burden (8). These results, therefore, suggest a role for the helminth-induced Th2 response in modulating the immunopathology of a concurrent bacterial infection.
To effectively control infection, appropriately developed and functionally polarized subsets of Th effector cells are required. Dendritic cells (DCs) are professional APCs that stimulate primary immune responses by inducing the activation and differentiation of Th cells. In addition to presenting Ag peptides to T cells via the MHC class II complex, DCs provide other signals required for differentiation of Th cells into Th1 and Th2 cells (12–14). Although the differentiation of naive CD4+ T cells into Th effector cells has been suggested to be regulated by several factors, including the type of APCs, Ag dose, and host genetics, it is the cytokine milieu present at the time of priming that has been implicated as the major determining factor (15). Microorganisms, such as bacteria, viruses, and protozoa parasites, can trigger the production of IL-12, IL-18, and IL-27 from APCs, which directly primes CD4+ T cells to express a Th1 phenotype (16–23). Furthermore, DCs pulsed with helminth parasite Ag, such as Schistosoma mansoni egg Ag, the schistosome glycan lacto-N-fucopentaose III, or Nip-postrongylus brasiliensis excretory/secretory Ag, prime mice for a Th2 response (24–26).
We provide evidence to indicate that intestinal helminth infection can induce DC activation and IL-10 production, and we demonstrate that adoptive transfer of parasite-primed DCs results in the impairment of host protection against C. rodentium with an enhancement of C. rodentium-mediated intestinal injury. The deleterious effects of the parasite-primed DCs on host protection against the bacterial infection and C. rodentium-mediated intestinal inflammatory response could be eliminated or significantly reduced when DC IL-10 was neutralized in mice that received parasite-primed DCs or in mice that received parasite-primed IL-10-deficient DCs. These results point to a unique role of parasite-primed DCs in the intestinal immune system in regulating the mucosal immune response to concurrent enteric bacterial infection and demonstrate that DC IL-10 response is required for parasite-primed DCs to exert their immune modulatory effects on the host response to enteric bacterial infection. These data, therefore, demonstrate that an enteric helminth infection not only can alter host immune response to concurrent enteric bacterial infection but also can promote a bacteria-induced intestinal inflammation through a novel mechanism that requires DC priming and an IL-10 response.
Materials and Methods
Mice
Six- to 8-wk-old female BALB/c ByJ and IL-10-deficient BALB/c mice were purchased from The Jackson Laboratory and were fed autoclaved food and water and maintained in a specific pathogen-free facility at Massachusetts General Hospital (Charlestown, MA).
H. polygyrus infection and parasite Ag preparation
H. polygyrus were propagated as previously described and stored at 4°C until use (27, 28). Mice were inoculated orally with 200 third-stage larvae. Seven days following parasitic infection, a subset of the H. polygyrus-infected mice were fed C. rodentium.
C. rodentium infection and Ag preparation
Mice were orally inoculated with C. rodentium (strain DBS100; American Type Culture Collection). Bacteria were grown overnight in Luria broth and resuspended in PBS before infecting the mice (0.5 ml/mouse, ~5 × 108 CFU of C. rodentium). To assess the clearance of C. rodentium, fecal pellets were collected from each mouse weekly during the course of the experiment. Fecal pellets were weighed, homogenized, serially diluted, and plated onto selective MacConkey agar. After overnight incubation at 37°C, bacterial colonies were counted as described by Vallance et al. (29). C. rodentium Ag was prepared by collecting an overnight culture of C. rodentium in Luria broth. The bacterial culture was washed with PBS (three times) and sonicated on ice. The homogenate was then centrifuged (14,000 rpm) at 4°C for 30 min. Supernatants were then collected and protein concentration determined. Aliquots were stored at −20°C until use.
Lymphocyte isolation
Mice were sacrificed 2 wk post-C. rodentium infection. Lymphocyte suspensions were prepared from the mesenteric lymph nodes (MLN) by pressing the cells through a 70-μM nylon cell strainer (Falcon; BD Labware) in complete DMEM (contains 10% FCS, 10 mM HEPES, 2 mM L-glutamine, 100 U penicillin/ml, 100 μg of streptomycin/ml, 50 μM 2-ME, 0.1 mM nonessential amino acids, and 1 mM sodium pyruvate; Invitrogen Life Technologies). Cells (5 × 106 cells/ml) were cultured in 24-well plates in the presence or absence of C. rodentium Ag (50 μg/ml), and culture supernatants were collected 72 h later and stored at −20°C until assayed for cytokine production.
Measurement of MLN cytokine production
Th1 (IFN-γ) and Th2 (IL-4) cytokines were assayed using ELISA as previously described (8). ELISA capture (BVD4-1D11, IL-4; R4-6A2, IFN-γ) and biotinylated second Abs (BVD6-24G2, IL-4; XMG1.2, IFN-γ) were purchased from BD Pharmingen. Standard curves were obtained using recombinant murine IFN-γ and IL-4 (Genzyme).
DC isolation and adoptive transfer
Spleens and MLN from H. polygyrus-infected mice (7 days post-H. polygyrus inoculation) and noninfected mice (BALB/c and IL-10-deficient BALB/c) were collected aseptically into complete DMEM as previously discussed. The tissues were digested with collagenase (type IV; Worthington Biochemical) at 200 U/ml for 45 min at room temperature. The low density fraction of the cell suspension was obtained by gradient centrifugation in an OptiPrep gradient (Invitrogen Life Technologies). DCs were purified by positive selection over a magnetic cell-sorting column (MACS) using microbead-conjugated anti-CD11c mAb (Miltenyi Biotec). Highly purified CD11c+ DCs (98.5 ± 1.5%, determined using FACS) were transferred into recipient mice (2–3 × 106 cells per mouse) via tail vein injection. DC recipients were infected with C. rodentium (5 × 108 CFU of C. rodentium) 24 h after DC transfer. In a subset of adoptive transfer experiments, DCs were prelabeled with the fluorescent dye CFSE (Molecular Probes) as previously described (30). Briefly, purified DCs were resuspended at 107 cells/ml in PBS containing 0.1% BSA with 10 μM CFSE for 10 min at 37°C. Similarly, the labeled DCs were washed and transferred (via tail vein injection) to BALB/c recipients (2–3 × 106 cells per mouse).
To examine the contribution of IL-10-secreting DCs primed by H. polygyrus to the altered host response to C. rodentium infection, we adoptively transferred parasite-primed DCs into normal BALB/c mice and neutralized IL-10 with anti-IL-10 mAb (100–150 μg per mouse at each injection, JESS-2A5; BD Pharmingen) or control Ab (rat IgG1) i.p. at the time of DC transfer, at the time of C. rodentium infection, and at 24 h after the bacterial infection. We also performed adoptive transfer experiments using DCs obtained from H. polygyrus-infected IL-10-deficient mice.
DC cytokine expression
To determine the impact of H. polygyrus infection on cytokine response of CD11c+ DCs, DCs were purified as described. RNA was prepared, and expression of IL-4, IL-10, and IL-12 was measured using real-time PCR as described.
Detection of colonic cytokine expression
Cytokine mRNA expression in colon tissues was determined by real-time PCR (8). Total cellular RNA was isolated from frozen colonic tissue (distal part of colon) using TRIzol (Invitrogen Life Technologies) according to the manufacturer’s instructions. cDNA was synthesized using 2 μg of target RNA (Ready-to-Go kit; Pharmacia Biotech). cDNA samples were then subjected to real-time PCR. Mouse IL-4, IL-10, IL-12, and IFN-γ real-time PCR probes, and primer pairs were purchased from BioSource International. Amplification of GAPDH was included for each experimental sample as control to account for the differences in the amount of total RNA loaded in each reaction. Target cytokine gene expression was normalized between different samples based on the values of the expression of the GAPDH gene. All experimental samples were amplified in triplicate.
Histopathological examination
At necropsy, colonic tissues were collected, frozen in Tissue-Tek OCT compound (Miles) and then stored at −80°C. Five micrometer sections were cut on a 2800 Frigocut cryostat (Reichert Jung) and were stained with H&E. Three tissue samples from the proximal, middle, and distal parts of the colon were prepared. The sections were analyzed in a blinded fashion. Colonic pathology was scored using a modified histology scoring system based on previously published methods (8, 31, 32). The scoring system consists of two parts: 1) determination of the infiltration of inflammatory cells into the colon, in which the scores range from 0 to 4 (0, normal cell pattern; 1, scattered inflammatory cells in the lamina propia (LP); 2, increased numbers of inflammatory cells in the LP; 3, confluence of inflammatory cells extending into the submucosa; and 4, transmural extension of the infiltrative inflammatory cells) and 2) evaluation of colon tissue damage that also ranged from 0 to 4 (0, normal tissue pattern; 1, minimal inflammation and colonic crypt hyperplasia; 2, mild colonic crypt hyperplasia with or without focal invasion of epithelium; 3, obvious colonic crypt hyperplasia, invasion of epithelium and goblet cell depletion; and 4, extensive mucosal damage and extension through deeper structures of the bowel wall). The total colon pathology score equals the inflammatory cell score plus the tissue damage score. The results presented show the histological evaluation of the distal part of the colon.
To assess the systemic effect on the host of H. polygyrus infection, C. rodentium infection, and concurrent infection, as well as parasite-primed CD11c+ DCs, the body weight and survival of the infected mice were monitored throughout the experimental period. Body weight is represented as the percentage of the initial weight of individual mice (10–15 mice pooled from three experiments) at each time point.
Immunofluorescence microscopy
Colon tissues were frozen in Tissue-Tek OCT compound as described. Sections (5-μm) were cut on a cryostat and fixed in ice-cold acetone. The sections were then washed with PBS and blocked with PBS-1% BSA and avidin/biotin blocking agent (Vector Laboratories). The tissue sections were incubated with different Abs. To examine intestinal CD4+ T cells, the sections were stained with anti-mouse CD4 FITC (BD Pharmingen). To study DCs, the sections were stained with biotin-labeled anti-mouse CD11c (Endogen). After washing in PBS tissue sections were incubated with streptavidin-Cy3 (Cedarlane Laboratories). Sections were analyzed by immunofluorescence microscopy.
Statistical analysis
All results were expressed as the mean ± SEM. Statistical differences were determined using a two-tailed Student’s t test with GraphPad Prism (GraphPad Software). Variability within the colonic histopathology scores was determined using a nonparametric one-way ANOVA (Kruskal-Wallis test) with GraphPad Prism (GraphPad Software). A value for p < 0.05 was considered significant.
Results
Coinfection with H. polygyrus induces CD11c+ DC expansion in colonic lymphoid follicles and results in an enhanced C. rodentium-mediated colitis
C. rodentium infection in most adult mice is self-limited with little morbidity and mortality. Mice infected with C. rodentium show Citrobacter-associated alterations such as soft stools, a hunched posture, disturbed body hair, and body weight loss. In contrast to C. rodentium infection alone, mice coinfected with H. polygyrus and C. rodentium developed a more severe disease pathology associated with extensive body weight loss (Fig. 1A) and gastrointestinal tract bleeding. Furthermore, ~30% of the coinfected mice developed an anal prolapse 10 days postbacterial inoculation. C. rodentium infection typically induces histopathological change in the colon of mice, which are similar to those induced in many murine models of colitis, including colonic hyperplasia, goblet cell depletion, and mucosal erosion (Fig. 1B). Consistent with our previous findings (8), the results from this study provide evidence to suggest that coinfection of mice with H. polygyrus and C. rodentium results in a marked enhancement of tissue injury in the colon (Fig. 1B).
FIGURE 1.

More severe disease pathology is shown in coinfected mice. Coinfection with H. polygyrus and C. rodentium induces significant body weight loss (A) and colonic inflammation (B). BALB/c mice were infected with H. polygyrus (200 L3) and inoculated with C. rodentium (5 × 108 CFU) 7 days later. A, Body weight changes of mice that were infected with C. rodentium (○), H. polygyrus (■), both H. polygyrus and C. rodentium (▲), and normal control (□) mice during the course of the experiment (2 wk) are shown. Data shown are pooled from three independent experiments and are expressed as the body weight change as a percentage of the individual mouse initial body weight ± SE (n = 10–15) at each time point. B, Coinfection with intestinal helminth parasite promotes C. rodentium-mediated colonic injury. Colon tissue was removed from uninfected mice or mice infected with H. polygyrus, C. rodentium, or both at 2 wk postbacterial infection, frozen in Tissue-Tek OCT compound and stained with H&E. Magnification, ×100. C and D, Helminth coinfection promotes C. rodentium-associated CD4+ T cell infiltration into colonic LP. E and F, Helminth coinfection results in an expansion of CD11c+ DCs in colonic lymphoid follicle. Five-micrometer sections of frozen colonic tissue (in OCT) were cut and fixed in ice-cold acetone. After washing with PBS, the sections were blocked with PBS-1% BSA. The tissue sections were incubated with anti-CD4 FITC (green) and anti-CD11c Ab, followed by Cy3-labeled hamster IgG. The sections were analyz+ ed by immunofluorescent microscopy. All images were digitized and cropped in Adobe Photoshop LE 5.0 (Adobe Systems). C and E, The mean number of positive cells is detected in each high-power field (×200) by counting 10 fields from each sample (samples from three mice per group were counted).
Prior reports have demonstrated that intestinal colonization with C. rodentium induces a significant CD3+ and CD4+ T cell infiltration into the colonic LP, which stimulates a predominant Th1-type immune response in the intestine characterized by an up-regulation of IFN-γ, TNF-α, and IL-12 (9, 10, 29, 33). Therefore, we next sought to analyze this response during coinfection with the intestinal helminth, H. polygyrus, which predominately resides in the duodenum of the small intestine for the colonic mucosal CD4+ T cell infiltration during concurrent infection. As shown in Fig. 1, C and D, we found that infection with C. rodentium alone evoked a CD4+ T cell infiltration into the colonic LP, whereas coinfection of C. rodentium with H. polygyrus enhanced such a CD4+ T cell cellular response. Our results further demonstrate that infection with both C. rodentium and H. polygyrus induced not only an expansion of the colonic lymphoid follicle but also initiated changes in the cell population of the colonic lymphoid follicle as exemplified by the expansion of CD11c+ DCs (Fig. 1, E and F). Subsequent analysis of the cellular abundance of CD11c+ DCs in the expanded colonic lymphoid follicles revealed that infection with H. polygyrus (H. polygyrus alone as well as coinfection) is associated with a significant augmentation in the number of CD11c+ DCs (Fig. 1, E and F). This finding suggests that H. polygyrus infection may induce expansion and/or migration of DC11c+ DC into colonic epithelial lymphoid follicles. Therefore, a numerical increase and functional alteration in DCs might significantly influence Ag uptake, processing, and presentation, as well as the T cell response initiated in the intestinal mucosa.
Influence of H. polygyrus infection on CD11c+ DC responses
DCs initiate immunity by the activation of naive T cells that require two signals: signal one is derived through the recognition of peptide-MHC complexes by the Ag-specific TCR and signal two is derived via the induction of costimulatory molecules (CD80, CD86) on APCs. We have shown that H. polygyrus infection induces up-regulation of costimulatory molecule expression on APCs, including CD11c+ DCs (Ref. 30 and data not shown), which provides the second signal required to induce T cell activation and proliferation. Given the fact that the cytokine response of DCs can have a significant impact on CD4 T cell differentiation, we next examined the influence of an intestinal helminth infection on DC cytokine responses. To this end, we purified CD11c+ DCs from mice with or without H. polygyrus infection (Fig. 2A), and our FACS analysis showed that these CD11c+ cells were MHC class II positive, confirming that they are, indeed, DCs. We then examined DC cytokine expression by real-time PCR. Our results demonstrate that CD11c+ DCs isolated from H. polygyrus-infected mice have an elevated level of IL-10 expression and, to a lesser extent, IL-4 expression (Fig. 2B). H. polygyrus exposure, however, has no effect on the DC IL-12 response because similar levels of IL-12 expression were detected in purified CD11c+ DCs from normal as well as H. polygyrus-infected mice (Fig. 2B). Thus, an up-regulation of cytokine IL-10 expression in DCs may play an important role in the initiation of a productive Th2 cell response or a regulatory immune response.
FIGURE 2.
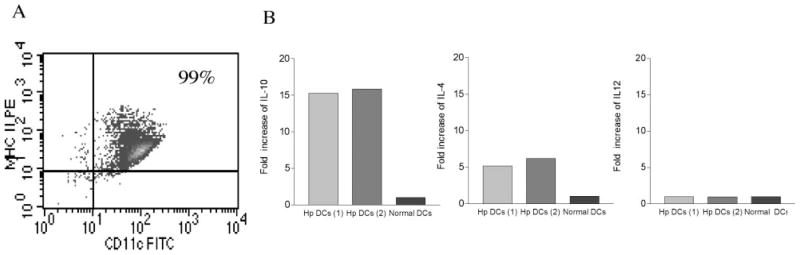
H. polygyrus induces DC IL-10 expression. A, FACS data show the purified CD11c+ DCs are MHC class II-positive. B, Cytokine expression of CD11c+ DCs (pooled from spleen and MLN) from H. polygyrus-infected mice by real-time PCR. Two preparations of DCs obtained from H. polygyrus-infected mice are shown. Values are mean fold increase compared with the baseline obtained from normal control animals.
Adoptive transfer of H. polygyrus-primed CD11c+ DCs results in increased morbidity and mortality in mice infected with C. rodentium
Thus far, our data substantiated the notion that coinfection of mice with helminth and C. rodentium leads to a significant increase in intestinal inflammation. Although the parasite-driven Th2-polarized mucosal immune response could have played a role in the impairment of host protection against bacterial infection in the coinfected host (8), it is unclear what is the relative contribution of APCs, in particular CD11c+ DCs, that precedes the Th cell activation for the alteration in the ability of a host to respond to concurrent infected pathogens. To examine the impact of helminth infection on the functional alterations of APCs and to demonstrate the role of parasite-primed DCs in the modulation of the host response to concurrent enteric pathogens, we established an adoptive transfer model. In this model, highly purified, helminth-primed CD11c+ DCs were obtained from H. polygyrus-infected mice and transferred into normal BALB/c mice that were then infected with C. rodentium. As expected, we found that mice infected with C. rodentium showed an initial drop in body weight, and as the infection subsided, the weight of these infected mice began to recover (Fig. 3A). Mice that were adoptively transferred with DCs isolated from normal mice displayed a delay in weight loss after C. rodentium infection, and by 2 wk postinfection the body weight recovered to the same level as C. rodentium-infected alone. In contrast, mice that received parasite-primed DC and were then infected with C. rodentium exhibited a progressive and significant reduction in body weight that presented at 2 wk postbacterial infection (Fig. 3A). Likewise, there was an increase in mortality in the recipient mice that had been adoptively transferred with parasite-primed CD11c+ DCs and then infected with C. rodentium (Fig. 3B). Taken together, these results suggest that helminth-primed DCs have the capacity to alter host responses to enteric bacterial infection and to enhance C. rodentium-induced disease.
FIGURE 3.

Adoptive transfer of H. polygyrus-primed CD11c+ DCs induces body weight loss and enhances C. rodentium-induced disease status. A, Body weight changes of mice that were infected with C. rodentium (◆), with H. polygyrus (■), H. polygyrus-primed DC recipient mice infected with C. rodentium (●), normal DC recipient mice infected with C. rodentium (*), normal DC recipient mice infected with both H. polygyrus and C. rodentium (▲), and normal control (□) during the course of the experiments. Data shown are pooled from three experiments and are the percentage of initial body weight ± SE (n = 10–15) at each time point). B, Survival curve. There was no mortality in C. rodentium-infected mice or mice that received normal DC. However, there is a 20% mortality at 2 wk postbacterial infection in mice that received parasite-primed DCs.
Adoptive transfer of H. polygyrus-primed CD11c+ DCs enhances C. rodentium-mediated colonic pathology replicating the intestinal injury noted in mice coinfected with the helminth
To further examine the influence of helminth-primed DCs on the host immune response to enteric bacterial infection using the adoptive transfer model, we next examined the contribution of parasite-primed DCs to the induction and modulation of bacteria-mediated colonic pathology. Two weeks after bacterial infection, the mice were sacrificed and the colon was removed so that comparisons could be made among the different groups (C. rodentium infection alone, H. polygyrus infection, coinfection, noninfected controls, normal DC-transferred C. rodentium-infected, and H. polygyrus DC-transferred C. rodentium-infected) at macroscopic and microscopic levels. At the macroscopic level, the thickness of the colon in coinfected mice and in mice that received H. polygyrus-primed CD11c+ DCs was more pronounced than that observed in mice with C. rodentium infection. In fact, colonic thickness of coinfected and the DC recipient mice was indistinguishable. No visible changes were observed in the small intestine and cecum in any of the mice.
Microscopic examination demonstrated that mice that received parasite-primed DCs and were infected with C. rodentium (2 wk postbacterial infection) (Fig. 4A) developed a more severe colonic pathology (including colonic crypt elongation, massive cellular infiltration of the colonic LP, epithelial erosions, and edema of the gut wall) as compared with the C. rodentium-infected mice (Fig. 4B) or the C. rodentium-infected normal DC recipient mice (Fig. 4C). Indeed, the scores for inflammation and intestinal damage were significantly higher in C. rodentium-infected parasite-primed DC recipients as compared with mice only infected with C. rodentium (Fig. 4G). In addition, microscopic analysis of colonic tissue of coinfected mice at 2 wk postinfection exhibited a similar level of intestinal pathology as compared with mice that received helminth-primed CD11c+ DCs (Fig. 4D). Thus, our results suggest that the contribution of CD11c+ DCs that are primed by helminth infection to the enhancement of bacteria-mediated intestinal injury suggesting that in addition to an elevated Th2 response, an ongoing helminth infection can significantly alter host responsiveness to concurrent bacterial infection and modulate bacteria-mediated disease by exerting its impact on DCs.
FIGURE 4.

Colonic histology of different groups at 2 wk postbacterial infection. A, Mice received CD11c+ DCs isolated from H. polygyrus-infected mice were then infected with C. rodentium. B, C. rodentium-infected mice. C, Mice adoptively transferred with CD11c+ DCs from normal BALB/c mice were then infected with C. rodentium. D, Mice coinfected with H. polygyrus and C. rodentium. E, Normal mice. F, H. polygyrus-infected mice. The results show that adoptive transfer of parasite-primed CD11c+ DCs enhances C. rodentium-mediated colonic pathology. G, Disease score of colonic inflammation is shown. The scores are assessed by determination of infiltration of inflammatory cells (range from 0 to 4), together with the evaluation of colon tissue damage (also a range from 0 to 4), with 0 scored normal and a score of 4 showing the most disease. The data shown are measurements of individual mouse (n = 8–12 per group) pooled from two to three independent experiments.
Additional experiments were performed to determine whether adoptive transfer of CD11c+ DCs could migrate to the intestinal mucosa. In this study, we labeled purified CD11c+ DCs from parasite-infected or uninfected mice with CFSE and transferred this cell population to normal BALB/c mice. The recipient mice were sacrificed 3 days after DC transfer, and the MLN, Peyer’s patch (PP), segments of small intestine and colon, and spleen were collected. Immunohistological analysis revealed that the adoptively transferred parasite-primed CD11c+ cells and DC from normal donor mice were readily and equally detectable in gut-associated lymphoid tissue (GALT), as demonstrated by the identification of CFSE-labeled CD11c+ cells in colonic lymphoid follicles, PP, and in MLN (Fig. 5). Low numbers of CFSE-labeled cells were detected in LP of small and large intestine. There were very few transferred DCs found in the spleen (data not shown). Taken together, these results suggest that the adoptively transferred DCs have the ability to migrate to the GALT and that parasite priming does not appear to influence this ability.
FIGURE 5.

Detection of the adoptively transferred DCs in GALT of recipient mice. A, FACS data show CFSE-labeled CD11c+ DCs used for the adoptive transfer experiments. B, Detection of CFSE-labeled (green) CD11c+ DCs (Cy3, red) in the PP, MLN, and colonic lymphoid follicle. Arrows indicate some of the double stained cells (yellow). CD11c+ DCs were purified from H. polygyrus-infected (7 days postinfection), and normal mice and were adoptively transferred to normal BALB/c mice. The recipient mice were sacrificed 3 days later. The tissue sections (PP, MLN, colon) were prepared and stained with anti-CD11c-biotin and followed by streptavidin-Cy3. The sections were analyzed by immunofluorescent microscopy.
Adoptive transfer of H. polygyrus-primed CD11c+ cells alters the dynamics of concurrent colonic bacterial infection
We next determined whether parasite-primed DCs can alter the host response which, in turn, might influence the bacterial dynamics of the C. rodentium-infected mice. Thus, bacterial colonization as well as proliferation and clearance were measured. As shown in Fig. 6, the bacterial output was significantly higher in mice that received parasite-primed DC as well as in mice that were coinfected with C. rodentium and H. polygyrus at an early stage of infection (1 wk postinfection) (Fig. 6). Such results indicate that parasite-primed DCs may enhance the initial colonization of C. rodentium on the colonic epithelial surface. The significantly higher levels of bacteria shed in feces were detected in both parasite-primed DC recipient mice and coinfected mice throughout the experimental period (up to 3 wk postinfection) compared with infection alone (Fig. 6). Furthermore, by the 3 wk postbacterial infection, those mice that were infected with C. rodentium alone almost completely cleared the infection, whereas the H. polygyrus-DC mice, along with the mice that were coinfected, remained heavily colonized with C. rodentium. In contrast, transfer of DC isolated from normal mice had no effect on fecal bacterial output in the recipient mice. These results suggest that adoptive transfer of CD11c+ DCs, which were primed by helminth infection, like the coinfection experiments with H. polygyrus, can alter host defense against bacterial infection, thereby leading to an increase in bacterial colonization and proliferation and to a decrease in bacterial clearance.
FIGURE 6.

Mice that received H. polygyrus-primed DCs and then infected with C. rodentium have a higher bacterial output in the fecal pellets. The data shown are the number of bacteria recovered from fecal samples of C. rodentium-infected mice and mice with adoptive transfer of DCs at 1, 2, and 3 wk postinfection. The data are represented as the mean ± SEM (n = 5–10 mice) at each time point.
IL-10 is required for the effects of parasite-primed DCs on the response to C. rodentium
Thus far we have shown that parasite-primed DCs play a role in immune modulation of host response to C. rodentium infection and intestinal pathology. Further analysis of CD11c+ DCs from mice infected with H. polygyrus show an enhanced IL-10 expression. We next examined whether the DC IL-10 response induced by parasite priming could contribute to the suppression of host defense against C. rodentium infection as well as to the enhancement of C. rodentium-meditated colitis. To test this possibility, we performed adoptive transfer experiments with two different approaches: one approach involved neutralization of donor DC IL-10, whereas the other used IL-10-deficient DCs as donor cells (isolated from IL-10 knockout mice).
In our neutralization experiments, we adoptively transferred parasite-primed DCs into normal BALB/c mice and subsequently neutralized IL-10 with an anti-IL-10 blocking mAb in vivo given simultaneously with the DC transfer and the C. rodentium infection. As shown in Fig. 7A, neutralizing the IL-10 cytokine response in mice receiving parasite-primed DCs led to a significant reduction in C. rodentium-associated body weight loss at 2 wk postbacterial infection (Fig. 7A). Whole body weight of mice receiving parasite-primed DCs and treated with anti-IL-10 mAb was similar to that of C. rodentium-infected mice alone, whereas overall body weight was significantly lower in mice receiving parasite-primed DCs and treated with anti-IL-10 than in mice that received the H. polygyrus-primed DCs in the absence of anti-IL-10 treatment (Fig. 7A). Neutralizing the IL-10 response also enhanced survival in the mice that were adoptively transferred with parasite-primed DCs (data not shown). The influence of the DC IL-10 response induced by the helminth H. polygyrus on the host response to bacterial infection was further demonstrated by a significant increase in bacterial clearance in C. rodentium-infected mice (Fig. 7B).
FIGURE 7.

Alterations induced by adoptive transfer of parasite-primed DCs in mice can be reversed by anti-IL-10 treatment at the time of C. rodentium infection. A, Body weight changes of mice that were infected with C. rodentium (■), received H. polygyrus-primed DC and were then infected with C. rodentium treated with control Ab (▼), parasite-primed DC recipients, infected with C. rodentium and treated with anti-IL-10 (□), infected with both H. polygyrus and C. rodentium (●), adoptively transferred DCs obtained from IL-10-deficient, H. polygyrus-infected mice (△) and normal control (○) during the course of the experiments. Data shown are pooled from two to three experiments and are the percentage of change of initial body weight ± SE (n = 10–15 mice) at each time point. B, C. rodentium output in fecal pellets. The data shown are the number of bacteria recovered from fecal samples of C. rodentium-infected mice (◆) with adoptive transfer of DCs (○) and treated with anti IL-10 (●) at 1, 2, and 3 wk postinfection. The data are represented as the mean ± SEM (n = 5–10 mice) at each time point.
Microscopic examination of colonic tissues revealed that mice that received the parasite-primed DCs developed a more severe colitis (Fig. 8B) as compared with mice only infected with C. rodentium (Fig. 8A). The influence of parasite-primed DCs on C. rodentium-induced intestinal inflammation was completely abolished when the DC IL-10 was neutralized at the same time the bacterial infection was initiated. This result was evidenced by the observation that there was a significant decrease in C. rodentium-induced colonic pathology in the mice that received helminth-primed DC treated with anti-IL-10 mAb compared with pathology in the parasite-primed DC recipient treated with control Ab (Fig. 8). These results suggest that the DC IL-10 response triggered by the helminth infection can significantly contribute to the modulation of the host immune response during an intestinal bacterial infection.
FIGURE 8.

Colonic histopathology shows that adoptive transfer of CD11c+ DCs that lack IL-10 fails to promote C. rodentium-induced colitis. A, Colon section from C. rodentium-infected mice. B, Colon section of parasite-primed DC recipient with C. rodentium infection. C, Colonic histopathology of mice that received parasite-primed DC infected with C. rodentium and treated with anti-IL-10 mAb. D, Colon section of the parasite-primed IL-10-deficient (IL-10−/−) DC recipient infected with C. rodentium. Duplicate samples are presented from each group. E, Disease score of colonic inflammation as described in Fig. 3.
The results from the neutralization experiments, however, do not exclude the possibility that the anti-IL-10 treatment acted directly on the recipient cells. Our second approach, therefore, involved using IL-10-deficient DCs as donor cells. This approach allowed us to further document the role of the DC IL-10 response, triggered by helminth infection, in the initiation and regulation of host protective immunity against enteric bacterial infections as well as in the modulation of bacterial-mediated intestinal injury. In these experiments, CD11c+ DCs were isolated from H. polygyrus-infected IL-10-deficient BALB/c mice as previously described. They were then adoptively transferred into normal BALB/c mice, which were subsequently infected with C. rodentium. The systemic influence of the C. rodentium infection on IL-10-deficient DC recipient mice was initially examined by measuring C. rodentium infection-associated loss in body weight. In a manner similar to mice that had received parasite-primed DCs and were treated with anti-IL-10 mAb, we found that IL-10-deficient DC recipient mice had a significant reduction in body weight loss as compared with mice that received parasite-primed wild-type DCs (Fig. 7A). Colonic inflammation was assessed at 2 wk postbacterial infection, and as expected, there was a substantial reduction in colonic inflammation in these mice that received H. polygyrus IL-10-deficient DCs (Fig. 8D). The reduction in colonic inflammation was somewhat less pronounced in IL-10-deficient DC recipient mice as compared with mice receiving H. polygyrus-primed DCs and anti-IL-10 treatment. Collectively, these results establish a role for the DC IL-10 response triggered by helminth infection in altering the host response to concurrent enteric bacterial infection and in promoting bacteria-mediated intestinal inflammation.
Contribution of parasite-primed IL-10 secreting DCs to the regulation of cytokine responses in intestinal mucosa
C. rodentium infection in mice induces a Th1-type immune response in the intestine, which is characterized by an up-regulation of IFN-γ, TNF-α, and IL-12 (29, 33). Mice lacking either IFN-γ, IL-12, or the receptor for TNF showed a reduced resistance to infection and enhanced intestinal injury (33, 34). Such studies imply that Th1 cytokines play an important role in host protective immunity to C. rodentium infection. Our observations that a DC expansion in the colonic mucosa of coinfected mice together with an up-regulation in the DC-IL-10 expression induced by helminth infection led us to hypothesize that DC primed by the small intestinal helminthic parasite may have an influence on local intestinal immune responses to bacterial infection. To test this hypothesis, we examined bacteria-specific cytokine production of MLN cells by isolating the MLN cells from C. rodentium-infected mice that received different DC (i.e., helminth-primed DC, helminth-primed IL-10-deficient DC, normal DC) or no DC, and from mice that were adoptively transferred with parasite-primed DC and treated with anti-IL-10 Ab. Our results showed that, as expected, C. rodentium infection in mice induced a Th1 response as evidenced by the induction of IFN-γ production of MLN cells, and that adoptive transfer of DC from helminth-infected mice resulted in a significant reduction in bacteria-specific IFN-γ production (Fig. 9). Such an impairment of bacteria-specific IFN-γ production was not observed in mice that received normal DC or helminth-primed DC followed by treatment with anti-IL-10 Ab or helminth-primed IL-10-deficient DC (Fig. 9). Our results further showed that transfer of helminth-primed DC failed to stimulate a detectable level of C. rodentium Ag-specific Th2 cytokine production (IL-4) by the MLN cells in vitro (data not shown).
FIGURE 9.
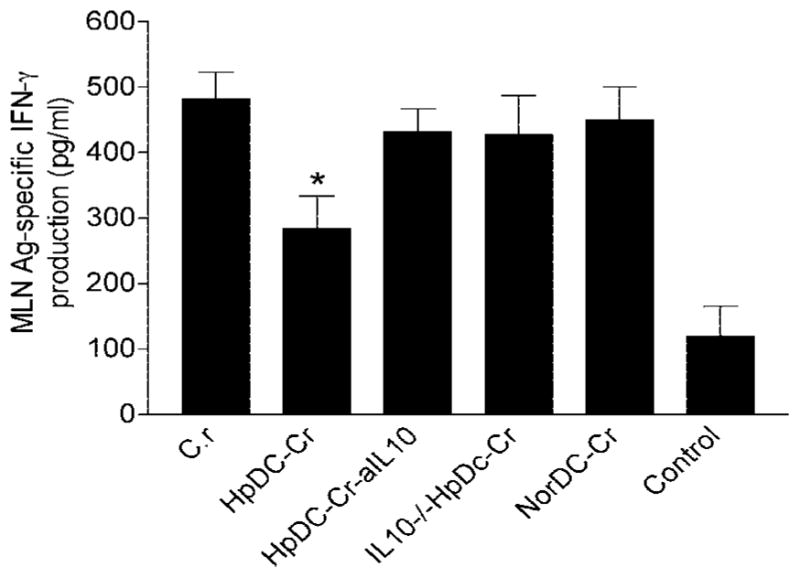
Adoptive transfer of helminth-primed DC results in an inhibition of bacterial Ag-specific IFN-γ production by MLN. MLN cells were cultured with and without C. rodentium Ag (10 μg/ml) for 72 h. Th1 (IFN-γ and Th2 (IL-4) cytokine production was measured using ELISA. The data show that C. rodentium infection induces Ag-specific IFN-γ production and that adoptive transfer of DCs from helminth-infected mice causes a significant suppression of bacteria-specific IFN-γ response. No detectable bacteria-specific Th2 (IL-4) response was found in any groups (data not shown).
To examine the impact of helminth-primed IL-10 secreting DC in local intestinal immune response to C. rodentium infection, we next compared the expression of both Th1 (IFN-γ) and Th2 (IL-4) cytokines in colon tissues under different infection conditions (C. rodentium infection alone, H. polygyrus DC-transferred C. rodentium-infected, or parasite-primed IL-10-deficient DC recipient with C. rodentium infection). As shown in Fig. 10, left, the colon of C. rodentium-infected mice expressed elevated levels of IFN-γ, which indicates the development of a predominant Th1 response. However, the expression of the Th1 cytokine (IFN-γ) was significantly suppressed in colonic tissue from parasite-primed DC recipients. Additionally, the transfer of parasite-primed IL-10-deficient DCs partially reversed the colonic IFN-γ expression. By contrast, the colon of mice that received parasite-primed DCs and were infected with C. rodentium produced an enhanced Th2 cytokine response as demonstrated by the up-regulation of IL-4 (Fig. 10, right). Yet, IL-10-deficient DCs were less able to elicit a Th2 response (Fig. 10, right). These results suggest that DC priming by prior helminth infection can play an important role in shaping the nature of local mucosal immune responses, contributing to the alteration in host response to concurrent enteric bacterial infections.
FIGURE 10.
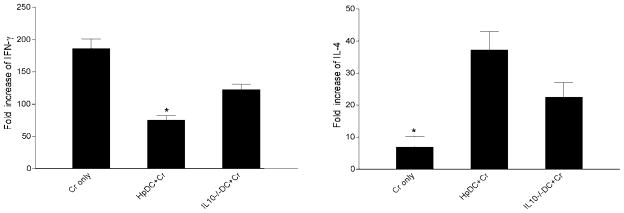
Cytokine mRNA expression (IFN-γ and IL-4) in colon tissue as measured by real-time PCR at 2 wk postbacterial infection. Values are mean fold increase compared with the baseline obtained from normal control animals. The data shown are from one of two experiments performed, showing similar results. *, p < 0.05 (n = 3–5 mice per group).
Discussion
Cross-regulatory suppression of the Th1 responses by a strong Th2 response has been considered to play a role in modulating diseases that are characterized by a Th1 response (4–7). Recently, we have shown that coinfection with a Th2-stimulating H. polygyrus results in a significantly impaired host protection against C. rodentium leading to the development of a more severe bacteria-mediated colitis (8). However, the interactions and the immune regulation between helminth infection and other concurrent exposed pathogens, Ags, or vaccines are more complex and are not fully understood. In the current study, we have expanded our previous observations and examined the hypothesis that the DC activation and cytokine response induced by a helminth may influence the host response to subsequent enteric bacterial infection.
We show that H. polygyrus infection induces DC activation and IL-10 expression. Adoptive transfer of the helminth-primed CD11c+ DCs into normal BALB/c mice that are then infected with C. rodentium produce a significant suppression of host defense mechanisms to bacterial infection, resulting in an increase in the bacterial load, a delay in bacterial clearance, and the development of a more severe colitis. These observations are similar to the results from mice coinfected with the helminth and C. rodentium. Our results further indicate that the capacity of parasite-primed DCs to alter host responses to C. rodentium infection is largely IL-10-dependent. This result is evidenced by the observation that neither adoptive transfer of the parasite-primed DCs that lack IL-10 nor transfer of parasite-primed DCs with its IL-10 response being neutralized in the recipient mice is able to induce an enhanced C. rodentium-mediated colitis. These results suggest that DCs that are primed by the helminth can play an important role in the modulation of host response to concurrent bacterial infection through IL-10-mediated mechanisms. This immune regulatory capacity of helminth-primed DCs to alter the host response to concurrent bacterial infection may be associated with the potential of these DCs to inhibit the development of protective bacterial Ag-specific Th1 response or to induce a Th2 polarized response as well as the immune regulatory response. Thus, priming of the DCs is an important means by which ongoing intestinal helminth infection modulates host responses to concurrent enteric bacterial infection, as well as the disease the bacteria induce.
In GALT, DCs are present in PP, MLNs, and LP. The dense and highly dynamic DC populations at mucosal sites play key regulatory roles in acquiring and presenting Ags and regulating host protective immune responses (35–37). In SCID mice, which develop colitis by transfer of CD45RBhighCD4+ T cell populations, an increased number of DCs along with an increased proportion of activated DCs were detected in MLNs (38). A suppression in the accumulation of activated DCs by cotransfer of regulatory T cells was found to inhibit the immune pathology in this murine model of colitis (38). Consistent with these observations, we also found a marked increase in CD11c+ DCs in colonic lymphoid follicles, which were also enlarged during the C. rodentium infection. Lymphoid follicle hyperplasia has been observed in colonic inflammation (39). Furthermore, we observed that coinfection with H. polygyrus led to a more pronounced CD11c+ DC recruitment and/or expansion in colonic lymphoid tissues of coinfected mice that developed the most severe colitis. Therefore, an increased number of DCs and an increase in size of the colonic lymphoid follicle in the intestinal mucosa might reflect an augmented local Ag handling and the priming of Th cells. In coinfected mice, DC expansion may also influence the outcome of a productive T cell response to concomitant bacterial infection in the intestinal mucosa.
A direct role of parasite-primed DCs in modulating host response to an enteric bacterial infection and in enhancing the pathogenesis of the bacteria was demonstrated by the results from our adoptive transfer experiments. We found that although both DCs isolated from helminth-infected and the uninfected mice can migrate to the intestinal mucosa of the recipient mice equally well, it is the mice that received parasite-primed CD11c+ DCs, and were subsequently infected with C. rodentium, that display an enhanced bacterial infection and intestinal injury. Recent evidence suggests that there is considerable heterogeneity within the DC pool (40, 41). However, it is still unclear whether helminth infection induces a specific subset of DCs and whether these subsets of DCs are responsible for the modulation of host immune responses to concomitant infections. DC activation and cytokine production have been suggested to play a role in the initiation of a Th1 or Th2 polarized response (42) and in the generation of regulatory cells (43). DCs can be activated following exposure to microbial Ags through ligation of a TLR, leading to the up-regulation of functional surface molecules and the release of cytokines such as IL-12, IL-18, and IL-27. This results in induction of Th1 immunity (17–23). However, in this context, it is less clear how helminth Ag activates DC.
Prior studies have shown that DCs pulsed with the helminth Ag, such as S. mansoni egg Ag or the schistosome glycan lacto-N-fucopentaose III (24), induce a Th2-biased response (25). It has also been shown that murine bone marrow-derived DCs pulsed with the helminth N. brasiliensis excretory/secretory Ag can, on transfer to naive recipients, prime mice for Th2 responsiveness (26). However, in these studies, no significant increase in Th2-inducing cytokines (IL-4, IL-10) was detected in DCs that were exposed to the parasite Ags, although cytokine production of DC was suggested to play a role in determining Th cell differentiation and in the generation of regulatory cells (43). It was also suggested that the polarization of the Th1 and Th2 response did not depend on DC secretion of the polarizing cytokines IL-12/IL-4 but instead correlated with state of DC activation induced by different parasite Ags (42).
Although it remains controversial, signals derived from Ag-primed DCs have been thought to be required for the differentiation of naive Th cells into Th1 and Th2 cells in addition to Ag-specific MHC-peptide complexes and costimulatory molecules (B7.1 and B7.2, signal 2) (12–14). The induction of cytokine production by DCs in response to helminth infection can have a direct result on the Th response. Our results in this study, provide evidence to suggest that Th2-stimulating H. polygyrus infection induces the activation of CD11c+ DCs and up-regulates the DC IL-10 response and to a much lesser extent IL-4 expression. Thus, our data support the contention that both the IL-10 and IL-4 response in DCs may initiate a Th2-polarized response (44–46). Furthermore, our results indicate that H. polygyrus infection had no effect on the DC IL-12 response, which is in agreement with the observation from a different helminth model showing that DCs from mice infected with schistosomes did not exhibit an elevated level of IL-12 (47). In fact, this failure to produce IL-12 by DCs has also been linked to its capacity to induce Th2 response (48). Our data, therefore, suggests that parasite-primed DCs may contribute to a dysfunctional intestinal mucosa immune response by creating a cytokine environment that fails to develop a protective Th1 response against C. rodentium. Indeed, our analysis of bacterial specific cytokine response showed that adoptive transfer of helminth-primed DC resulted in a suppression of Th1 response (IFN-γ) in the MLN and an induction of the development of a Th2-biased cytokine response in the colon tissue of recipient mice (Fig. 10), contributing to an impaired host resistance against C. rodentium infection.
Our data also provide direct evidence to suggest that the DC IL-10 response can be a possible mechanism by which helminth-primed DCs modulate host responses to concurrent C. rodentium infection. This correlation is evidenced by the inability of parasite-primed IL-10-deficient DCs or parasite-primed DCs, for which IL-10 response was blocked by anti-IL-10 treatment, to induce an enhanced C. rodentium-mediated intestinal injury in the recipient mice that are subsequently infected with C. rodentium. These observations correlated with results showing that blocking of DC IL-10 (by transferring IL-10-deficient DC or anti-IL-10 treatment of the recipient mice that received parasite-primed DC) resulted in a restoration of mucosal Th1 response (Fig. 9). These results, therefore, suggest a central role of the DC IL-10 response triggered by the helminth infection in regulating host immune and pathological responses to C. rodentium infection. Such results substantiate the observation that neutralization of DC IL-10 enhances the ability of DCs to dramatically stimulate a Th1 response (IFN-γ) (44, 49). Therefore, our data provide strong evidence that DC IL-10 response initiated by the helminth infection is responsible for the altered host responses and enhanced C. rodentium-induced intestinal pathology.
Notably, we demonstrate that when mice were transferred with parasite-primed DCs isolated from IL-10-deficient mice and subsequently challenged with C. rodentium, the ability to cause a reduction in the severity of bacteria-induced colitis was less pronounced compared with anti-IL-10 treatment (Fig. 7). This difference may be explained by the fact that when parasite-primed DC recipient mice are treated with exogenous anti-IL-10 mAb the effects of cytokine IL-10 from both transferred DC and the recipient host can be blocked at the time of bacterial infection. However, under circumstances when parasite-primed, IL-10-deficient DCs were adoptively transferred to a normal recipient host, only IL-10 was absent in these transferred DCs. It is possible that other DC secreted cytokines (such as IL-4) might be involved in directing the mucosal Th response because our results indicated that helminth infection also induces a modest elevation of DC IL-4 expression. In the absence of IL-10, the helminth-induced DC IL-4 response may contribute to priming for a Th2 response that would impair host protective Th1 immunity to enteric bacterial infection.
In addition to initiating a Th2-polarized immune response (44), which inhibits protective Th1 responses against bacterial infection, IL-10-producing DC may also be associated with the activation of other regulatory immune responses during helminth infection. Recently, an IL-10-producing B220+ DC cell population has been shown to be involved in activating regulatory immune responses (50). Although it is still not fully understood how a helminth parasite induces host regulatory immune mechanisms, helminth-induced regulatory T cell responses have been suggested to be involved in the suppression of immune-mediated disorders (3). There is also a growing body of evidence to suggest that regulatory T cells play a significant role in modulating the severity of microbial infection (51–55) as well as in the prevention of intestinal inflammation through secretion of TGF-β and IL-10 (56). Regardless, it is possible that both Th2 and regulatory T cell responses can be initiated by helminth infection, both of which may contribute to suppression of Th1 response development (57). Therefore, our results indicate that helminth-primed DCs are involved in damping of host protective Th1 responses, which in turn may allow for increased colonization and proliferation of the microorganisms and a delayed clearance of the infecting bacteria leading to more severe tissue damage and disease.
In conclusion, our investigation provides evidence suggesting that H. polygyrus infection alters the host response to a concurrent enteric bacterial infection and promotes a C. rodentium-mediated intestinal injury via a novel mechanism that requires DC activation and IL-10 expression. A better understanding of immunoregulation in the intestinal mucosa may provide new information for establishing more effective preventive and therapeutic approaches to the treatment of both Th1- and Th2-mediated diseases and for a design of effective intestinal vaccines.
Acknowledgments
We thank Dr. Bobby Cherayil for critical review of the manuscript.
Footnotes
This work was supported in part by a Hood Foundation research grant, a First Award from the Crohn’s and Colitis Foundation of America (to H.N.S.), the National Institutes of Health Grants RO1 DK70260, RO1 HD31852, and PO1 DK-33506 (to W.A.W.), and by the Clinical Nutrition Research Center, Harvard Medical School Grant P30 DK 40561. H.N.S. is a recipient of the National Institutes of Health Award KO1 DK059996. C.-C.C. is sponsored by Chang Gung University and Chang Gung Children’s Hospital, Taoyuan, Taiwan.
Abbreviations used in this paper: EPEC, enteropathogenic Escherichia coli; DC, dendritic cell; MLN, mesenteric lymph node; PP, Peyer’s patch; LP, lamina propria; GALT, gut-associated lymphoid tissue.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Nokes C, Cooper ES, Robinson BA, Da B. Geohelminth infection and academic assessment in Jamaican children. Trans R Soc Trop Med Hyg. 1991;85:272–273. doi: 10.1016/0035-9203(91)90052-z. [DOI] [PubMed] [Google Scholar]
- 2.Crompton DWT, Nesheim MC. Nutritional impact of intestinal helminthiasis during the human life cycle. Annu Rev Nutr. 2002;22:35–59. doi: 10.1146/annurev.nutr.22.120501.134539. [DOI] [PubMed] [Google Scholar]
- 3.Yazdanbakhsh M, Kremsner PG, van Ree R. Allergy, parasites, and the hygiene hypothesis. Science. 2002;296:490–494. doi: 10.1126/science.296.5567.490. [DOI] [PubMed] [Google Scholar]
- 4.Elliott DE, Li J, Blum A, Metwali A, Qadir K, Urban JF, Jr, Weinstock JV. Exposure to schistosome eggs protects mice from TNBS-induced colitis. Am J Physiol. 2003;284:G385–G391. doi: 10.1152/ajpgi.00049.2002. [DOI] [PubMed] [Google Scholar]
- 5.Fox JG, Beck P, Dangler CA, Whary MT, Wang TC, Shi HN, Nagler-Anderson C. Concurrent enteric helminth infection modulates inflammation and gastric immune responses and reduces helicobacter-induced gastric atrophy. Nat Med. 2000;6:536–542. doi: 10.1038/75015. [DOI] [PubMed] [Google Scholar]
- 6.Khan WI, Blennerhasset PA, Varghese AK, Chowdhury SK, Omsted P, Deng Y, Collins SM. Intestinal nematode infection ameliorates experimental colitis in mice. Infect Immun. 2002;70:5931–5937. doi: 10.1128/IAI.70.11.5931-5937.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moreels TG, Nieuwendijk RJ, De Man JG, De Winter BY, Herman AG, Van Marck EA, Pelckmans PA. Concurrent infection with Schistosoma mansoni attenuates inflammation induced changes in colonic morphology, cytokine levels, and smooth muscle contractility of trinitrobenzene sulphonic acid induced colitis in rats. Gut. 2004;53:99–107. doi: 10.1136/gut.53.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen CC, Louie S, McCormick B, Walker WA, Shi HN. Concurrent infection of an intestinal helminth parasite impairs host resistance to enteric Citrobacter rodentium and enhances Citrobacter-induced colitis in mice. Infect Immun. 2005;73:5468–5481. doi: 10.1128/IAI.73.9.5468-5481.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Higgins LM, Frankel G, Connerton I, Gonçalves NS, Dougan G, MacDonald TT. Role of bacterial intimin in colonic hyperplasia and inflammation. Science. 1999;285:588–591. doi: 10.1126/science.285.5427.588. [DOI] [PubMed] [Google Scholar]
- 10.Higgins LM, Frankel G, Douce G, Dougan G, MacDonald TT. Citrobacter rodentium infection in mice elicits a mucosal Th1 cytokine response and lesions similar to those in murine inflammatory bowel disease. Infect Immun. 1999;67:3031–3039. doi: 10.1128/iai.67.6.3031-3039.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barthold SW, Coleman GL, Jacoby RO, Livestone EM, Jonas AM. Transmissible murine colonic hyperplasia. Vet Pathol. 1978;15:223–236. doi: 10.1177/030098587801500209. [DOI] [PubMed] [Google Scholar]
- 12.Abbas AK, Murphy KM, Sher A. Functional diversity of helper T lymphocytes. Nature. 1996;383:787–793. doi: 10.1038/383787a0. [DOI] [PubMed] [Google Scholar]
- 13.Kalinski P, Hilkens CM, Wierenga EA, Kapsenberg ML. T-cell priming by type-1 and type-2 polarized dendritic cells: the concept of a third signal. Immunol Today. 1999;20:561–567. doi: 10.1016/s0167-5699(99)01547-9. [DOI] [PubMed] [Google Scholar]
- 14.de Jong EC, Vieira PL, Kalinski P, Schuitemaker JH, Tanaka Y, Wierenga EA, Yazdanbakhsh M, Kapsenberg ML. Microbial compounds selectively induce Th1 cell-promoting or Th2 cell-promoting dendritic cells in vitro with diverse Th cell-polarizing signals. J Immunol. 2002;168:1704–1709. doi: 10.4049/jimmunol.168.4.1704. [DOI] [PubMed] [Google Scholar]
- 15.Constant SL, Bottomly K. Induction of Th1 and Th2 CD4+ T cell responses: the alternative approaches. Annu Rev Immunol. 1997;15:297–322. doi: 10.1146/annurev.immunol.15.1.297. [DOI] [PubMed] [Google Scholar]
- 16.Hsieh CS, Macatonia SE, Tripp CS, Wolf SF, O’Garra A, Murphy KM. Development of TH1 CD4+ T cells through IL-12 produced by. Listeria-induced macrophages. Science. 1993;260:547–549. doi: 10.1126/science.8097338. [DOI] [PubMed] [Google Scholar]
- 17.Agrawal S, Agrawal A, Doughty B, Gerwitz A, Blenis J, Van Dyke T, Pulendran B. Cutting edge: different Toll-like receptor agonists instruct dendritic cells to induce distinct Th responses via differential modulation of extracellular signal-regulated kinase-mitogen-activated protein kinase and c-Fos. J Immunol. 2003;171:4984–4989. doi: 10.4049/jimmunol.171.10.4984. [DOI] [PubMed] [Google Scholar]
- 18.Pflanz S, Timans JC, Cheung J, Rosales R, Kanzler H, Gilbert J, Hibbert L, Churakova T, Travis M, Vaisberg E, et al. IL-27, a heterodimeric cytokine composed of EBI3 and p28 protein, induces proliferation of naive CD4+ T cells. Immunity. 2002;16:779–790. doi: 10.1016/s1074-7613(02)00324-2. [DOI] [PubMed] [Google Scholar]
- 19.Robinson DS, O’Garra A. Further checkpoints in Th1 development. Immunity. 2002;16:755–758. doi: 10.1016/s1074-7613(02)00331-x. [DOI] [PubMed] [Google Scholar]
- 20.Heufler C, Koch F, Stanzl U, Topar G, Wysocka M, Trinchieri G, Enk A, Steinman R, Romani N, Schuler G. Interleukin-12 is produced by dendritic cells and mediates T helper 1 development as well as interferon-γ production by T helper 1 cells. Eur J Immunol. 1996;26:659–668. doi: 10.1002/eji.1830260323. [DOI] [PubMed] [Google Scholar]
- 21.Edwards AD, Manickasingham SP, Sporri R, Diebold SS, Schulz O, Sher A, Kaisho T, Akira S, Reis e Sousa C. Microbial recognition via Toll-like receptor-dependent and -independent pathways determines the cytokine response of murine dendritic cell subsets to CD40 triggering. J Immunol. 2002;169:3652–3660. doi: 10.4049/jimmunol.169.7.3652. [DOI] [PubMed] [Google Scholar]
- 22.Medzhitov R, Janeway C., Jr The Toll receptor family and microbial recognition. Trends Microbiol. 2000;8:452–456. doi: 10.1016/s0966-842x(00)01845-x. [DOI] [PubMed] [Google Scholar]
- 23.Barton GM, Medzhitov R. Control of adaptive immune responses by Toll-like receptors. Curr Opin Immunol. 2002;14:380–383. doi: 10.1016/s0952-7915(02)00343-6. [DOI] [PubMed] [Google Scholar]
- 24.Thomas PG, Carter MR, Atochina O, Da’Dara AA, Piskorska D, McGuire E, Harn DA. Maturation of dendritic cell 2 phenotype by a helminth glycan uses a Toll-like receptor 4-dependent mechanism. J Immunol. 2003;171:5837–5841. doi: 10.4049/jimmunol.171.11.5837. [DOI] [PubMed] [Google Scholar]
- 25.MacDonald AS, Straw AD, Bauman B, Pearce EJ. CD8-dendritic cell activation status plays an integral role in influencing Th2 response development. J Immunol. 2001;167:1982–1988. doi: 10.4049/jimmunol.167.4.1982. [DOI] [PubMed] [Google Scholar]
- 26.Balic A, Harcus Y, Holland MJ, Maizels RM. Selective maturation of dendritic cells by Nippostrongylus brasiliensis-secreted proteins drives Th2 immune responses. Eur J Immunol. 2004;34:3047–3059. doi: 10.1002/eji.200425167. [DOI] [PubMed] [Google Scholar]
- 27.Shi HN, Ingui CJ, Dodge I, Nagler-Anderson C. A helminth-induced mucosal Th2 response alters nonresponsiveness to oral administration of a soluble antigen. J Immunol. 1998;160:2449–2455. [PubMed] [Google Scholar]
- 28.Shi HN, Scott ME, Stevenson MM, Koski KG. Zinc deficiency impairs T cell function in mice with primary infection of Heligomosomoides polygyrus (Nematoda) Parasite Immunol. 1994;16:339–350. doi: 10.1111/j.1365-3024.1994.tb00359.x. [DOI] [PubMed] [Google Scholar]
- 29.Vallance BA, Deng W, Knodler LA, Finlay BB. Mice lacking T and B lymphocytes develop transient colitis and crypt hyperplasia yet suffer impaired bacterial clearance during Citrobacter rodentium infection. Infect Immun. 2002;70:2070–2081. doi: 10.1128/IAI.70.4.2070-2081.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shi HN, Liu HY, Nagler-Anderson C. Enteric infection acts as an adjuvant for the response to a model food antigen. J Immunol. 2000;165:6174–6182. doi: 10.4049/jimmunol.165.11.6174. [DOI] [PubMed] [Google Scholar]
- 31.Cartland BR, Rivera-Nieves J, Moskaluk CA, Matsumoto S, Cominelli F, Klaus L. Antibody blockade of ICAM-1 and VCAM-1 ameliorates inflammation in the SAMP-1/Yit adoptive transfer model of Crohn’s disease in mice. Gastroenterology. 2001;121:1428–1436. doi: 10.1053/gast.2001.29568. [DOI] [PubMed] [Google Scholar]
- 32.Loher F, Schmall K, Freytag P, Landauer N, Hallwachs R, Bauer C, Siegmund B, Rieder F, Lehr HA, Dauer M, et al. The specific type-4 phosphodiesterase inhibitor mesopram alleviates experimental colitis in mice. J Pharmacol Exp Ther. 2003;305:549–556. doi: 10.1124/jpet.102.039529. [DOI] [PubMed] [Google Scholar]
- 33.Gonçalves NS, Ghaem-Maghami M, Monteleone G, Frankel G, Dougan G, Lewis DJ, Simmons CP, MacDonald TT. Critical role for tumor necrosis factor α in controlling the number of lumenal pathogenic bacteria and immunopathology in infectious colitis. Infect Immun. 2001;69:6651–6659. doi: 10.1128/IAI.69.11.6651-6659.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Simmons CP, Gonçalves NS, Ghaem-Maghami M, Bajaj-Elliott M, Clare S, Neves B, Frankel G, Dougan G, MacDonald TT. Impaired resistance and enhanced pathology during infection with a noninvasive, attaching-effacing enteric bacterial pathogen, Citrobacter rodentium, in mice lacking IL-12 or IFN-γ. J Immunol. 2002;168:1804–1812. doi: 10.4049/jimmunol.168.4.1804. [DOI] [PubMed] [Google Scholar]
- 35.McWilliam AS, Nelson D, Thomas JA, Holt PG. Rapid dendritic cell recruitment is a hallmark of the acute inflammatory response at mucosal surfaces. J Exp Med. 1994;179:1331–1336. doi: 10.1084/jem.179.4.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.MacPherson GG, Jenkins CD, Stein MJ, Edwards C. Endotoxin-mediated dendritic cell release from the intestine: characterization of released dendritic cells and TNF dependence. J Immunol. 1995;154:1317–1322. [PubMed] [Google Scholar]
- 37.Stagg AJ, Hart AL, Knight SC, Kamm MA. The dendritic cell: its role in intestinal inflammation and relationship with gut bacteria. Gut. 2003;52:1522–1529. doi: 10.1136/gut.52.10.1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Malmström V, Shipton D, Singh B, Al-Shamkhani A, Puklavec MJ, Barclay AN, Powrie F. CD134L expression on dendritic cells in the mesenteric lymph nodes drives colitis in T cell-restored SCID mice. J Immunol. 2001;166:6972–6981. doi: 10.4049/jimmunol.166.11.6972. [DOI] [PubMed] [Google Scholar]
- 39.Demetter P, Van Huysse JA, De Keyser F, Van Damme N, Verbruggen G, Mielants H, De Vos M, Veys EM, Cuvelier CA. Increase in lymphoid follicles and leukocyte adhesion molecules emphasizes a role for the gut in spondyloarthropathy pathogenesis. J Pathol. 2002;198:517–522. doi: 10.1002/path.1235. [DOI] [PubMed] [Google Scholar]
- 40.Vremec D, Pooley J, Hochrein H, Wu L, Shortman K. CD4 and CD8 Expression by dendritic cell subtypes in mouse thymus and spleen. J Immunol. 2000;164:2978–2986. doi: 10.4049/jimmunol.164.6.2978. [DOI] [PubMed] [Google Scholar]
- 41.Liu YJ. Dendritic cell subsets and lineages, and their functions in innate and adaptive immunity. Cell. 2001;106:259–262. doi: 10.1016/s0092-8674(01)00456-1. [DOI] [PubMed] [Google Scholar]
- 42.Jankovic D, Kullberg MC, Caspar P, Sher A. Parasite-induced Th2 polarization is associated with down-regulated dendritic cell responsiveness to Th1 stimuli and a transient delay in T lymphocyte cycling. J Immunol. 2004;173:2419–2427. doi: 10.4049/jimmunol.173.4.2419. [DOI] [PubMed] [Google Scholar]
- 43.Moser M, Murphy KM. Dendritic cell regulation of Th1-Th2 development. Nat Immunol. 2000;1:199–205. doi: 10.1038/79734. [DOI] [PubMed] [Google Scholar]
- 44.Iwasaki A, Kelsall BL. Freshly isolated Peyer’s patch, but not spleen, dendritic cells produce interleukin 10 and induce the differentiation of T helper type 2 cells. J Exp Med. 1999;190:229–239. doi: 10.1084/jem.190.2.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kelleher P, Maroof A, Knight SC. Retrovirally induced switch from production of IL-12 to IL-4 in dendritic cells. Eur J Immunol. 1999;29:2309–2318. doi: 10.1002/(SICI)1521-4141(199907)29:07<2309::AID-IMMU2309>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 46.Manickasingham SP, Edwards AD, Schulz O, Reis e Sousa C. The ability of murine dendritic cell subsets to direct T helper cell differentiation is dependent on microbial signals. Eur J Immunol. 2003;33:101–107. doi: 10.1002/immu.200390001. [DOI] [PubMed] [Google Scholar]
- 47.Straw AD, MacDonald AS, Denkers EY, Pearce EJ. CD154 plays a central role in regulating dendritic cell activation during infections that induce Th1 or Th2 responses. J Immunol. 2003;170:727–734. doi: 10.4049/jimmunol.170.2.727. [DOI] [PubMed] [Google Scholar]
- 48.Cervi L, MacDonald AS, Kane C, Dzierszinski F, Pearce EJ. Cutting edge: dendritic cells copulsed with microbial and helminth antigens undergo modified maturation, segregate the antigens to distinct intracellular compartments, and concurrently induce microbe-specific Th1 and helminth-specific Th2 responses. J Immunol. 2004;172:2016–2020. doi: 10.4049/jimmunol.172.4.2016. [DOI] [PubMed] [Google Scholar]
- 49.Igietseme JU, Ananaba GA, Bolier J, Bowers S, Moore T, Belay T, Eko FO, Lyn D, Black CM. Suppression of endogenous IL-10 gene expression in dendritic cells enhances antigen presentation for specific Th1 induction: potential cellular vaccine development. J Immunol. 2000;164:4212–4219. doi: 10.4049/jimmunol.164.8.4212. [DOI] [PubMed] [Google Scholar]
- 50.Burke F, Stagg AJ, Bedford PA, English N, Knight SC. IL-10-producing B220+ CD11c− APC in mouse spleen. J Immunol. 2004;173:2362–2372. doi: 10.4049/jimmunol.173.4.2362. [DOI] [PubMed] [Google Scholar]
- 51.Aseffa A, Gumy A, Launois P, MacDonald HR, Louis JA, Tacchini-Cottier F. The early IL-4 response to Leishmania major and the resulting Th2 cell maturation steering progressive disease in BALB/c mice are subject to the control of regulatory CD4+CD25+ T cells. J Immunol. 2002;169:3232–3241. doi: 10.4049/jimmunol.169.6.3232. [DOI] [PubMed] [Google Scholar]
- 52.Xu D, Liu H, Komai-Koma M, Campbell C, McSharry C, Alexander J, Liew FY. CD4+CD25+ regulatory T cells suppress differentiation and functions of Th1 and Th2 cells, Leishmania major infection, and colitis in mice. J Immunol. 2003;170:394–399. doi: 10.4049/jimmunol.170.1.394. [DOI] [PubMed] [Google Scholar]
- 53.Hisaeda H, Maekawa Y, Iwakawa D, Okada H, Himeno K, Kishihara K, Tsukumo S, Yasutomo K. Escape of malaria parasites from host immunity requires CD4+CD25+ regulatory T cells. Nat Med. 2004;10:29–30. doi: 10.1038/nm975. [DOI] [PubMed] [Google Scholar]
- 54.Mendez S, Reckling SK, Piccirillo CA, Sacks D, Belkaid Y. Role for CD4+CD25+ regulatory T cells in reactivation of persistent leishmaniasis and control of concomitant immunity. J Exp Med. 2004;200:201–210. doi: 10.1084/jem.20040298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.O’Garra A, Vieira P. Regulatory T cells and mechanisms of immune system control. Nat Med. 2004;10:801–805. doi: 10.1038/nm0804-801. [DOI] [PubMed] [Google Scholar]
- 56.Groux H, O’Garra A, Bigler M, Rouleau M, Antonenko S, de Vries JE, Roncarolo MG. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature. 1997;389:737–742. doi: 10.1038/39614. [DOI] [PubMed] [Google Scholar]
- 57.McKee AS, Pearce EJ. CD25+CD4+ cells contribute to Th2 polarization during helminth infection by suppressing Th1 response development. J Immunol. 2004;173:1224–1231. doi: 10.4049/jimmunol.173.2.1224. [DOI] [PubMed] [Google Scholar]