

Abstract
Aplastic anemia is a bone marrow failure syndrome that causes pancytopenia and can lead to life-threatening complications. Bone marrow transplantation remains the standard of care for younger patients and those with a good performance status but many patients may not have a suitable donor. Immunosuppressive therapy is able to resolve cytopenias in a majority of patients with aplastic anemia but relapses are not uncommon and some patients remain refractory to this approach. Patients may require frequent blood and platelet transfusion support which is expensive and inconvenient. Life-threatening bleeding complications still occur despite prophylactic platelet transfusion. Thrombopoietin (TPO) mimetics, such as romiplostim and eltrombopag, were developed to treat patients with refractory immune thrombocytopenia but are now being investigated for the treatment of bone marrow failure syndromes. TPO is the main regulator for platelet production and its receptor (c-Mpl) is present on megakaryocytes and hematopoietic stem cells. Trilineage hematopoietic responses were observed in a recent clinical trial using eltrombopag in patients with severe aplastic anemia refractory to immunosuppression suggesting that these agents can provide a new therapeutic option for enhancing blood production. In this review, we discuss these recent results and ongoing investigation of TPO mimetics for aplastic anemia and other bone marrow failure states like myelodysplastic syndromes. Clonal evolution or progression to acute myeloid leukemia remains a concern when using these drugs in bone marrow failure and patients should only be treated in the setting of a clinical trial.
Keywords: Aplastic anemia, Eltrombopag, Thrombopoietin (TPO) mimetics, Bone marrow failure, Thrombocytopenia, Myelodysplastic syndromes (MDS)
Introduction
Aplastic anemia is a rare, life-threatening bone marrow failure disorder characterized by pancytopenia and a hypocellular bone marrow. Most cases are due to autoimmune attack of marrow stem and progenitor cells leading to pancytopenia [1]. Allogeneic bone marrow transplantation offers the opportunity for cure in younger patients, but most are not suitable candidates for transplantation due to advanced age or lack of a histocompatible donor. Comparable long-term survival in severe aplastic anemia is attainable with standard immunosuppressive treatment (IST). However, of those patients treated with IST, one-quarter to one-third will not respond, and 30–40 % of responders relapse. These patients can have persistent thrombocytopenia, require regular platelet transfusions, which are expensive and inconvenient, and are at risk for further serious bleeding complications.
Pathophysiology of thrombocytopenia in aplastic anemia
Thrombocytopenia is a major cause of morbidity and mortality in patients with aplastic anemia. At presentation, virtually all patients with aplastic anemia are thrombocytopenic: platelet counts of <50,000 or 20,000/μL are diagnostic criteria for moderate and severe aplastic anemia, respectively. Decreased hematopoietic stem and progenitor cell numbers and function, resulting in impaired megakaryocytopoiesis and insufficient mature platelet production, are the causes of thrombocytopenia.
The clinical efficacy of immunosuppression and much laboratory data suggest that the ultimate mechanism leading to hematopoietic stem and progenitor depletion is immune-mediated attack and destruction [1, 2]. The frequency of the most primitive hematopoietic cells is profoundly decreased and, even with clinical response to immunosuppressive therapy, can remain persistently low, as reflected by studies utilizing long-term culture-initiating cells (LT-ICs) and cobblestone formation. Stem cell numbers are markedly diminished and transcriptome analysis demonstrates a pro-apoptotic profile for CD34+ cells from aplastic anemia patients compared to healthy controls, signifying stem cells under stress [3, 4]. This severely diminished population of stem cells subsequently leads to a paucity of megakaryocytes capable of producing platelets.
Clinical consequences of thrombocytopenia
Thrombocytopenia in aplastic anemia causes bleeding, usually petechiae and ecchymoses of the skin and mucous membranes, epistaxis and gingival hemorrhage. Bleeding is not typically observed until the platelet count falls below 10–20,000/μL. Life-threatening bleeding can occur in the presence of accompanying physical lesions related to the underlying aplastic anemia or treatment with immunosuppression, such as corticosteroid-related gastritis, or neutropenia-related fungal infection of the lungs [5, 6]. Intracranial hemorrhage is most feared, as this complication can be life threatening or severely debilitating if not promptly treated [7]. Introduction of routine platelet transfusions in patients with aplastic anemia led to a clear reduction of bleeding complications. Yet, even in the modern era when platelet transfusions are widely used both prophylactically and therapeutically, in long-term follow-up studies as many as 10 % of patients presenting with aplastic anemia still eventually die of bleeding [8]. There is also evidence for asymptomatic hemorrhage, such as cerebral microbleeds, although their clinical significance is currently unknown [9].
Treatment of thrombocytopenia
The main treatment for thrombocytopenia related to bone marrow failure has been platelet transfusion support, while awaiting a response to IST or engraftment following allogeneic stem cell transplantation. Months typically are required for platelet counts to respond following IST, and time from diagnosis to a sibling or particularly a matched unrelated transplant can be even longer. Routine practice is to provide prophylactic platelet transfusions to avoid significant bleeding in these intervals. Most guidelines recommend transfusing patients with thrombocytopenia prophylactically when platelets fall to <10,000/μL, or in patients with fevers or a bleeding history,<20,000/μL [10]. However, it is important to realize that the clinical evidence supporting transfusion thresholds was gathered primarily from patients with hematologic malignancies undergoing chemotherapy or stem cell transplantation, not aplastic anemia, and these thresholds remain controversial.
Using these thresholds for transfusion typically requires patients to receive transfusions once or twice weekly due to the short half-life of platelets. The frequency of transfusions is dependent on the residual platelet production, the half-life of the platelets transfused, and whether the patient is significantly alloimmunized. Splenomegaly and platelet sequestration are not generally a problem in AA. Prior to the introduction of leukocyte depleted blood products in the 1980s, a significant proportion of patients developed HLA alloimmunization and became refractory to platelet transfusions. In addition, studies demonstrated that the number of pre-transplant transfusions was associated with an increased risk of graft failure [11] and death [12] following hematopoietic stem cell transplant. Since the introduction of universal leukoreduction of blood products, the incidence of HLA alloimmunization appears to be low, although there are no formal studies in AA. In the trial to reduce alloimmunization to platelets (TRAP), patients with acute myelogenous leukemia (AML) were less likely to become refractory to platelet transfusions if they received filtered apheresis platelets [13]. However, it is our experience that even patients with alloimmunization and severe thrombocytopenia can survive without catastrophic bleeding for many years with HLA-matched platelets.
Hematopoietic growth factors
The clinical trials of hematopoietic growth factors, mainly erythropoietic stimulating agents (ESAs) and G-CSF and also GM-CSF and IL-3, to treat aplastic anemia have been disappointing. There is little rationale for use of cytokines that act on committed hematopoietic progenitors in specific differentiated lineages. First, since receptors for these cytokines are not present on primitive stem cells, and progenitors and lineage committed precursors able to respond are missing or profoundly reduced, treatment at best might be expected to shorten time to recovery of blood counts following therapy, once immunosuppression or transplantation allows some recovery of stem cell numbers. Second, endogenous erythropoietin and G-CSF levels are extremely elevated in SAA [14, 15]. Whereas in myelodysplastic syndrome (MDS), low endogenous levels of erythropoietin are present in a small proportion of patients but predict responsiveness to ESAs [16, 17]. Clinical trials have examined GM-CSF, G-CSF, stem cell factor (SCF) and IL-3, as single agents or added to standard immunosuppressive therapy for aplastic anemia. In this setting, G-CSF has been widely studied in combination with horse ATG plus CsA. This regimen has not been shown to improve outcomes in several randomized trials [18–20] and the most recent European guidelines for the management of aplastic anemia only support the use of a short course of G-CSF in the setting of severe systemic infections, but advise against their routine use entirely [10]. A recent meta-analysis of 19 individual trials concluded that there was no impact on survival or response rate with the addition of these cytokines to standard immunosuppressive regimens [21].
TPO receptor agonists for aplastic anemia
Thrombopoietin (TPO) is the principal endogenous regulator of platelet production. TPO is a potent endogenous cytokine that binds to its receptor (c-Mpl) on megakaryocyte progenitors and stimulates a number of signal transduction events promoting megakaryocyte proliferation, and maturation and platelet release [22]. TPO levels are greatly increased in aplastic anemia [23]. Conversely, in immune thrombocytopenia (ITP) TPO levels are within the normal range or only moderately increased, and this finding suggested the utility of a TPO receptor agonist in this clinical condition [23, 24]. However, the regulation of endogenous TPO production is not yet fully understood. In the “sponge” model, following constitutive synthesis by the liver, TPO is passively regulated by its binding to TPO receptors on platelets and megakaryocytes [25].
Multiple lines of evidence support a pleiotropic role for TPO in hematopoiesis, beyond its obvious function as the primary endogenous factor controlling platelet production. First, TPO receptors are present on primitive hematopoietic stem and progenitor cells [26]. Second, hematopoietic repopulating assays performed in mice support a role for TPO in hematopoietic stem cell survival, selfrenewal, and expansion [27, 28]. Third, TPO/c-Mpl signaling in humans plays a role in regulating quiescent hematopoietic stem cells [29]. Fourth, humans with bi-allelic mutations in c-Mpl develop congenital amegakaryocytic thrombocytopenia (CAMT); a disorder characterized by severe thrombocytopenia presenting shortly after birth. Patients with CAMT typically have absent or greatly diminished mega-karyocytes in the bone marrow and are at high risk for developing aplastic anemia [30]. Fifth, the equivalent Mpl knockout mouse model (Mpl(−/−)) demonstrates reduced numbers of hematopoietic stem and progenitor cells [27, 31, 32]. And finally, a homozygous nonsense mutation in the Mpl gene has been reported in association with familial aplastic anemia [33].
Although TPO levels are very elevated in bone marrow failure syndromes [23, 34], the hypothesis that supraphysiologic pharmacologic levels might be therapeutic is being explored with TPO receptor agonists. The lack of benefit of other growth factors, such as EPO and G-CSF, may be because these cytokines act on more committed myeloid progenitors. The ability for TPO to act on less differentiated progenitor cells supports a hypothetical role in treating bone marrow failure. Eltrombopag (Promacta), is a synthetic, nonpeptide, oral thrombopoietin mimetic that noncompetitively binds to c-Mpl in a region distinct from that of thrombopoietin. Upon binding to c-Mpl, eltrombopag activates signaling through JAK-STAT (Janus-associated kinase-signal transducers) and MAPK (mitogen-activated protein kinase) pathways. In a nonrandomized phase II trial conducted at the National Institutes of Health, 25 patients with severe aplastic anemia and thrombocytopenia refractory to immunosuppression were treated with eltrombopag [35]. All patients were transfusion dependent for platelets and had failed a median of 2 rounds of IST (range 1–4) prior to enrollment. The starting dose of eltrombopag was 50 mg daily (the typical starting dose in ITP), and the dose was escalated every 2 weeks by 25 mg if the platelet count remained below 20,000/μL to a maximum of 150 mg daily. All but one patient reached this maximum dose since eltrombopag was well tolerated and had few side effects. The primary endpoint was improvement in blood counts with defined responses for each lineage after 12 weeks. 44 % (11/25) of patients had a hematological response after 12–16 weeks, with responses in all 3 lineages seen. The majority of responders became transfusion independent for platelets (9/11) with an average eventual increase in platelet count of 44,000/μL. Remarkably, clinically significant increases in the erythroid lineage were observed in six patients (6/11) and nine patients had a neutrophil response (9/11). Of note, while responses were detected by 3–4 months on therapy, maximal and trilineage responses were slower, occurring in responding patients remaining on drug in an extension phase of the trial (Fig. 1). These clinical results support the hypothesis that c-Mpl stimulation can directly improve trilineage hematopoiesis.
Fig. 1. Multilineage hematologic responses to eltrombopag (venn diagrams show the number of patients with unilineage, bilineage, and trilineage hematologic responses). Adapted from Olnes et al. [35].

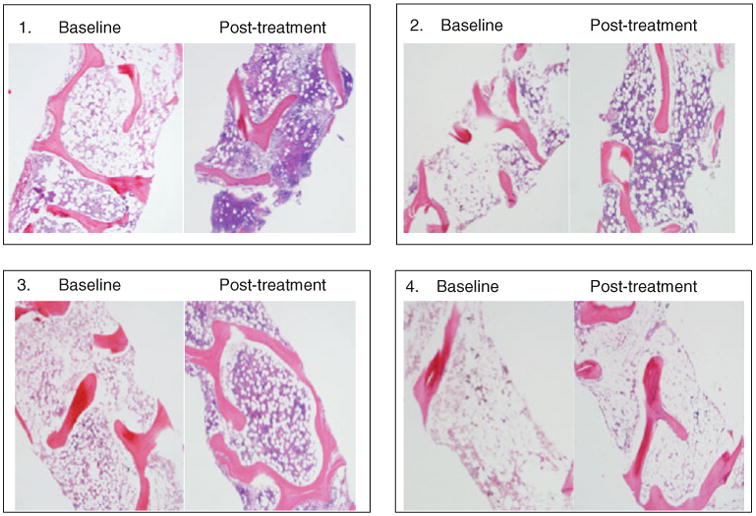
Serial bone marrow biopsies including a pre-treatment bone marrow biopsy were performed in order to monitor response and toxicity. Three of four patients with hematologic responses who were followed for more than 8 months had normalization of bone marrow cellularity that mirrored the normalization of their peripheral blood counts (Fig. 2). Previous studies using TPO mimetics to treat ITP suggested chronic TPO receptor stimulation might lead to bone marrow fibrosis [36]. However, in the current study for aplastic anemia there was no evidence to indicate myelofibrosis, even among patients who responded and underwent serial bone marrow biopsies every 6 months for up to 30 months. In ITP, patients treated with TPO mimetics have supranormal levels of megakaryocytes in order to keep up with platelet destruction, and fibrosis is likely related to release of fibrogenic cytokines by megakaryocytes.
Fig. 2. Bone marrow cellularity in patients with trilineage responses to eltrombopag. Adapted from Olnes et al. [35].
As expected, measurements of serum thrombopoietin levels of patients during the treatment were markedly elevated compared to healthy controls, but they remained unchanged over time. Analysis of leukocytes' telomere length and immunophenotypic panel of T cell subsets were also unrevealing for significant differences pre- or post-eltrombopag, or between that of responders and nonresponders.
While strategies are ongoing investigating this agent in naïve patients with severe aplastic anemia, in moderate aplastic anemia and in myelodysplastic syndromes, ours is currently the only published study of the use of a TPO mimetic for acquired bone marrow failure.
TPO receptor agonists for thrombocytopenia in myelodysplastic syndromes
Preclinical research showing that eltrombopag stimulates megakaryopoiesis in patients with myelodysplastic syndromes (MDS)[37], while potentially decreasing malignant cells [38] provide a rationale for testing TPO agonists in this setting. Furthermore, TPO levels are also elevated in MDS, but to a lesser extent as in aplastic anemia [39]. Romiplostim has been tested in patients with lower risk MDS and thrombocytopenia [40]. This single-arm study reported durable platelet responses in 46 % of patients with decreased bleeding events and platelet transfusion frequency. There were two cases (5 %) of progression to acute myeloid leukemia (AML) and four cases (9 %) of increases in blasts counts, which reversed on discontinuation of romiplostim. A subsequent randomized, double-blind, placebo controlled trial treating patients with low to intermediate risk-1 disease was terminated early due to more cases of AML progression in the treatment arm. In total 219 patients were randomized 2:1 to treatment with romiplostim (147 patients) to placebo (72 patients). There were 11 patients with progression to AML, 10 on the treatment arm versus 2 on the placebo arm, giving a hazard ratio (HR) of 2.51 for patients treated with romiplostim. Circulating myeloblast numbers increased to greater than 10 % in 28 patients, with 25 of these on the treatment arm. Eight of these patients were diagnosed with AML. A warning was subsequently added to the prescribing information, based on these results of increased blast cell counts and increased risk of progression to AML in patients with MDS. On long-term follow-up of this study published in abstract form and presented at ASH 2012 [41], however, two more cases of AML developed in the placebo arm that had not been included in the previous analysis; with a median follow-up of 17.8 months, AML rates were 8.9% for romiplostim and 8.5% for placebo with a HR of 1.2 for those on drug to progress to AML. Of interest, twelve of the 22 cases of AML were in patients who were RAEB-1.
Standard therapy for MDS includes hypomethylating agents and lenalidomide [42]. These agents are effective with about half of patients responding but have significant toxicity and commonly cause cytopenias [43, 44]. Romiplostim has been used in combination with standard therapy for low-risk MDS in an attempt to reduce complications from thrombocytopenia and to prevent course delays or dose reductions. A phase 2 study investigated adding romiplostim to patients with low- or intermediate-risk MDS receiving azacytadine [45]. Forty patients were randomized to either 500 or 750 lg of romiplostim or placebo subcutaneously once weekly during 4 cycles of azacytadine. There was no statistically significant difference in the primary endpoint of clinically significant thrombocytopenic events (CSTEs) defined as grade 3 or 4 thrombocytopenia (platelet count <50,000/μL), starting on day 15 of the first cycle or by a platelet transfusion at any time during the treatment period between groups although numbers were small. However, the higher dose of romiplostim did raise median platelet counts during cycle 3 on day 1 and at the nadir compared to placebo.
Romiplostim has been used in combination with the hypomethylating agent decitabine [46]. Patients with low-or intermediate-risk MDS were randomized to either placebo or romiplostim at 750 μg. The primary endpoint was CSTEs as defined in the romiplostim/azacytadine study. Similar to that study the numbers were too small to detect significant differences between the groups although there were trends towards higher platelet counts at the beginning of treatment cycles, lower rates of platelet transfusion and less bleeding events in the romiplostim treated group. These two studies suggest a modest clinical benefit to combining hypomethylating agents with romiplostim with no added toxicity. Neither of these small studies were adequately powered to detect a significant difference between placebo and romiplostim treatment groups and larger clinical trials may be needed.
Lenalidomide therapy while useful in patients with low-risk MDS causes thrombocytopenia in 44–74 % of patients [47, 48], which may lead to dose reduction and treatment interruptions. Combination therapy with a TPO agonist could, hypothetically, result in improved outcomes if dosing schedules could be maintained. This approach was investigated in a phase 2, placebo controlled, multicenter study which randomized 39 patients with low or intermediate-1 risk MDS to placebo, romiplostim at 500 or 750 μg dose while receiving four cycles of lenalidomide [49]. In the romiplostim groups, there was a trend towards higher percentage of patients achieving MDS treatment responses and lower percentage of patients who received transfusions during the four cycles. No romiplostim dose response was seen probably because of small numbers. There was a decreased risk of reduction or delay in lenalidomide dosing with 50 % of patients receiving placebo, 36 % in the 500 μg and 15 % in the 750 μg group requiring a reduction. This study also had too few patients to come to clear conclusions regarding the utility of combination therapy.
Preliminary data using eltrombopag in MDS have recently been reported orally and in abstract form at ASH 2012. Promising results were described from an ongoing Italian multicenter trial, which enrolled patients with low and intermediate-1 IPSS risk disease and platelet counts less than 30,000/μL [50]. By exclusion criteria, patients were ineligible for or refractory to other treatments. Eltrombopag commenced at 50 mg and was increased every 2 weeks to a maximum of 300 mg. At a median follow-up of 6 months, five of ten patients on drug had had a complete response as defined by a platelet count greater than 100,000μL and absence of bleeding. One patient in the treatment group had a response (R) defined as an increase in platelet count of ≥30,000/μL if baseline is >20,000/μL or if baseline <20,000/μL an increase of <20,000/μL and increase by at least 100 %, not due to transfusions. Just one patient had a response in the placebo arm and was called an ‘unstable response.’ There were no progressions in the treatment arm and one in the placebo arm. Eltrombopag has also been used in patients with advanced MDS, and preliminary results were presented in poster form at ASH 2012 by an independent group of investigators [51]. 17 patients with advanced MDS (int-2 to high risk by WHO criteria) or AML with thrombocytopenia were treated with eltrombopag for 8 weeks with dose increases if no platelet response was seen. 24 % of patients had a response. In all 11 patients with post baseline bone marrow examinations there were no clinically meaningful increase in blasts from pre-treatment. As these results have yet to be published in a peer reviewed journal, the data should be regarded as preliminary and not definitive.
Risk of clonal evolution or progression
Patients with aplastic anemia have a rate of clonal evolution of about 15 % over 10 years [52]. There is a concern that, as TPO agonists stimulate stem cells, they may also stimulate dysplastic or leukemic stem cells and precipitate or accelerate clonal evolution in these patients. Currently, it is unclear whether treatment with TPO receptor agonists may increase, or decrease, the risk of clonal evolution to myelodysplasia and leukemia in bone marrow failure. Despite the concern that some leukemia blast cells express TPO receptors, in vivo and in vitro studies thus far suggest eltrombopag inhibits proliferation of leukemic cells while continuing to stimulate megakaryopoiesis [38, 53, 54]. More clinical data are necessary to determine if this is more than just a theoretical risk. In the only published study using TPO agonists in aplastic anemia clonal evolution to monosomy 7 developed in 2 patients who did not have a response [35]. Long-term follow-up data will be crucial to investigate whether this is a valid concern in this group of drugs. Routine utilization of these drugs to treat patients with aplastic anemia or MDS is premature and not advised: we strongly warn against their use in routine practice until there is more information on long-term outcomes and risks.
Future directions
Current NIH protocols are testing eltrombopag in combination with immunosuppression for treatment-naïve severe aplastic anemia, and as a single agent in patients with moderate aplastic anemia and myelodysplastic syndrome. The majority of the hematologic responses observed following initial treatment with standard immunosuppression (h-ATG/CsA) for SAA are partial, with only a few patients achieving normal blood counts. The explanation for partial recovery and relapse are not fully understood, but incomplete elimination of autoreactive T cells and insufficient stem cell reserve are both possible. Furthermore, 10–15 % of SAA patients treated with standard immunosuppression will eventually develop an abnormal karyotype, with monosomy 7 being most common, which portends progression to myelodysplasia and leukemia. Clonal evolution is less frequent in complete responders to immunosuppression. Although horse ATG/CsA represented a major advance in the treatment of SAA, refractoriness, incomplete responses, relapse, and clonal evolution limit the success of this modality. Thus, newer regimens are needed to address these limitations, and provide a better alternative to stem cell transplantation.
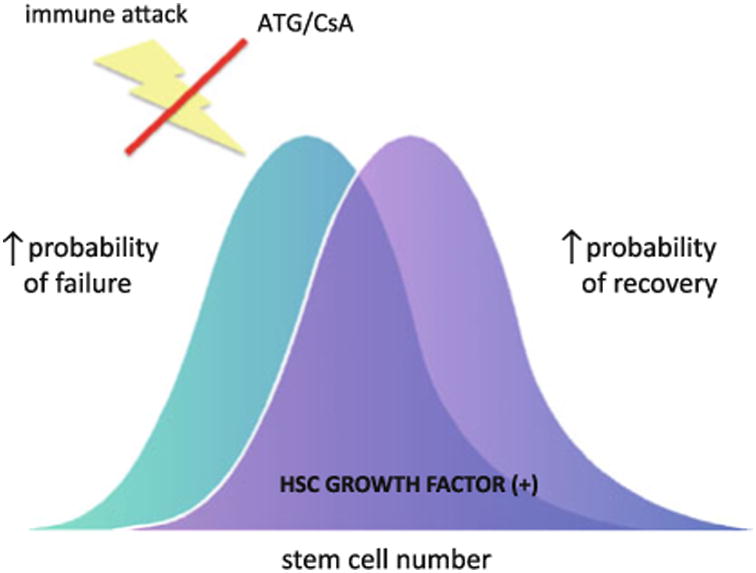
One approach to augment the quality of hematologic responses to immunosuppression therapy is to improve underlying stem cell function (Fig. 3). Previous attempts to improve responses in SAA with hematopoietic cytokines, including erythropoietin, G-CSF, and stem cell factor, have proved unsuccessful. TPO has stimulatory effects on more primitive multilineage progenitors and stem cells in vitro and in animal models. The recent demonstration that eltrombopag can stimulate trilineage responses even amongst patients with profoundly refractory SAA, supports a role for TPO receptor agonists in stimulating stem cells. Further studies are necessary to investigate whether exogenous treatment with eltrombopag allows normalization of HSC numbers and/or function despite high levels of endogenous TPO, and why some patients appear to have a persistent deficit in HSC function despite absence of active immune attack. Eltrombopag specifically binds to the human and chimpanzee TPO receptor excluding mice as a useful model to study broad differences between eltrombopag and TPO. However, using an in vivo NOD/SCID mouse xenotransplant model, eltrombopag has been shown to selectively expand hematopoietic stem/progenitor cells in human umbilical cord blood [55]. Furthermore, signal transduction pathways favored stimulation of earlier HSC/HPC populations by eltrombopag.
Fig. 3. Potential Role for eltrombopag to augment responsiveness to ATG/CsA.
Footnotes
Conflict of interest The authors declare that they have no conflicts of interest or competing financial or personal relationships that could inappropriately influence the content of this article.
References
- 1.Young NS, Calado RT, Scheinberg P. Current concepts in the pathophysiology and treatment of aplastic anemia. Blood. 2006;108:2509–19. doi: 10.1182/blood-2006-03-010777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goto M, Kuribayashi K, Takahashi Y, Kondoh T, Tanaka M, Kobayashi D, Watanabe N. Identification of autoantibodies expressed in acquired aplastic anaemia. Br J Haematol. 2013;160:359–62. doi: 10.1111/bjh.12116. [DOI] [PubMed] [Google Scholar]
- 3.Maciejewski JP, Selleri C, Sato T, Anderson S, Young NS. A severe and consistent deficit in marrow and circulating primitive hematopoietic cells (long-term culture initiating cells) in acquired aplastic anemia. Blood. 1996;88:1983–91. [PubMed] [Google Scholar]
- 4.Zeng W, Chen G, Kajigaya S, Nunez O, Charrow A, Billings EM, Young NS. Gene expression profiling in CD34 cells to identify differences between aplastic anemia patients and healthy volunteers. Blood. 2004;103:325–32. doi: 10.1182/blood-2003-02-0490. [DOI] [PubMed] [Google Scholar]
- 5.Hochsmann B, Moicean A, Risitano A, Ljungman P, Schrezenmeier H. Supportive care in severe and very severe aplastic anemia. Bone Marrow Transplant. 2013;48:168–73. doi: 10.1038/bmt.2012.220. [DOI] [PubMed] [Google Scholar]
- 6.Munoz J, Hughes A, Guo Y. Mucormycosis-associated intracranial hemorrhage. Blood Coagulation Fibrinolysis Int J Haemostasis Thrombosis. 2013;24:100–1. doi: 10.1097/MBC.0b013e32835a72df. [DOI] [PubMed] [Google Scholar]
- 7.Yamasaki M, Akagi K, Niinomi K, Kinoshita S, Kitawaki T, Yoshioka K. Intracranial hemorrhage associated with aplastic anemia. No to Hattatsu Brain Dev. 1989;21:215–21. [PubMed] [Google Scholar]
- 8.Viollier R, Passweg J, Gregor M, Favre G, Kuhne T, Nissen C, Gratwohl A, Tichelli A. Quality-adjusted survival analysis shows differences in outcome after immunosuppression or bone marrow transplantation in aplastic anemia. Ann Hematol. 2005;84:47–55. doi: 10.1007/s00277-004-0930-3. [DOI] [PubMed] [Google Scholar]
- 9.Sharma S, Malhotra P, Lal V, Singh P, Varma N, Varma S. Asymptomatic cerebral bleeds in patients with aplastic anemia. Ann Hematol. 2012;91:1187–91. doi: 10.1007/s00277-012-1448-8. [DOI] [PubMed] [Google Scholar]
- 10.Marsh JC, Ball SE, Cavenagh J, Darbyshire P, Dokal I, Gordon-Smith EC, Keidan J, Laurie A, Martin A, Mercieca J, et al. Guidelines for the diagnosis and management of aplastic anaemia. Br J Haematol. 2009;147:43–70. doi: 10.1111/j.1365-2141.2009.07842.x. [DOI] [PubMed] [Google Scholar]
- 11.Champlin RE, Horowitz MM, van Bekkum DW, Camitta BM, Elfenbein GE, Gale RP, Gluckman E, Good RA, Rimm AA, Rozman C, et al. Graft failure following bone marrow transplantation for severe aplastic anemia: risk factors and treatment results. Blood. 1989;73:606–13. [PubMed] [Google Scholar]
- 12.Piccin A, O'Marcaigh A, Smith O, O'Riordan J, Crowley M, Vandenberg E, Gardiner N, McCann S. Outcome of bone marrow transplantation in acquired and inherited aplastic anaemia in the Republic of Ireland. Ir J Med Sci. 2005;174:13–9. doi: 10.1007/BF03169141. [DOI] [PubMed] [Google Scholar]
- 13.Slichter SJ, Davis K, Enright H, Braine H, Gernsheimer T, Kao KJ, Kickler T, Lee E, McFarland J, McCullough J, et al. Factors affecting posttransfusion platelet increments, platelet refractoriness, and platelet transfusion intervals in thrombocytopenic patients. Blood. 2005;105:4106–14. doi: 10.1182/blood-2003-08-2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Watari K, Asano S, Shirafuji N, Kodo H, Ozawa K, Takaku F, Kamachi S. Serum granulocyte colony-stimulating factor levels in healthy volunteers and patients with various disorders as estimated by enzyme immunoassay. Blood. 1989;73:117–22. [PubMed] [Google Scholar]
- 15.Urabe A, Mitani K, Yoshinaga K, Iki S, Yagisawa M, Ohbayashi Y, Takaku F. Serum erythropoietin titers in hematological malignancies and related diseases. International journal of cell cloning. 1992;10:333–7. doi: 10.1002/stem.5530100604. [DOI] [PubMed] [Google Scholar]
- 16.Ferrini PR, Grossi A, Vannucchi AM, Barosi G, Guarnone R, Piva N, Musto P, Balleari E. A randomized double-blind placebo-controlled study with subcutaneous recombinant human erythropoietin in patients with low-risk myelodysplastic syndromes. Br J Haematol. 1998;103:1070–4. doi: 10.1046/j.1365-2141.1998.01085.x. [DOI] [PubMed] [Google Scholar]
- 17.Musto P, Lanza F, Balleari E, Grossi A, Falcone A, Sanpaolo G, Bodenizza C, Scalzulli PR, La Sala A, Campioni D, et al. Darbepoetin alpha for the treatment of anaemia in low-intermediate risk myelodysplastic syndromes. Br J Haematol. 2005;128:204–9. doi: 10.1111/j.1365-2141.2004.05288.x. [DOI] [PubMed] [Google Scholar]
- 18.Tichelli A, Schrezenmeier H, Socie G, Marsh J, Bacigalupo A, Duhrsen U, Franzke A, Hallek M, Thiel E, Wilhelm M, et al. A randomized controlled study in newly diagnosed severe aplastic anemia patients receiving antithymocyte globulin (ATG), cyclosporine, with or without G-CSF: a study of the SAA Working Party of the EBMT. Blood. 2011 doi: 10.1182/blood-2010-08-304071. [DOI] [PubMed] [Google Scholar]
- 19.Kojima S, Hibi S, Kosaka Y, Yamamoto M, Tsuchida M, Mugishima H, Sugita K, Yabe H, Ohara A, Tsukimoto I. Immunosuppressive therapy using antithymocyte globulin, cyclosporine, and danazol with or without human granulocyte colony-stimulating factor in children with acquired aplastic anemia. Blood. 2000;96:2049–54. [PubMed] [Google Scholar]
- 20.Marsh JC, Ganser A, Stadler M. Hematopoietic growth factors in the treatment of acquired bone marrow failure states. Semin Hematol. 2007;44:138–47. doi: 10.1053/j.seminhematol.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 21.Gurion R, Gafter-Gvili A, Paul M, Vidal L, Ben-Bassat I, Yeshurun M, Shpilberg O, Raanani P. Hematopoietic growth factors in aplastic anemia patients treated with immunosuppressive therapy systematic review and meta-analysis. Haematologica. 2009;94:712–9. doi: 10.3324/haematol.2008.002170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaushansky K. The molecular mechanisms that control thrombopoiesis. J Clin Investig. 2005;115:3339–47. doi: 10.1172/JCI26674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Emmons RV, Reid DM, Cohen RL, Meng G, Young NS, Dunbar CE, Shulman NR. Human thrombopoietin levels are high when thrombocytopenia is due to megakaryocyte deficiency and low when due to increased platelet destruction. Blood. 1996;87:4068–71. [PubMed] [Google Scholar]
- 24.Hirayama Y, Sakamaki S, Matsunaga T, Kuga T, Kuroda H, Kusakabe T, Sasaki K, Fujikawa K, Kato J, Kogawa K, et al. Concentrations of thrombopoietin in bone marrow in normal subjects and in patients with idiopathic thrombocytopenic purpura, aplastic anemia, and essential thrombocythemia correlate with its mRNA expression of bone marrow stromal cells. Blood. 1998;92:46–52. [PubMed] [Google Scholar]
- 25.Kuter DJ, Rosenberg RD. The reciprocal relationship of thrombopoietin (c-Mpl ligand) to changes in the platelet mass during busulfan induced thrombocytopenia in the rabbit. Blood. 1995;85:2720–30. [PubMed] [Google Scholar]
- 26.Zeigler FC, de Sauvage F, Widmer HR, Keller GA, Donahue C, Schreiber RD, Malloy B, Hass P, Eaton D, Matthews W. In vitro megakaryocytopoietic and thrombopoietic activity of c-mpl ligand (TPO) on purified murine hematopoietic stem cells. Blood. 1994;84:4045–52. [PubMed] [Google Scholar]
- 27.Kimura S, Roberts AW, Metcalf D, Alexander WS. Hematopoietic stem cell deficiencies in mice lacking c-Mpl, the receptor for thrombopoietin. Proc Natl Acad Sci USA. 1998;95:1195–200. doi: 10.1073/pnas.95.3.1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qian H, Buza-Vidas N, Hyland CD, Jensen CT, Antonchuk J, Mansson R, Thoren LA, Ekblom M, Alexander WS, Jacobsen SE. Critical role of thrombopoietin in maintaining adult quiescent hematopoietic stem cells. Cell Stem Cell. 2007;1:671–84. doi: 10.1016/j.stem.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 29.Yoshihara H, Arai F, Hosokawa K, Hagiwara T, Takubo K, Nakamura Y, Gomei Y, Iwasaki H, Matsuoka S, Miyamoto K, et al. Thrombopoietin/MPL signaling regulates hematopoietic stem cell quiescence and interaction with the osteoblastic niche. Cell Stem Cell. 2007;1:685–97. doi: 10.1016/j.stem.2007.10.020. [DOI] [PubMed] [Google Scholar]
- 30.Geddis AE. Congenital amegakaryocytic thrombocytopenia. Pediatr Blood Cancer. 2011;57:199–203. doi: 10.1002/pbc.22927. [DOI] [PubMed] [Google Scholar]
- 31.Heckl D, Wicke DC, Brugman MH, Meyer J, Schambach A, Busche G, Ballmaier M, Baum C, Modlich U. Lentiviral gene transfer regenerates hematopoietic stem cells in a mouse model for Mpl-deficient aplastic anemia. Blood. 2011;117:3737–47. doi: 10.1182/blood-2010-09-308262. [DOI] [PubMed] [Google Scholar]
- 32.Alexander WS, Roberts AW, Nicola NA, Li R, Metcalf D. Deficiencies in progenitor cells of multiple hematopoietic lineages and defective megakaryocytopoiesis in mice lacking the thrombopoietic receptor c-Mpl. Blood. 1996;87:2162–70. [PubMed] [Google Scholar]
- 33.Walne AJ, Dokal A, Plagnol V, Beswick R, Kirwan M, de la Fuente J, Vulliamy T, Dokal I. Exome sequencing identifies MPL as a causative gene in familial aplastic anemia. Haematologica. 2012;97:524–8. doi: 10.3324/haematol.2011.052787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hsu HC, Lee YM, Tsai WH, Jiang ML, Ho CH, Ho CK, Wang SY. Circulating levels of thrombopoietic and inflammatory cytokines in patients with acute myeloblastic leukemia and myelodysplastic syndrome. Oncology. 2002;63:64–9. doi: 10.1159/000065722. [DOI] [PubMed] [Google Scholar]
- 35.Olnes MJ, Scheinberg P, Calvo KR, Desmond R, Tang Y, Dumitriu B, Parikh AR, Soto S, Biancotto A, Feng X, et al. Eltrombopag and improved hematopoiesis in refractory aplastic anemia. N Engl J Med. 2012;367:11–9. doi: 10.1056/NEJMoa1200931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kuter DJ, Mufti GJ, Bain BJ, Hasserjian RP, Davis W, Rutstein M. Evaluation of bone marrow reticulin formation in chronic immune thrombocytopenia patients treated with romiplostim. Blood. 2009;114:3748–56. doi: 10.1182/blood-2009-05-224766. [DOI] [PubMed] [Google Scholar]
- 37.Mavroudi I, Pyrovolaki K, Pavlaki K, Kozana A, Psyllaki M, Kalpadakis C, Pontikoglou C, Papadaki HA. Effect of the non-peptide thrombopoietin receptor agonist eltrombopag on megakaryopoiesis of patients with lower risk myelodysplastic syndrome. Leuk Res. 2011;35:323–8. doi: 10.1016/j.leukres.2010.06.029. [DOI] [PubMed] [Google Scholar]
- 38.Will B, Kawahara M, Luciano JP, Bruns I, Parekh S, Erickson Miller CL, Aivado MA, Verma A, Steidl U. Effect of the non-peptide thrombopoietin receptor agonist Eltrombopag on bone marrow cells from patients with acute myeloid leukemia and myelodysplastic syndrome. Blood. 2009;114:3899–908. doi: 10.1182/blood-2009-04-219493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Feng X, Scheinberg P, Wu CO, Samsel L, Nunez O, Prince C, Ganetzky RD, McCoy JP, Jr, Maciejewski JP, Young NS. Cytokine signature profiles in acquired aplastic anemia and myelodysplastic syndromes. Haematologica. 2011;96:602–6. doi: 10.3324/haematol.2010.030536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kantarjian H, Fenaux P, Sekeres MA, Becker PS, Boruchov A, Bowen D, Hellstrom-Lindberg E, Larson RA, Lyons RM, Muus P, et al. Safety and efficacy of romiplostim in patients with lower risk myelodysplastic syndrome and thrombocytopenia. J Clin Oncol. 2010;28:437–44. doi: 10.1200/JCO.2009.24.7999. [DOI] [PubMed] [Google Scholar]
- 41.Kantarjian HM, Mufti GJ, Fenaux P, Sekeres MA, Szer J, Platzbecker U, Kuendgen A, Gaidano G, Wiktor-Jedrzejczak W, Bennett JM, et al. Treatment with the thrombopoietin (tpo)-receptor agonist romiplostim in thrombocytopenic patients (Pts) with low or intermediate-1 (int-1) risk myelodysplastic syndrome (MDS): Follow-up AML and survival results of a randomized, double-blind, placebo (PBO)-controlled study. Blood. 2012;120:8–11. [Google Scholar]
- 42.Santini V. Novel therapeutic strategies: hypomethylating agents and beyond. Hematology Am Soc Hematol Educ Program. 2012;2012:65–73. doi: 10.1182/asheducation-2012.1.65. [DOI] [PubMed] [Google Scholar]
- 43.Santini V, Fenaux P, Mufti GJ, Hellstrom-Lindberg E, Silverman LR, List A, Gore SD, Seymour JF, Backstrom J, Beach CL. Management and supportive care measures for adverse events in patients with myelodysplastic syndromes treated with azacitidine*. Eur J Haematol. 2010;85:130–8. doi: 10.1111/j.1600-0609.2010.01456.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van den Bosch J, Lubbert M, Verhoef G, Wijermans PW. The effects of 5-aza-2′-deoxycytidine (Decitabine) on the platelet count in patients with intermediate and high-risk myelodysplastic syndromes. Leuk Res. 2004;28:785–90. doi: 10.1016/j.leukres.2003.11.016. [DOI] [PubMed] [Google Scholar]
- 45.Kantarjian HM, Giles FJ, Greenberg PL, Paquette RL, Wang ES, Gabrilove JL, Garcia-Manero G, Hu K, Franklin JL, Berger DP. Phase 2 study of romiplostim in patients with low- or intermediate-risk myelodysplastic syndrome receiving azacitidine therapy. Blood. 2010;116:3163–70. doi: 10.1182/blood-2010-03-274753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Greenberg PL, Garcia-Manero G, Moore M, Damon L, Roboz G, Hu K, Yang AS, Franklin J. A randomized controlled trial of romiplostim in patients with low- or intermediate-risk myelodysplastic syndrome receiving decitabine. Leuk Lymphoma. 2013;54:321–8. doi: 10.3109/10428194.2012.713477. [DOI] [PubMed] [Google Scholar]
- 47.List A, Kurtin S, Roe DJ, Buresh A, Mahadevan D, Fuchs D, Rimsza L, Heaton R, Knight R, Zeldis JB. Efficacy of lenalidomide in myelodysplastic syndromes. N Engl J Med. 2005;352:549–57. doi: 10.1056/NEJMoa041668. [DOI] [PubMed] [Google Scholar]
- 48.List A, Dewald G, Bennett J, Giagounidis A, Raza A, Feldman E, Powell B, Greenberg P, Thomas D, Stone R, et al. Lenalidomide in the myelodysplastic syndrome with chromosome 5q deletion. N Engl J Med. 2006;355:1456–65. doi: 10.1056/NEJMoa061292. [DOI] [PubMed] [Google Scholar]
- 49.Wang ES, Lyons RM, Larson RA, Gandhi S, Liu D, Matei C, Scott B, Hu K, Yang AS. A randomized, double-blind, placebo-controlled phase 2 study evaluating the efficacy and safety of romiplostim treatment of patients with low or intermediate-1 risk myelodysplastic syndrome receiving lenalidomide. J Hematol Oncol. 2012;5:71. doi: 10.1186/1756-8722-5-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Oliva EN, Santini V, Zini G, Palumbo GA, Poloni A, Cortelezzi A, Voso MT, Molteni A, Sanpaolo G, Marino A, et al. Efficacy and safety of eltrombopag for the treatment of thrombocytopenia of low and intermediate-1 IPSS risk myelodysplastic syndromes: interim analysis of a prospective, randomized, single-blind, placebo-controlled trial (EQoL-MDS) Blood. 2012;120:20. [Google Scholar]
- 51.Mittelman M, Assouline S, Briasoulis E, Alonso A, Delgado RG, O'Gorman P, Kim HJ, Yoon SS, Zaritskey A, Flynn CM, et al. Eltrombopag treatment of thrombocytopenia in advanced myelodysplastic syndromes and acute myeloid leukemia: results of the 8-week open-label part of an ongoing study. ASH Annual Meeting Abstracts. 2012;120:3822–8. [Google Scholar]
- 52.Maciejewski JP, Selleri C. Evolution of clonal cytogenetic abnormalities in aplastic anemia. Leuk Lymphoma. 2004;45:433–40. doi: 10.1080/10428190310001602363. [DOI] [PubMed] [Google Scholar]
- 53.Roth M, Will B, Simkin G, Narayanagari S, Barreyro L, Bartholdy B, Tamari R, Mitsiades CS, Verma A, Steidl U. Eltrombopag inhibits the proliferation of leukemia cells via reduction of intracellular iron and induction of differentiation. Blood. 2012;120:386–94. doi: 10.1182/blood-2011-12-399667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mavroudi I, Pyrovolaki K, Pavlaki K, Kozana A, Psyllaki M, Kalpadakis C, Pontikoglou C, Papadaki HA. Effect of the non-peptide thrombopoietin receptor agonist eltrombopag on megakaryopoiesis of patients with lower risk myelodysplastic syndrome. Leuk Res. 2011;35:323–8. doi: 10.1016/j.leukres.2010.06.029. [DOI] [PubMed] [Google Scholar]
- 55.Sun H, Tsai Y, Nowak I, Liesveld J, Chen Y. Eltrombopag, a thrombopoietin receptor agonist, enhances human umbilical cord blood hematopoietic stem/primitive progenitor cell expansion and promotes multi-lineage hematopoiesis. Stem Cell Res. 2012;9:77–86. doi: 10.1016/j.scr.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]