

Abstract
Objectives
To examine the medical status of children with biliary atresia (BA) with their native livers after hepatic portoenterostomy (HPE) surgery.
Study design
The Childhood Liver Disease Research and Education Network (ChiLDREN) database was utilized to examine subjects with BA living with their native livers 5 or more years after HPE and to describe the prevalence of subjects with BA with an “ideal” outcome, defined as no clinical evidence of chronic liver disease, normal liver biochemical indices (aspartate aminotransferase, alanine aminotransferase, gamma glutamyl transpeptidase, platelet count, total bilirubin, International Normalized Ratio, and albumin) and normal Health-Related Quality of Life (HRQOL) 5 or more years after HPE.
Results
Children with BA (n=219; 43% male) with median age 9.7 years were studied. Median age at HPE was 56 (range 7-125) days. Median age- and sex-adjusted height and weight Z-scores at 5 year follow-up were 0.487 (interquartile range [IQR]: -0.27 to 1.02) and 0.00 (IQR: -0.74 to 0.70), respectively. During the 12 preceding months, cholangitis and bone fractures occurred in 17% and 5.5%, respectively. HRQOL was reported normal by 53% of patients. However, only 1.8% met the study definition of “ideal” outcome. Individual tests of liver synthetic function (TB, Alb, and INR) were normal in 75%, 85% and 73% of the study cohort.
Conclusion
Cholangitis and fractures in long-term survivors underscore the importance of ongoing medical surveillance. Over 98% of this North American cohort of subjects with BA living with native livers 5 or more years after HPE have clinical or biochemical evidence of chronic liver disease.
Keywords: Outcomes, Health status, Health-related quality of life, cholangitis
Biliary atresia (BA) is a progressive fibro-obliterative cholangiopathy presenting only in infancy with a prevalence ranging from 1 in 5,000 to 18,000 newborns1. Hepatoportoenterostomy (HPE; the Kasai procedure) provides a means of relieving extrahepatic biliary obstruction and permitting bile flow, but is not a curative procedure2, 3. Despite HPE being performed in a timely fashion, liver transplantation (LT) is ultimately required for the majority of patients during childhood4, 5. Although single center 6-8 and multi-center 9-11 characterizations of the health status and medical outcomes in patients with BA who have undergone LT are available, the long-term outcomes in older children with BA living with their native livers have not been examined in detail in large patient populations, and are limited to small single-center retrospective experiences12-18. Progression to chronic liver disease was avoided in only 11% of 244 ten-year survivors of HPE performed at King's College Hospital over the 12 year period of 1979 to 199119. Detailed information from a contemporary multi-center population of older subjects with BA surviving with their native livers currently does not exist, and would aid clinicians in providing important and generalizable information to families about school-aged children with BA.
Established in 2002, the Childhood Liver Disease Research and Education Network (ChiLDREN; originally known as the Biliary Atresia Research Consortium [BARC]) is an NIDDK/NIH-funded cooperative research consortium at 15 clinical sites in the United States and Canada with the goal of understanding the etiology, pathogenesis, course and outcomes of BA as well as other pediatric cholestatic conditions. Herein, the objectives of this cross-sectional study were to utilize enrollment data available within the ChiLDREN database to characterize the medical status of a multi-center cohort of subjects with BA who were 5 years of age or greater at time of study enrollment and still living with their native livers following HPE and to identify the percentage of subjects with BA who have no clinical findings of chronic liver disease, normal health related quality of life (HRQOL) and normal laboratory indices at least 5 years after HPE.
Methods
The Biliary Atresia Study of Infants and Children (BASIC) is one of the ongoing longitudinal studies within ChiLDREN with a specific aim to determine natural history and outcomes of older non-transplanted children with BA. Patient inclusion criteria for the BASIC protocol (ClinicalTrials.gov study ID: NCT00345553) were subjects with: (1) a confirmed diagnosis of BA determined by chart review, including review of pertinent diagnostic biopsy reports, radiologic reports and surgical reports; (2) age ≥6 months; and (3) either native livers or post- liver transplantation; and iv) a parent/guardian willing to provide informed consent (and, when appropriate, the subject is willing to assent). For this present cross-sectional analysis, subjects were identified from the BASIC database with patient age between 5 and 17.99 years at time of BASIC enrollment, and at least one year of follow-up at a ChiLDREN study site in the year preceding BASIC enrollment. Importantly, to reduce the bias of assessing patients with BA with severe disease that were unlikely to have long-term survival with native liver, subjects were excluded from this analysis if they were already listed for LT at time of BASIC enrollment.
All of the participating ChiLDREN centers had institutional review board and/or research ethics board approval for this study20. Written informed consent was obtained from parents and/or guardians and assent obtained from subjects age 7 years and older.
At BASIC entry, demographic, clinical and laboratory data were collected, physical examinations performed, and past medical histories (including medical events in the previous 12 months abstracted from the medical record) were recorded. Definitions and specific criteria for complications of chronic liver disease were standardized within the ChiLDREN protocol. Diagnosis of cholangitis required presence of fever of >38 degrees Celsius without other obvious clinical source of infection; new onset of acholic stools, right upper quadrant pain or tenderness; and both elevation of direct bilirubin by 25% and at least >1 mg/dL above previous baseline; however, positive bacterial or fungal culture (of blood or liver) was not required. Hepatopulmonary syndrome (HPS) required documentation of hypoxemia with pulse oximetry (transcutaneous saturation) levels of less than 94% and evidence of intrapulmonary shunting by contrast echocardiography with agitated saline21.
Data collected included sex, race, date of birth (DOB), ethnicity, date of HPE, associated congenital malformations22, and medications. Physical exam findings included weight, height, head circumference, anthropometry, liver and spleen size on examination. Laboratory indices included complete blood count (CBC) with differential, International Normalized Ratio (INR), liver biochemical results and basic metabolic panel. Growth data were expressed as Z-scores relative to published age-adjusted normative values23.
Given the healthcare complexities involved in the care of a child with chronic medical condition, an overall framework for outcome assessment based on a hierarchy of outcome measures can provide caregivers and parents a measure of the observed versus expected health status of the individual patient24. Understanding the extent to which the health of the infant with BA is restored to normal following HPE is important as a benchmark by which future interventions can be measured. For the purposes of this present analysis, “ideal” clinical outcome in BA (criteria variables provided in Table II) as derived by the ChiLDREN investigators was defined as the combination of i) normal liver biochemical test values, ii) absence of selected clinical complications of chronic liver disease (in the entire life of the subject), iii) absence of the need for additional medications specifically indicated for underlying liver disease; and iv) normal self-reported HRQOL. These criteria were dichotomized to facilitate a “yes” or “no” answer from review of the BASIC data collection forms. Normal liver laboratory values were defined as: serum total bilirubin (TB) ≤ 1.5 mg/dL, aspartate aminotransferase (AST) ≤45 IU/L, alanine aminotransferase (ALT) ≤ 40 IU/L, γ-glutamyltranspeptidase (GGT) ≤ 55 U/L, albumin (Alb)≥ 3.3 g/dL, INR ≤ 1.3, and platelet count >150 × 109/L. Absence of clinical chronic liver disease was defined as no report of ascites, hepatopulmonary syndrome, variceal bleeding and/or pathological bone fractures, in conjunction with the presence of age-adjusted growth (both weight and height) Z-scores > -2.0, and no reported use of cholangitis prophylactic antibiotics. Normal HRQOL was defined as PedsQL™ 4.0 Generic Core Scale total score scores >69.7 for child self-report or >65.4 for parent proxy – threshold scores representing one standard deviation below the population mean25, 26. Given the paucity of literature on good outcomes for patients with BA, we elected to perform a secondary analysis on our multi-center contemporareous cohort using the criteria for good patient outcome reported by Hadzic et al. (TB<1.2 mg/dL (20 umol/L), AST <50 IU/L, Alb>3.5 g/dL, INR <1.2, and platelet count >150 × 109)19.
Table 2. The “Ideal” subject with BA surviving with native livers for at least 5 years after HPE.
| Medical Variable: Result reported at 5 year visit | Patient data available, n | Patients who answered “yes” to variable as phrased n (%) | Patients missing data n (%) |
|---|---|---|---|
| Stability of Native livers – “Normal” Lab Indices | |||
| 1. TB level normal (≤ 1.5 mg/dl) | 197 | 164 (74.9%) | 22 (10%) |
| 2. AST level normal (≤ 45 IU/L) | 196 | 75 (34.2%) | 23 (10.5%) |
| 3. ALT level normal (≤ 40 IU/L) | 199 | 75 (34.2%) | 20 (9.1%) |
| 4. GGT level normal (≤ 55 u/l) | 186 | 82 (37.4%) | 33 (15.1%) |
| 5. INR normal (≤ 1.3) | 167 | 160 (73.1%) | 52 (23.7%) |
| 6. Serum Alb level normal (≥ 3.3 g/dl) | 196 | 187 (85.4%) | 23 (10.5%) |
| 7. Platelet count (≥ 150 × 103/mm3) | 173 | 78 (35.6%) | 46 (21%) |
| TOTAL # Subjects with ALL above Lab Indices normal | 186 | 16 (7.3%) | 33 (15.1%) |
| Absence of known complications of chronic liver disease | |||
| 8. Ascites | 209 | 172 (78.5%) | 10 (4.6%) |
| 9. Hepatopulmonary syndrome | 201 | 198 (90.4%) | 18 (8.2%) |
| 10. Variceal bleeding | 198 | 178 (81.3%) | 21 (9.6%) |
| 11. Fractures | 192 | 159 (72.6%) | 27 (12.3%) |
| 12. Age-adjusted weight z-score > -2 | 205 | 204 (93.2%) | 14 (6.4%) |
| 13. Age-adjusted height z-score > -2 | 204 | 197 (90%) | 15 (6.8%) |
| Absence of need for additional medications | |||
| 14. No use of cholangitis prophylaxis | 219 | 201 (91.8%) | 0 |
| HRQOL | |||
| 15. PedsQL Total Score ≥ 69.7 (normal) | 161 | 117 (53.4%) | 58 (26.5%) |
| >5-year native livers survivor with “ideal” outcomes* | 192 | 4 (1.8%) | 27 (12.3%) |
“Ideal” Outcomes defined as the presence of: i) normal liver biochemical tests; ii) absence of complications of chronic liver disease; iii) not receiving medications for liver disease; and iv) normal HRQOL
Statistical Analyses
This cross-sectional analysis used data collected at enrollment on subjects who were 5 years through 17.99 years at BASIC entry, and did not include longitudinal follow-up data. For dichotomous and categorical response variables, counts and percentages are reported. For continuous response variables, descriptive statistics (mean, standard deviation, minimum, median and maximum) are reported. When continuous response variables are dichotomized (e.g., when TB ≤ 1.5 mg/dl is classified as “ideal” outcome, and TB >1.5 mg/dl is classified as “not ideal” outcome), counts and percentages are reported for the dichotomized version. Parallel child self-report and parent proxy-report versions of the PedsQL™ Generic Core Scales were reviewed, with higher PedsQL™ Total Scale scores (scale of 0 to 100) indicating better HRQOL. All statistical analyses were conducted using SAS v9.3 (SAS Institute, Cary, North Carolina).
Results
As of May 1, 2012, 432 subjects with BA living with their native livers were enrolled in BASIC from May 2006 to May 2012. Excluded from further evaluation were 187 subjects enrolled before age 5 years (n=163) or after age 18 years (n=24). Among the remaining 245 subjects, we further excluded those already listed for LT at time of study enrollment (n=26). The remaining 219 (43% male, 63% white) subjects with BA with their native livers (median patient age 9.7 [range 5.1-17.9] years) were included in this analysis (Table I). Median subject age at HPE was 56 (range 7-125) days, and associated malformations included polysplenia in 5 (2.3%), congenital heart disease in 23 (10.5%), and other gastrointestinal malformations in 25 (11.4%), including abdominal heterotaxia in 3 (1.4%), malrotation in 3 (1.4%), midline liver in 2(0.9%), preduodenal portal vein in 2 (0.9%), and right-sided stomach in 1 (0.5%) subject.
Table 1. Characteristics of Study Cohort of 219 subjects with BA.
| Characteristic | N (%) |
|---|---|
|
| |
| # patients | 219 |
|
| |
| Sex | |
| -male | 95 (43.4) |
| -female | 124 (56.6) |
|
| |
| Race | |
| -White | 139 (63.5) |
| -Black | 31 (14.1) |
| -Asian | 23 (10.5) |
| -Other | 26 (11.9) |
|
| |
| Age at Kasai (days) | |
| - N | 183 |
| - Mean ± SD | 56.4 ± 23.19 |
| - Median (Range) | 56 (7–125) |
|
| |
| Current age at evaluation (years) | |
| - N | 219 |
| - Mean ± SD | 10.5 ± 3.98 |
| - Median (Range) | 9.7 (5.1–17.9) |
|
| |
| Polysplenia syndrome | 5 (2.3) |
|
| |
| Other congenital malformations present | |
| -Cardiac | 23 (10.5%) |
| -Gastrointestinal | 25 (11.4%) |
|
| |
| Biochemical* characteristics at enrollment; median (IQR) | |
| -TB, mg/dL | 0.8 (0.4–1.2) |
| -AST, IU/L | 55 (38–99) |
| -ALT, IU/L | 54 (30–98) |
| -Alb, g/dL | 4.2 (3.8–4.5) |
| -INR | 1.1 (1.0–1.2) |
| -platelet count, ×109/L | 138 (85–209) |
| -GGT, IU/L | 67 (29–161) |
Normal liver laboratory values were defined as: serum total bilirubin (TB) ≤ 1.5 mg/dL, aspartate aminotransferase (AST) ≤45 IU/L, alanine aminotransferase (ALT) ≤ 40 IU/L, γ-glutamyltranspeptidase (GGT) ≤ 55 U/L, albumin ≥ 3.3 6g/dL, INR ≤ 1.3, and platelet count >150 × 109/L
Physical examination
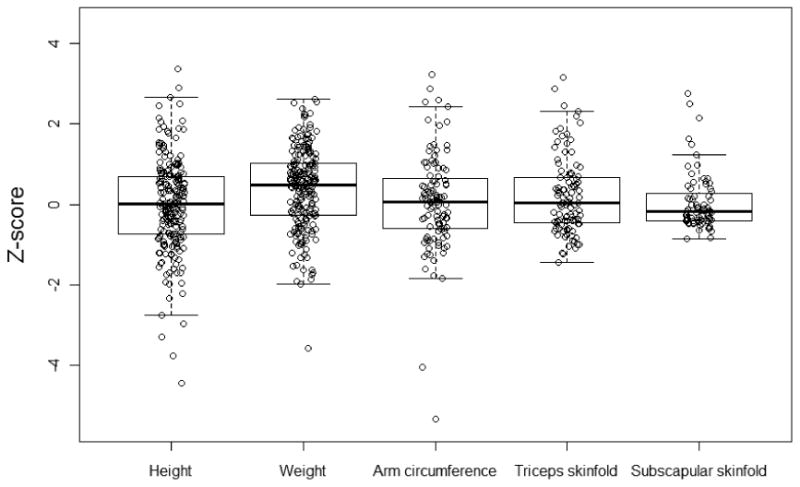
The median weight and height Z-scores at BASIC enrollment were 0.49 (interquartile range [IQR] -0.27, 1.02) and 0.00 (IQR: -0.74 to 0.70), respectively. Only 1 (0.5%) and 7 (3.2%) children had age-adjusted weight Z-score or height Z-score less than -2.0 (Table II). Nine (4.4%) of 205 subjects had a weight Z-score greater than 2.0. Mean Z-scores for right mid-arm circumference (0.06), right triceps skinfold thickness (0.03), and right subscapular skinfold thickness (-0.18) were within the range of published norms for healthy children (Figure 1). Median spleen size measured below the left costal margin was 2 cm (IQR: 0 to 6). Ninety-seven (44%) subjects had spleen size ≤2 cm below the costal margin.
Figure 1.
Boxplots of age-adjusted anthropometric Z-scores in subjects with BA. From top to bottom, the five horizontal lines represent the largest data point, which is not more than 1.5 times the interquartile range from the box, the 3rd quartile, the median, the 1st quartile, and the smallest data point which is no more than 1.5 times the interquartile range from the box, respectively.
Biochemical Outcomes
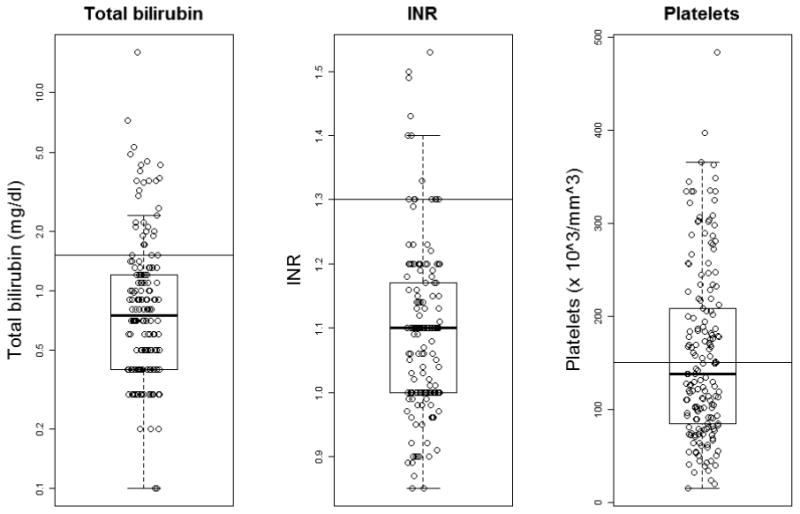
At BASIC study enrollment, median serum values included: total bilirubin 0.8 (IQR: 0.4 to 1.2) mg/dL; AST 55 (IQR: 38 to 99) IU/L; ALT 54 (IQR: 30 to 98) IU/L; albumin 4.2 (IQR: 3.8 to 4.5) g/dL; INR 1.1 (IQR: 1 to 1.2); and platelet count 138 (IQR: 85 to 209) ×109/L. The median GGT of this cohort was 67 (IQR: 29 to 161) U/L. Individual tests of liver synthetic function (TB, Alb, and INR) were normal in 75%, 85% and 73% of the study cohort. Platelet count was ≥150 ×109/L in 78 (35.6%) subjects. Figure 2 shows the distribution of TB, INR and platelet counts for the study cohort. Sixteen (7.3%) study subjects had normal values for the study biochemical composite of TB, AST, ALT, GGT, INR, Alb and platelet counts (Table II).
Figure 2.
Boxplots of serum total bilirubin, INR, and platlet count. From top to bottom, the five horizontal lines represent the largest data point, which is not more than 1.5 times the interquartile range from the box, the 3rd quartile, the median, the 1st quartile, and the smallest data point which is no more than 1.5 times the interquartile range from the box, respectively.
Complications
136 (62.1%) of our study cohort experienced cholangitis at least once since HPE. Multiple cholangitis episodes occurred in 11 subjects (7 patients with 2 episodes, and 4 patients with 3 episodes). Cholangitis occurred within the 12 months preceding BASIC enrollment in 36 (17%) of subjects, at a median subject age of 7.6 years. Ascites and variceal hemorrhage were seen in 17% and 9% of this study cohort. Thirty-three (15%) subjects with BA experienced a bone fracture since HPE, with fracture site available in 14 instances. Fractures affecting the upper extremity (forearm, arm, buckle wrist, hand, radial bone, arm, or thumb) were most common (8/14). Twelve (36%) fractures occurred within the 12 months preceding BASIC enrollment, of which 2 were receiving vitamin D supplementation at enrollment. There was no correlation between the presence of a bone fracture within the preceding year and serum TB levels.
Treatment and Care Interventions
Medications received by this cohort at enrollment included vitamin supplementation (132, 60.3%), ursodeoxycholic acid (122, 55.7%), non-selective beta blockers (7, 3.2%) and diuretics (1, 0.5%). Eighteen (8%) subjects were receiving antibiotics for cholangitis prophylaxis. Among the 36 (16%) subjects who experienced cholangitis in the preceding 12 months, 8 (22%) were receiving antibiotic prophylaxis and 17 (47%) ursodeoxycholic acid. Only 7 (21%) of the 33 subjects who reported a history of a bone fracture reported taking vitamin D supplements.
HRQOL
117 (53.4%) of our cohort of >5 year survivors with native livers had a self-reported PedsQL Total Scale Score greater than 69.7 (>-1 standard deviation below the mean score of healthy children), amongst which 41 (35%) had not experienced any clinical complications of chronic liver disease. Parent proxy-report was substituted for five subjects for whom patient self-report was not completed.
Composite profile of the subject with BA with native liver 5 years after HPE
Table II lists the clinical, biochemical, medication history and HRQOL criteria for identifying subjects who had achieved an “ideal” clinical outcome. These criteria are dichotomized to facilitate a “yes” or “no” response to the presence or absence of physical examination findings and selected laboratory data-points meeting specified target ranges. Four (1.8%) subjects fulfilled the study criteria for an “ideal clinical outcome” with “yes” to all 15 elements (Table II). Answering “no” to 1 or more variables (125 subjects) and the presence of 1 or more missing data elements (90 subjects) contributed to the remaining 215 (98.2%) subjects enrolled in BASIC who did not meet the criteria for the “ideal clinical outcome” at 5 or more years after HPE (Table III; available at www.jpeds.com).
Table 3. (Online):Characterization of 129 Study Participants with Complete Data Available for “Ideal” Clinical Outcome Composite Determination.
| All Lab Indices Normal | Absent known CLD complications | Absence of need for additional medication | Normal HRQOL | FREQ |
|---|---|---|---|---|
| 2 | ||||
| 3 | ||||
| 12 | ||||
| 31 | ||||
| 4 | ||||
| 6 | ||||
| 16 | ||||
| 46 | ||||
| 2 | ||||
| 2 | ||||
| 1 | ||||
| 4 |
Each shaded cell represents the presence of a post-HPE subcategory required for the study criteria of “ideal” clinical outcome in BA patients. A total of 129 subjects had information on all four categories. Achieving 4 shaded cells (normal lab indices, absence of known complications of chronic liver disease (CLD), absence of medication requirements, and normal HRQOL) are required for the composite “ideal outcome” status – and achieved in 4 patients. Patients with <4 categories (n=90) were ineligible for ideal outcome determination, and thus are not represented in this Table.
Given our multi-center contemporaneous cohort, we analyzed our cohort utilizing the variables previously published by Hadzic et al19 in their 12 year review of patients with BA living with native liver 10 years after HPE performed at King's College Hospital in the United Kingdom. Twenty-two (10%) subjects in our cohort met this single center study's research definition of no clinical evidence of chronic liver disease and normal serum TB, AST, Alb, INR, and platelet count. This group of subjects did not have a significantly different median age at HPE when compared with our overall series (51.5 versus 57 days, p>0.05).
Discussion
This multi-center cross-sectional analysis of a contemporary cohort of 219 subjects with BA who were not listed for LT and living with their native livers 5 or more years after HPE, with a median age of 9.7 years, demonstrates a number of important findings. Normal growth was achievable in over 90%. Although tests of liver synthetic function were normal in 75%, other liver biochemical abnormalities and clinical complications of chronic liver disease were experienced by over 90% by 5 years following HPE. Almost half of subjects self-reported impaired HRQOL. These findings provide benchmark data that will be useful for designing interventional study protocols, for stratification estimates and sample size calculations for clinical trials, and for counseling parents about expectations following HPE in BA for those children who escape the early need for LT.
Failure to thrive in BA is one of the more common indications for LT at a younger age5 and is an independent risk factor for pre-transplant mortality, post-transplant mortality, and liver graft failure27. Our group previously reported that early growth failure after HPE is associated with LT or death by 24 months of age28. In the present study, we found mean height and weight Z-scores to be normal at > 5 years after HPE. Acknowledging organomegaly and ascites as potential confounding variables with the risk of falsely representing a normal weight-for-age, the normal growth status was corroborated by concurrent normal Z-scores for mid-arm circumference, triceps skinfolds, and subscapular skinfolds (data not shown); furthermore few subjects in our group had ascites. These findings support previous reports that growth failure in children with successful HPE is uncommon, and that improving nutritional status may slow disease progression29, 30. Amongst this cohort of native liver survivors, only 4.4% of subjects were obese, in contrast to post-LT recipients, where up to 19% have been found to be obese31.
Cholangitis has been reported to occur in up to 60% of children with BA, and most commonly in the first 1 to 2 years after HPE32-34. However, over 37% of adult survivors with BA with native livers were diagnosed with cholangitis (median age at follow-up 24.7 years old)13, 14, 35. In our study, utilizing strict a priori-determined protocol definitions, an episode of cholangitis was diagnosed in 63% of children followed at least 5 years post HPE, with over a quarter (n=36) of these subjects experiencing an episode in the 12 months preceding enrollment into the BASIC study. Thus, the risk of cholangitis after HPE continues throughout early childhood32, as it does in survivors with BA over the age of 15 years15, 17. Wu et al32 previously reported that 50% of cholangitis episodes occurred in patients with BA not receiving prophylactic antibiotics. In our study, oral antibiotics for cholangitis prophylaxis were used in only 8.2% of all subjects with BA, with only 8 receiving antibiotic prophylaxis among the 36 (28%) who reported cholangitis within the preceding year. Because the current variation of clinical practice among ChiLDREN investigators ranges from discontinuation of prophylactic antibiotics at 2 years post HPE to never stopping antibiotics, there may be an opportunity for more effective prevention of cholangitis episodes. Because the efficacy of antibiotic prophylaxis has not been clearly determined at this age32, 36, 37, further prospective, randomized studies are indicated to provide the needed evidence upon which to establish best practices.
Pre-LT fracture prevalence rates in infants with BA have been reported to range between 8% and 35%38-41. Patients with BA are at risk for vitamin D deficiency; contributing factors include malabsorption of fat-soluble vitamins in the setting of cholestasis, lack of sunlight exposure, and poor hepatic 25-hydroxylation42, 43. We found that 14.6% of our study cohort was taking supplemental vitamin D. Although vitamin D deficiency was previously reported in 21-37% of patients with BA in the first year of life44, suggesting that current strategies for vitamin D supplementation in patients with BA are insufficient, little is known about vitamin D status in the older survivor with BA. Thirty-three (15%) of our >5-year native livers survivors had at least one bone fracture, with average age at time of fractures of 6.5 years for female and 6.4 years for males. A population based study in Finland found an overall annual fracture incidence of 163 per 10,000 for children ages 0 to 15 years, with a peak in fracture incidence at 10 years in girls and 14 years in boys45. Another study from northern Sweden reported an incidence of fractures of 240 per 10,000 among children and adolescents, with one-third of all children sustaining at least one fracture before the age of 17 years 46. Thus, our cohort appears to be at increased risk for bone fractures, an outcome that requires further investigation to develop evidence-based recommendations for prevention.
Portal hypertension (PHT) is a common consequence of BA and can lead to significant morbidity and mortality. Our group has previously characterized manifestations of PHT, and found definite PHT to be present in 49% of subjects enrolled in BASIC using an operational clinical definition of PHT (platelet count <150,000 and clinical splenomegaly, defined as >2 cm below the costal margin, or history of clinical complications including ascites and/or variceal hemorrhage and/or hepatopulmonary syndrome) 47. Failure to thrive and significant derangements of laboratory variables were not empirically observed in those with definite PHT. In our present study cohort, clinically detectable splenomegaly, thrombocytopenia, ascites and variceal hemorrhage were seen in 56%, 43%, 17% and 9% of patients with BA with native livers 5 or more years after HPE. Longitudinal follow-up of this group of patients will be helpful towards understanding the natural history and factors that predict progression of PHT and the development of adverse outcomes.
With reports of patients with BA entering their adult years with their native livers 13-15, 17, strategies started as early as possible to most fully optimize both the quality and quantity of life years will be important for families and health care provider teams. Given the complexities of chronic medical conditions, an outcome measure hierarchy may be applicable to assessing outcome in BA24. In this study, we specifically targeted historical data points, physical examination findings, and biochemical tests results routinely obtained in the ambulatory clinic surveillance of subjects with BA living with their native livers, in addition to HRQOL assessments obtained as per research protocol. Although 98% of five-year native livers survivors who were not listed for LT had clinical or biochemical evidence of chronic liver disease, it was intriguing that over half of these subjects still reported normal HRQOL, emphasizing the need to consider patient-reported subjective outcome variables in assessing clinical outcomes. Our group recently reported HRQOL in subjects with BA surviving with their native livers was similar to subjects with BA post-LT, although psychosocial and school functioning were significantly impaired in both groups compared with healthy children48. The similarities observed between the subjects with BA with their native livers and pediatric LT recipients were mainly driven by psychosocial problems rather than issues with physical health. Indeed, the importance of psychosocial problems, increasingly identified as “hidden morbidities” across an array of pediatric conditions, will continue to significantly impact generic HRQOL.
Older patients with BA surviving with their native livers in the United Kingdom were previously reported to be underperforming on social functioning compared with normative data; however lack of power due to sample size prevented achievement of statistical significance 16. Of 244 ten-year survivors of HPE at King's College Hospital, 11% had a normal physical examination and normal values for 5 liver laboratory tests. These authors concluded that progression to chronic liver disease is not inevitable in patients with BA. In our present study cohort, 10% of our subjects met these secondary analysis criteria for good outcome. However, inclusion of absence of complications of liver disease and acceptable HRQOL in our study reduced this to less than 2% of subjects with optimal outcomes. This 2% of subjects ranged in age from 6 to 17 years old, suggesting there may not necessarily be a cumulative incidence of complications. One other reason for the lower percentage with ideal outcome in our study may be that children who are doing well may have less regular clinical follow-up visits and potentially be missed for study recruitment.
Potential limitations and biases of this study relate to the retrospective nature of baseline data collection for a longitudinal clinical study. Our study relied on the analysis of historical data collected at the time of enrollment into the study. We acknowledge that this study may present an overly optimistic view of BA – those who suffered the most severe complications underwent LT at an earlier age or who were listed for LT were not included in this analysis, which may include those listed for failure to thrive. In our attempt to remove bias towards sicker patients who are being evaluated for LT, an opposite bias may have been created. However, our goal was to examine detailed information from a contemporary population of older subjects with BA with prolonged survival to provide clinicians with evidence about clinical outcomes for school-aged children with BA. Despite rigorous data quality controls, missing and incomplete data were present for variables due to differences in institutions' standard of care or the availability of the data. This is seen particularly in our analysis of the “ideal” clinical outcome, with 90 participants failing to meet “ideal” criteria simply because of missing data. In addition, baseline enrollment data did not include liver or bile duct histology or radiographic assessments, so these data could not be used as criteria for prediction of outcomes. Finally, each BASIC site was queried to ensure all available data was captured.
In conclusion, this study provides benchmarks for clinical outcomes expected in school age children with BA surviving with their native livers who are not listed for liver transplantation. Over 98% of this North American cohort of subjects with BA living with native livers 5 or more years after HPE have clinical or biochemical evidence of chronic liver disease or its consequences. Despite these findings, over half of patients rated their QOL as good. These data should prove helpful in the design of clinical interventions, stratification for clinical trials and as a baseline for longitudinal assessment of the natural history of BA. Moreover, this information will be of value in guiding expectations of families with infants and younger children with BA. Optimizing outcome is the ultimate goal for the clinician caring for children and teenagers with BA. Meticulous multidisciplinary family-focused care post-HPE is critical in order to improve outcomes beyond these benchmarks.
Acknowledgments
Supported by the National Institute of Diabetes, Digestive, and Kidney Diseases (NIDDK; U01 DK061693 and U54 DK078377; individual grants available in the Appendix), National Center for Research Resources (NCRR; 5M01 RR00069; individual grants available in the Appendix), and the Eunice Kennedy Shriver National Institute of Child Health and Human Development (R01 HD045694).
List of Abbreviations
- BA
Biliary atresia
- HPE
Kasai hepatoportoenterostomy
- LT
liver transplantation
- AST
aspartate aminotransferase
- ALT
alanine aminotransferase
- GGT
gamma glutamyltranspeptidase
- INR
International Normalized Ratio
- HRQOL
Health-related quality of life
Appendix
Members of CHiLDREN include
Johns Hopkins School of Medicine (supported by NIDDK DK62503 and NCATS UL1TR000424), Baltimore, MD: Kathleen Schwarz, MD, Robert Anders, MD, PhD, Paul Colombani, MD, Kim Kafka, RN, BSN, Wikrom Karnsakul, MD, Laura Wachter; Ann & Robert H. Lurie Children's Hospital of Chicago (supported by NIDDK DK62436 and NCATS UL1TR000150), Chicago, IL: Peter Whitington, MD, Estella Alonso, MD, Lee Bass, MD, Elizabeth Kaurs, Sue Kelly, RN, BSN, Hector Melin-Aldana, MD, Denise Rizzo, PharmD, Ricardo Superina, MD; Cincinnati Children's Hospital Medical Center (supported by NIDDK DK 62497 and NCATS UL1TR000077), Cincinnati, OH: Jorge Bezerra, MD, Kevin Bove, MD, Julie Denlinger, Andrea Ferris, James Heubi, MD, Pinky Jha, Denise LaGory, RPh, Alexander Miethke, MD, Joseph Palermo, MD, Stacey Reed, Kenneth Setchell, PhD, Melissa Stamper, Greg Tiao, MD; Children's Hospital Colorado (supported by NIDDK U54DK078377 and DK62453, and NCATS UL1TR000154), Aurora, CO: Ronald Sokol, MD, Tim Byrne, Joan Hines, MPH, Michelle Hite, Frederick Karrer, MD, Mark Lovell, MD, Cara Mack, MD, Todd Miller, Michael Narkewicz, MD, Timothy Schardt, PharmD, BCPS, Frederick Suchy, MD, Shikha Sundaram, MD, Tracy Urban, Johan Van Hove, MD; Mount Sinai School of Medicine (supported by NIDDK DK62445 and NCATS UL1TR000067), New York, NY: Ronen Arnon, MD, Jamie Chu, MD, Ivy Cohen, Brandy Haydel, Kishore Iyer, MD, Margret Magid, MD, Sheetal Ramnath; The Children's Hospital of Philadelphia (supported by NIDDK DK62481 and NCATS UL1TR000003), Philadelphia, PA: Kathleen Loomes, MD, Lindsay Brown, Timothy Crisci, Jessi Erlichman, MPH, Alex Falsey, Alan Flake, MD, David Piccoli, MD, Elizabeth Rand, MD, Pierre Russo, MD, Nancy Spinner, PhD, Eileen Wu; Children's Hospital of Pittsburgh (supported by NIDDK DK62466 and NCATS UL1TR000005), Pittsburgh, PA: Benjamin Shneider, MD, Feras Alissa, MD, Amy Bookser, A'Delbert Bowen, MD, Kathy Bukauskas, RN, CCRC, Ronald Jaffe, MD, Douglas Lindblad, MD, George Mazariegos, MD, Roberto Ortiz-Aguayo, MD, David Perlmutter, MD, Sarangarajan Ranganathan, Erin Sandene, Stefan Scholz, MD, Rakesh Sindhi, MD, Donna Smith, Robert Squires, Jr., MD, Veena Venkat, MD, Jerry Vockley, MD, PhD; UCSF Children's Hospital (supported by NIDDK DK62500 and NCATS UL1TR000004), San Francisco, CA: Philip Rosenthal, MD, Laura Bull, PhD, Scott Fields, Shannon Fleck, Melvin Heyman, MD, Shinjiro Hirose, MD, Grace Kim, MD, Camille Langlois, John Roberts, MD; Washington University School of Medicine (supported by NIDDK DK62452 and NCATS UL1TR000448), St. Louis, MO: Yumirle Turmelle, MD, Laura Bontemps, Pat Dillon, MD, Kathleen Harris, Jeffrey Lowell, MD, FACS, Stacey Postma, Jonathan Rider, RPh, David Rudnick, MD, PhD, Janis Stoll, Alexander Weymann, MD, Frances White, MD; Texas Children's Hospital (supported by NIDDK DK62470), Houston, TX: Paula Hertel, MD, Zoe Apted, BA, Mary Brandt, MD, Jameisha Brown, Beth Carter, MD, Milton Finegold, MD, Richard Gibbs, PhD, Sanjiv Harpavat, MD, Tara McCartney, RPh; Saint Louis University School of Medicine, St. Louis, MO: Jeffery Teckman, MD; Vikki Kociela, BSN, CCRC; Riley Hospital for Children (supported by NIDDK DK84536), Indianapolis, IN: Jean Molleston, MD, Molly Bozic, MD, Beth Byam, RN, Oscar Cummings, MD, Ann Klipsch, RN, Cindy Sawyers, BSRT, Girish Subbarao, MD, Karen West; Seattle Children's Hospital, Seattle (supported by NIDDK DK84575), WA: Karen Murray, MD, Kara Cooper, Laura Finn, MD, Patrick Healey, MD, Simon Horslen, MD, Susan Jacob, Melissa Young; The Hospital for Sick Children, Toronto, Ontario, CA: Vicky Ng, MD, Catherine Chung, MD, Maria DeAngelis, Annie Fecteau, MD, Wardah Hannan, Kelsey Hunt, Binita Kamath, MD, Jacob Langer, MD, Constance O'Connor, Claudia Quammie, Stephanie Rankin, Krista Van Roestel; Children's Hospital Los Angeles (supported by NIDDK DK84538 and NCATS UL1TR000130), Los Angeles, CA: Kasper Wang, MD, Henri Ford, MD, MHA, Evelyn Franco, Catherine Goodhue, CPNP, Nanda Kerker, MD, Sonia Michail, MD, John Pech, Danny Thomas, MD, Larry Wang, MD, PhD; Children's Healthcare of Atlanta (supported by NIDDK DK84585), Atlanta, GA: Saul Karpen, MD, PhD, Carlos Abramowsky, MD, Matthew Clifton, Liezl de la Cruz-Tracey, CCRC, Nikita Gupta, MD, Dana Hankerson-Dyson, Jim Rhodes, PharmD, Rene Romero, MD, Bahig Shehata, MD, Rita Tory, Taieshia Turner-Green, Mariam Vos, MD, MSPH, Allison Wellons; King's College Hospital, London, UK: Richard Thompson; National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD: Sherry Brown, MS, Edward Doo, MD, Jay Hoofnagle, MD, Averell Sherker, MD, FRCP, Rebecca Torrance, RN, MSN; University of Michigan Data Coordinating Center (supported by NIDDK DK62456), Ann Arbor, MI: John Magee, MD, Donna DiFranco, Karen Jones, Beverley Marchant, Trivellore Raghunathan, PhD, Cathie Spino, DSc, Wen Ye, PhD
Footnotes
The authors declare no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chardot C. Biliary atresia. Orphanet J Rare Dis. 2006;1:28. doi: 10.1186/1750-1172-1-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sokol RJ, Mack C, Narkewicz MR, Karrer FM. Pathogenesis and outcome of biliary atresia: current concepts. J Pediatr Gastroenterol Nutr. 2003;37:4–21. doi: 10.1097/00005176-200307000-00003. [DOI] [PubMed] [Google Scholar]
- 3.Davenport M. Biliary atresia: clinical aspects. Semin Pediatr Surg. 2012;21:175–84. doi: 10.1053/j.sempedsurg.2012.05.010. [DOI] [PubMed] [Google Scholar]
- 4.McDiarmid SV, Anand R, Lindblad AS. Studies of Pediatric Liver Transplantation: 2002 update. An overview of demographics, indications, timing, and immunosuppressive practices in pediatric liver transplantation in the United States and Canada. Pediatr Transplant. 2004;8:284–94. doi: 10.1111/j.1399-3046.2004.00153.x. [DOI] [PubMed] [Google Scholar]
- 5.Shneider BL, Mazariegos GV. Biliary atresia: a transplant perspective. Liver Transpl. 2007;13:1482–95. doi: 10.1002/lt.21303. [DOI] [PubMed] [Google Scholar]
- 6.Chen CL, Concejero A, Wang CC, Wang SH, Lin CC, Liu YW, et al. Living donor liver transplantation for biliary atresia: a single-center experience with first 100 cases. Am J Transplant. 2006;6:2672–9. doi: 10.1111/j.1600-6143.2006.01528.x. [DOI] [PubMed] [Google Scholar]
- 7.Fouquet V, Alves A, Branchereau S, Grabar S, Debray D, Jacquemin E, et al. Long-term outcome of pediatric liver transplantation for biliary atresia: a 10-year follow-up in a single center. Liver Transpl. 2005;11:152–60. doi: 10.1002/lt.20358. [DOI] [PubMed] [Google Scholar]
- 8.Diem HV, Evrard V, Vinh HT, Sokal EM, Janssen M, Otte JB, et al. Pediatric liver transplantation for biliary atresia: results of primary grafts in 328 recipients. Transplantation. 2003;75:1692–7. doi: 10.1097/01.TP.0000062570.83203.A3. [DOI] [PubMed] [Google Scholar]
- 9.Barshes NR, Lee TC, Balkrishnan R, Karpen SJ, Carter BA, Goss JA. Orthotopic liver transplantation for biliary atresia: the U.S. experience. Liver Transpl. 2005;11:1193–200. doi: 10.1002/lt.20509. [DOI] [PubMed] [Google Scholar]
- 10.Ng V, Fecteau A, Shepherd R, Magee J, Bucuvalas J, Alonso E, et al. Outcomes of 5-year survivors of pediatric liver transplantation: report on 461 children from a north american multicentre registry. Pediatrics. 2008;122:e1128–e35. doi: 10.1542/peds.2008-1363. [DOI] [PubMed] [Google Scholar]
- 11.Ng VL, Alonso EM, Bucuvalas JC, Cohen G, Limbers CA, Varni JW, et al. Health status of children alive 10 years after pediatric liver transplantation performed in the US and Canada: report of the studies of pediatric liver transplantation experience. J Pediatr. 2012;160:820–6 e3. doi: 10.1016/j.jpeds.2011.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Altman RP, Lilly JR, Greenfeld J, Weinberg A, van Leeuwen K, Flanigan L. A multivariable risk factor analysis of the portoenterostomy (Kasai) procedure for biliary atresia: twenty-five years of experience from two centers. Ann Surg. 1997;226:348–53. doi: 10.1097/00000658-199709000-00014. discussion 53-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kumagi T, Drenth JP, Guttman O, Ng V, Lilly L, Therapondos G, et al. Biliary atresia and survival into adulthood without transplantation: a collaborative multicentre clinic review. Liver Int. 2012;32:510–8. doi: 10.1111/j.1478-3231.2011.02668.x. [DOI] [PubMed] [Google Scholar]
- 14.Shinkai M, Ohhama Y, Take H, Kitagawa N, Kudo H, Mochizuki K, et al. Long-term outcome of children with biliary atresia who were not transplanted after the Kasai operation: >20-year experience at a children's hospital. J Pediatr Gastroenterol Nutr. 2009;48:443–50. doi: 10.1097/mpg.0b013e318189f2d5. [DOI] [PubMed] [Google Scholar]
- 15.Lykavieris P, Chardot C, Sokhn M, Gauthier F, Valayer J, Bernard O. Outcome in adulthood of biliary atresia: a study of 63 patients who survivied for over 20 years with their native liver. Hepatology. 2005;41:366–71. doi: 10.1002/hep.20547. [DOI] [PubMed] [Google Scholar]
- 16.Howard ER, MacLean G, Nio M, Donaldson N, Singer J, Ohi R. Survival patterns in biliary atresia and comparison of quality of life of long-term survivors in Japan and England. J Pediatr Surg. 2001;36:892–7. doi: 10.1053/jpsu.2001.23965. [DOI] [PubMed] [Google Scholar]
- 17.Nio M, Ohi R, Hayashi Y, Endo N, Ibrahim M, Iwami D. Current status of 21 patients who have survived more than 20 years since undergoing surgery for biliary atresia. J Pediatr Surg. 1996;31:381–4. doi: 10.1016/s0022-3468(96)90742-3. [DOI] [PubMed] [Google Scholar]
- 18.Laurent J, Gauthier F, Bernard O, Hadchouel M, Odievre M, Valayer J, et al. Long-term outcome after surgery for biliary atresia. Study of 40 patients surviving for more than 10 years. Gastroenterology. 1990;99:1793–7. doi: 10.1016/0016-5085(90)90489-n. [DOI] [PubMed] [Google Scholar]
- 19.Hadzic N, Davenport M, Tizzard S, Singer J, Howard ER, Mieli-Vergani G. Long-term survival following Kasai portoenterostomy: is chronic liver disease inevitable? J Pediatr Gastroenterol Nutr. 2003;37:430–3. doi: 10.1097/00005176-200310000-00006. [DOI] [PubMed] [Google Scholar]
- 20.Shneider BL, Brown MB, Haber B, Whitington PF, Schwarz K, Squires R, et al. A multicenter study of the outcome of biliary atresia in the United States, 1997 to 2000. J Pediatr. 2006;148:467–74. doi: 10.1016/j.jpeds.2005.12.054. [DOI] [PubMed] [Google Scholar]
- 21.Machicao V, Fallon MB. Hepatopulmonary syndrome. Semin Respir Crit Care Med. 2012;33:11–6. doi: 10.1055/s-0032-1301730. [DOI] [PubMed] [Google Scholar]
- 22.Schwarz KB, Haber BH, Rosenthal P, Mack CL, Moore J, Bove K, et al. Extrahepatic Anomalies in Infants With Biliary Atresia: Results of a Large Prospective North American Multicenter Study. Hepatology. 2013 doi: 10.1002/hep.26512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Centers for Disease Control and Prevention. Growth Charts. Atlanta: 2010. [Google Scholar]
- 24.Porter ME. What is value in health care? N Engl J Med. 2010;363:2477–81. doi: 10.1056/NEJMp1011024. [DOI] [PubMed] [Google Scholar]
- 25.Varni JW, Seid M, Kurtin PS. PedsQL 4.0: reliability and validity of the Pediatric Quality of Life Inventory version 4.0 generic core scales in healthy and patient populations. Med Care. 2001;39:800–12. doi: 10.1097/00005650-200108000-00006. [DOI] [PubMed] [Google Scholar]
- 26.Varni JW, Burwinkle TM, Seid M, Skarr D. The PedsQL 4.0 as a pediatric population health measure: feasibility, reliability, and validity. Ambul Pediatr. 2003;3:329–41. doi: 10.1367/1539-4409(2003)003<0329:tpaapp>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 27.Utterson EC, Shepherd RW, Sokol RJ, Bucuvalas J, Magee JC, McDiarmid SV, et al. Biliary atresia: clinical profiles, risk factors, and outcomes of 755 patients listed for liver transplantation. J Pediatr. 2005;147:180–5. doi: 10.1016/j.jpeds.2005.04.073. [DOI] [PubMed] [Google Scholar]
- 28.DeRusso PA, Ye W, Shepherd R, Haber BA, Shneider BL, Whitington PF, et al. Growth failure and outcomes in infants with biliary atresia: a report from the Biliary Atresia Research Consortium. Hepatology. 2007;46:1632–8. doi: 10.1002/hep.21923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karrer FM, Price MR, Bensard DD, Sokol RJ, Narkewicz MR, Smith DJ, et al. Long-term results with the Kasai operation for biliary atresia. Arch Surg. 1996;131:493–6. doi: 10.1001/archsurg.1996.01430170039006. [DOI] [PubMed] [Google Scholar]
- 30.Valayer J. Conventional treatment of biliary atresia: long-term results. J Pediatr Surg. 1996;31:1546–51. doi: 10.1016/s0022-3468(96)90174-8. [DOI] [PubMed] [Google Scholar]
- 31.Sundaram SS, Alonso EM, Zeitler P, Yin W, Anand R. Obesity After Pediatric Liver Transplantation: Prevalence and Risk Factors. J Pediatr Gastroenterol Nutr. 2012;55:657–62. doi: 10.1097/MPG.0b013e318266243c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu ET, Chen HL, Ni YH, Lee PI, Hsu HY, Lai HS, et al. Bacterial cholangitis in patients with biliary atresia: impact on short-term outcome. Pediatr Surg Int. 2001;17:390–5. doi: 10.1007/s003830000573. [DOI] [PubMed] [Google Scholar]
- 33.Hung PY, Chen CC, Chen WJ, Lai HS, Hsu WM, Lee PH, et al. Long-term prognosis of patients with biliary atresia: a 25 year summary. J Pediatr Gastroenterol Nutr. 2006;42:190–5. doi: 10.1097/01.mpg.0000189339.92891.64. [DOI] [PubMed] [Google Scholar]
- 34.Ecoffey C, Rothman E, Bernard O, Hadchouel M, Valayer J, Alagille D. Bacterial cholangitis after surgery for biliary atresia. J Pediatr. 1987;111:824–9. doi: 10.1016/s0022-3476(87)80195-6. [DOI] [PubMed] [Google Scholar]
- 35.Nio M, Sano N, Ishii T, Sasaki H, Hayashi Y, Ohi R. Cholangitis as a late complication in long-term survivors after surgery for biliary atresia. J Pediatr Surg. 2004;39:1797–9. doi: 10.1016/j.jpedsurg.2004.08.021. [DOI] [PubMed] [Google Scholar]
- 36.Bu LN, Chen HL, Chang CJ, Ni YH, Hsu HY, Lai HS, et al. Prophylactic oral antibiotics in prevention of recurrent cholangitis after the Kasai portoenterostomy. J Pediatr Surg. 2003;38:590–3. doi: 10.1053/jpsu.2003.50128. [DOI] [PubMed] [Google Scholar]
- 37.Mones RL, DeFelice AR, Preud'Homme D. Use of neomycin as the prophylaxis against recurrent cholangitis after Kasai portoenterostomy. J Pediatr Surg. 1994;29:422–4. doi: 10.1016/0022-3468(94)90583-5. [DOI] [PubMed] [Google Scholar]
- 38.DeRusso PA, Spevak MR, Schwarz KB. Fractures in biliary atresia misinterpreted as child abuse. Pediatrics. 2003;112:185–8. doi: 10.1542/peds.112.1.185. [DOI] [PubMed] [Google Scholar]
- 39.Guichelaar MM, Schmoll J, Malinchoc M, Hay JE. Fractures and avascular necrosis before and after orthotopic liver transplantation: long-term follow-up and predictive factors. Hepatology. 2007;46:1198–207. doi: 10.1002/hep.21805. [DOI] [PubMed] [Google Scholar]
- 40.Hill SA, Kelly DA, John PR. Bone fractures in children undergoing orthotopic liver transplantation. Pediatr Radiol. 1995;25(Suppl 1):S112–7. [PubMed] [Google Scholar]
- 41.Hogler W, Baumann U, Kelly D. Endocrine and bone metabolic complications in chronic liver disease and after liver transplantation in children. J Pediatr Gastroenterol Nutr. 2012;54:313–21. doi: 10.1097/MPG.0b013e31823e9412. [DOI] [PubMed] [Google Scholar]
- 42.Compston JE. Hepatic osteodystrophy: vitamin D metabolism in patients with liver disease. Gut. 1986;27:1073–90. doi: 10.1136/gut.27.9.1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Klein GL, Soriano H, Shulman RJ, Levy M, Jones G, Langman CB. Hepatic osteodystrophy in chronic cholestasis: evidence for a multifactorial etiology. Pediatr Transplant. 2002;6:136–40. doi: 10.1034/j.1399-3046.2002.01060.x. [DOI] [PubMed] [Google Scholar]
- 44.Shneider BL, Magee JC, Bezerra JA, Haber B, Karpen SJ, Raghunathan T, et al. Efficacy of Fat-Soluble Vitamin Supplementation in Infants With Biliary Atresia. Pediatrics. 2012;130:e607–e14. doi: 10.1542/peds.2011-1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mayranpaa MK, Makitie O, Kallio PE. Decreasing incidence and changing pattern of childhood fractures: A population-based study. J Bone Miner Res. 2010;25:2752–9. doi: 10.1002/jbmr.155. [DOI] [PubMed] [Google Scholar]
- 46.Hedstrom EM, Svensson O, Bergstrom U, Michno P. Epidemiology of fractures in children and adolescents. Acta Orthop. 2010;81:148–53. doi: 10.3109/17453671003628780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shneider BL, Abel B, Haber B, Karpen SJ, Magee JC, Romero R, et al. Portal hypertension in children and young adults with biliary atresia. J Pediatr Gastroenterol Nutr. 2012;55:567–73. doi: 10.1097/MPG.0b013e31826eb0cf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sundaram SS, Alonso EM, Haber B, Magee JC, Fredericks E, Kamath B, et al. Health related quality of life in patients with biliary atresia surviving with their native liver. J Pediatr. 2013;163:1052–7 e2. doi: 10.1016/j.jpeds.2013.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]