

Abstract
Many proteins interact transiently with other proteins or are integrated into multi-protein complexes to perform their biological function. Bimolecular fluorescence complementation (BiFC) is an in vivo method to monitor such interactions in plant cells. In the presented protocol the investigated candidate proteins are fused to complementary halves of fluorescent proteins and the respective constructs are introduced into plant cells via agrobacterium-mediated transformation. Subsequently, the proteins are transiently expressed in tobacco leaves and the restored fluorescent signals can be detected with a confocal laser scanning microscope in the intact cells. This allows not only visualization of the interaction itself, but also the subcellular localization of the protein complexes can be determined. For this purpose, marker genes containing a fluorescent tag can be coexpressed along with the BiFC constructs, thus visualizing cellular structures such as the endoplasmic reticulum, mitochondria, the Golgi apparatus or the plasma membrane. The fluorescent signal can be monitored either directly in epidermal leaf cells or in single protoplasts, which can be easily isolated from the transformed tobacco leaves. BiFC is ideally suited to study protein-protein interactions in their natural surroundings within the living cell. However, it has to be considered that the expression has to be driven by strong promoters and that the interaction partners are modified due to fusion of the relatively large fluorescence tags, which might interfere with the interaction mechanism. Nevertheless, BiFC is an excellent complementary approach to other commonly applied methods investigating protein-protein interactions, such as coimmunoprecipitation, in vitro pull-down assays or yeast-two-hybrid experiments.
Keywords: Plant Biology, Issue 85, Tetratricopeptide repeat domain, chaperone, chloroplasts, endoplasmic reticulum, HSP90, Toc complex, Sec translocon, BiFC
Introduction
Studying the formation of protein complexes and their localization in plant cells in vivo is essential to investigate cellular networks, signaling and metabolic processes. BiFC allows visualization of protein-protein interactions in their natural environment directly within the living plant cell1-5.
In the BiFC approach the complementation of two nonfluorescent N- and C-terminal fragments of a fluorescent protein lead to a reconstituted fluorescent protein. Fragments of many different fluorescent proteins have been used to detect protein interactions, e.g. the green fluorescent protein (GFP) the chromophore of which is chemically formed by three distinct residues6. Fluorescent proteins can be halved within a loop or ß-strand to result in the two nonfluorescent fragments which can be fused to both proteins of interest. The assay can be used to detect interactions in any subcellular compartment in any aerobically growing organism or cells that can be genetically modified to express the fusion proteins. If the two proteins come into close proximity within the cell, fluorescence is reconstituted and can be monitored by microscopy without the addition of exogenous fluorophores or dyes3.
Tobacco (Nicotiana benthamiana) has proven to be a convenient model organism to visualize the interaction of plant proteins, since proteins can easily be expressed by utilizing agrobacterium-mediated transformation of tobacco leaves with the generated constructs. Agrobacteria use a so-called Ti plasmid (tumor inducing) coding for enzymes that mediate the transduction of the gene of interest into plant cells. BiFC is well applicable for soluble as well as for membrane proteins within all cellular compartments and has been successfully used over the past years to identify interacting proteins in vivo as well as to analyze interaction sites within the proteins7-9. Upon expression of the introduced genes, the interaction of the fluorescent proteins can be visualized directly in leaves, which is suitable for larger cellular structures, such as the endoplasmic reticulum (ER), the plasma membrane or chloroplasts. However, to monitor the localization in more refined structures, for example, the chloroplast envelope, it is advisable to visualize the fluorescence in protoplasts isolated from transformed tobacco leaves. A set of BiFC vectors containing either a C-terminal or an N-terminal fluorescent tag has to be used for the BiFC approach in plants10. The hereafter described protocol was used to study the interaction of cytosolic heat shock protein 90 (HSP90) with the tetratricopeptide repeat (TPR) domain containing docking proteins Toc64 and AtTPR7 residing in the chloroplast outer envelope and the endoplasmic reticulum, respectively11-13. For this purpose, HSP90 was fused to the C-terminal part of SCFP (SCFPC). The tag was N-terminally fused to the chaperone to ensure accessibility of the C-terminal MEEVD binding motif of HSP90 to clamp-type TPR domains. In parallel, the N-terminal part of Venus (VenusN) was fused to the cytosolic domains of the TPR domain containing docking proteins Toc64 and AtTPR7, respectively. As a negative control we cloned the soluble C-terminal part of SCFPC solely which resides in the cytosol and is therefore an appropriate control.
The fluorescent tags of the studied proteins have to face the same cellular compartment to allow close proximity and thereby reconstitution of the fluorescent signal. To determine the localization of the reconstituted fluorescent signal a marker protein fused to a different fluorescent tag can be cotransformed to demonstrate the subcellular localization of the interaction. An ER marker protein fused to mCherrry was transformed simultaneously in the case of the ER located AtTPR714. The autofluorescence of chlorophyll served as chloroplast marker in case of Toc64. By this not only the in vivo interaction of Toc64 and AtTPR7, respectively, with the cytosolic HSP90 chaperone can be monitored directly in the tobacco leaves but also the subcellular localization of the interaction can be investigated.
BiFC is well suited as a complementary approach to other methods studying protein-protein interactions. Compared to coimmunoprecipitation or in vitro pull-down experiments, for example, no specific antibodies have to be available for the proteins of interest, and the proteins do not have to be recombinantly expressed in vitro, which can be challenging, especially for membrane proteins. Moreover, also transient interactions can be monitored using BiFC, since the proteins are captured by the interaction of the fused fluorescent tags15.
Protocol
1. Transformation of BiFC Constructs in Agrobacteria
- Cloning of BiFC constructs
- Amplify the gene of interest from an appropriate template using oligonucleotides containing flanking attB-sites. Perform a PCR using a proofreading polymerase. Adapt the length of the annealing step to the designed primer combination and the length of the elongation step according to the fragment size. Check the PCR product by agarose gel electrophoresis and purify it by using a PCR Clean-up Kit.
- Perform the BP reaction with the obtained fragment and the entry vector using a BP recombinase. Mix 15-150 ng of the attB-PCR product with 150 ng of the entry vector, add 2 µl of the BP recombinase and fill up with TE buffer to a total volume of 8 µl. Incubate the reaction for 1 hr at RT and stop the reaction by adding 2 µl Proteinase K for 10 min at 37 °C.
- Transform the entire reaction into competent E. coli DH5α cells and screen the obtained colonies by colony PCR for correct insertion of the DNA fragment into the vector. DNA sequencing of positive plasmids is performed to verify the absence of mutations.
- Use the obtained entry clone for LR recombination with the appropriate destination vector using a LR recombinase. Mix 50-150 ng of the entry vector with 150 ng of the destination vector, add 1 µl LR recombinase and fill up with TE buffer to a total volume of 5 µl. Incubate the reaction for 1 hr at RT and stop the reaction by adding 1 µl Proteinase K for 10 min at 37 °C.
- Transform the entire reaction into competent E. coli DH5α cells and screen obtained colonies by colony PCR for correct insertion of the DNA fragment into the vector. DNA sequencing is not necessary at this step.
- Isolate plasmid DNA with a plasmid Mini kit to ensure a high degree of purity.
- Preparation of chemically competent Agrobacteria (strain AGL1, Rifampicin and Carbenicillin resistance)
- Streak out agrobacteria from a stock culture and grow for 24 hr at 28 °C.
- Inoculate 5 ml LB medium with a single colony and incubate overnight at 28 °C.
- Inoculate 50 ml LB medium with 2 ml of the overnight culture and grow for approximately 4 hr at 28 °C to an OD600 of 1.0.
- Centrifuge the cells at 3,000 x g for 15 min at 4 °C and resuspend the pellet in 1 ml sterilized ice-cold CaCl2 (10 mM). Keep cells on ice after this step.
- Prepare aliquots (100 µl) of the cells, freeze immediately in liquid nitrogen and store at -80 °C.
- Transformation of chemically competent Agrobacteria
- Thaw one aliquot of competent AGL1 cells on ice. Add 1-2 µg of plasmid DNA to the cells. Incubate for 5 min on ice, 5 min in liquid nitrogen and 5 min at 37 °C. Add 600 µl LB medium to the cells and shake at 650 rpm for 4 hr at 28 °C.
- Centrifuge the cells for 1 min at 8,000 x g and discard the supernatant. Resuspend the pellet in 50 µl of the remaining LB medium and plate on LB plates containing the appropriate antibiotics. Seal the plates and incubate for 2 days at 28 °C. Screen colonies for the presence of the plasmid by colony PCR.
- Inoculate one positive colony in 5 ml LB medium containing the appropriate antibiotics and incubate at 28 °C over night. Mix 500 µl of the overnight culture with 500 µl 50% sterilized glycerol and freeze at -80 °C.
- Inoculate the agrobacteria in LB medium containing the appropriate antibiotics directly from the glycerol stock for transient transformation of tobacco leaves.
2. Transient Transformation of Tobacco Leaves
- Agrobacteria growth
- Prepare the following stock solutions: Acetosyringone (150 mM, dissolve in 70% EtOH), store as aliquots at -20 °C. MES/KOH (0.1 M) pH 5.7, store at 4 °C (sterile filter through a 0.2 µM filter to prevent bacterial growth during longer storage), MgCl2 (1 M) stock, store at RT.
- Inoculate 10 ml LB medium containing the appropriate antibiotics with 50 µl of the AGL1 glycerol stock culture containing the plasmid of interest in a sterile 50 ml tube and incubate at 28 °C for at least 24 hr shaking at 190 rpm until an OD600 between 1.0-2.0 is reached.
- Centrifuge bacteria at 3,000 x g for 15 min. Resuspend the pellet in freshly made infiltration medium [MgCl2 (10 mM), MES/KOH (10 mM) pH 5.7, Acetosyringone (150 µM)] and adjust the suspension to an OD600 of 1.0.
- Incubate the agrobacteria cells in an overhead shaker for 2 hr in darkness. The cells can then be used for infiltration.
- Infiltration of tobacco leaves
- Use three week old tobacco (Nicotiana benthamiana) plants. Choose several older leaves for infiltration.
- Mix equal volumes of the agrobacteria carrying the constructs of interest (3 ml each). Take a 5 ml syringe without a needle for infiltration. Infiltrate the cell suspension carefully into the tobacco leaves by pressing the syringe on the bottom side of the leaves in several places.
- Water the plants and leave them in the darkness for two days.
3. Protoplast Preparation
Protoplast preparation of tobacco leaves was adapted from Koop et al.16 and slightly modified.
- Buffer preparation
- Prepare F-PCN medium: Macro-salts [KNO3 (1012 µg/ml), CaCl2•2H2O (440 µg/ml), MgSO4•7H2O (370 µg/ml), KH2PO4 (170 µg/ml), NH4-succinate [(20 mM; prepare a 2 M stock solution (succinate (236 µg/ml) and NH4Cl (106 µg/ml), adjust to pH 5.8 to dissolve)], Micro-salts [EDTA-Fe(III) x Na-salt (40 µg/ml), KJ (0.75 µg/ml), H3BO3 (3 µg/ml), MnSO4•H2O (10 µg/ml), ZnSO4•7H2O (2 µg/ml), Na2MoO4•7H2O (0.25 µg/ml), CuSO4•5H2O (0.025 µg/ml), CoCl2•6H2O (0.025 µg/ml)], MES (390 µg/ml), glucose (approximately 80 µg/ml) osmolarity 550 mOsm, pH 5.8 (KOH). Store in aliquots at -20 °C.
- Prepare F-PIN medium: All ingredients as F-PCN but instead of glucose use sucrose (approximately 110 µg/ml), osmolarity 550 mOsm, pH 5.8 (KOH). Store in aliquots at -20 °C.
- Prepare W5 medium: 150 mM NaCl, 125 mM CaCl2, 5 mM KCl, 2 mM MES, osmolarity 550-580 mOsm, pH 5.7 (KOH). Store at 4 °C (sterile filter through a 0.2 µm filter to prevent bacterial growth during longer storage).
- Prepare fresh enzyme solution for protoplast isolation (0.1 g cellulase, 0.03 g macerozym in 10 ml F-PIN). Incubate the solution at 55 °C for 10 min and cool to RT. Add 100 µl of 10% BSA to 10 ml solution.
- Isolation of protoplasts
- Place one infiltrated leaf into a Petri dish and add the fresh enzyme solution. Use a new razorblade to cut the leaf into approximately 0.5 cm2 sized pieces. Transfer the leaf-pieces with the enzyme solution into a vacuum-infiltration flask and vacuum infiltrate for approximately 20 sec until air bubbles emerge from the leaves (release vacuum very carefully).
- Shake the flask for 90 min at 40 rpm in darkness.
- Release protoplasts by shaking for 1 min at 90 rpm. Filter the solution through gauze (100 µM) into a 15 ml centrifugation tube (round bottom).
- Overlay the protoplast solution with 2 ml F-PCN buffer and centrifuge for 10 min at 70 x g (slow acceleration and deceleration) at RT.
- Intact protoplasts accumulate at the interface of enzyme solution and F-PCN. Take a wide orifice 1 ml pipette tip to transfer the intact protoplasts into a fresh centrifuge tube and fill up with W5 buffer. Centrifuge for 2 min at 100 x g (slow acceleration and deceleration) to pellet the protoplasts.
- Remove the supernatant carefully by using a pipette and resuspend pellet in approximately 200 µl W5 buffer, depending on the amount of protoplasts.
- Always use wide orifice tips to prevent rupturing of intact protoplasts.
4. Laser Scanning Microscopy
- Sample preparation
- Paste two small strips of sealant around a microscope slide (2 cm apart). Place 20 µl of the protoplast solution between the strips and carefully place a cover glass on top. The sealant strips make sure that the protoplasts are not squashed by the cover glass.
- For total leaf samples cut a 1 cm piece from the leaf and place it onto a microscope slide with the bottom side of the leaf facing upwards. Add approximately 30 µl H2O, place a cover glass on top and fix it tightly with adhesive tape on both sides.
- Confocal imaging and microscope settings
- Imaging is performed with a confocal laser scanning microscope from Leica, Type: TCS SP5. For magnification use an objective lens (HCX PL APO CS) with a magnitude of 63X with glycerol as imaging medium. Set the numerical aperture to 1.3. Use the Leica Application Suite/Advanced Fluorescence software for evaluation (see Supplemental Data S1).
- Set the Argon laser to 30% and the laser power at 488 nm to an intensity of 18% to monitor the reconstituted BiFC signal at 515 nm and set one PMT detector emission bandwidth from 495-550.
- To monitor chlorophyll autofluorescence set a second PMT detector emission bandwidth from 650-705.
- To monitor mCherry signal use the HeNe 561 laser, set the intensity of laser 561 to 18% and the emission bandwidth of a third PMT detector from 587-610.
- Make sure that pictures of all PMT detector channels are taken with the same gain settings (gain should be between 800-900 to exclude background signals).
- Take pictures in a format width/height of 1024 x 1024 pixels with a scan speed of 100 Hz.
- For Z-stackings use a maximum distance of 0.5 µm between each stack.
Representative Results
In this example we used the BiFC method to monitor the interaction of the cytosolic molecular chaperone HSP90 with the membrane docking proteins AtTPR7 and Toc64. AtTPR7 is part of the Sec translocon and interacts with cytosolic chaperones, which possibly deliver secretory preproteins for post-translational translocation to the ER membrane. Likewise, Toc64 at the chloroplast outer envelope acts in post-translational import by receiving HSP90 associated chloroplast preproteins. Both proteins comprise a cytosolic exposed TPR domain, which mediates interaction with the C-terminal MEEVD motif of HSP90.
Proteins were cloned by means of specific recombinases into suitable destination vectors fusing the TPR domain containing docking proteins to VenusN, ensuring that the fluorescent tag is attached to the cytosolic domain and does thus not hinder targeting and membrane insertion of the proteins. In the case of HSP90, SCFPC was fused to the N-terminus, as not to interfere with the C-terminal MEEVD motif (Figures 1 and 2).
AtTPR7 and HSP90 were cotransformed with an ER marker (mCherry) to verify the localization of the protein complex. The fluorescence was monitored in intact leaves with a laser scanning microscope. As a control SCFPC alone, which is located in the cytosol (like HSP90), was expressed along with AtTPR7 and the ER marker. Several leaves were checked for fluorescence and pictures were taken with identical microscope settings. In our experience a typical signal should be visible with gain settings at 800-900, whereas the negative control should only show very slight background fluorescence with these settings (Figure 3). A reconstituted signal for VenusN-AtTPR7 together with SCFPC-HSP90 at 515 nm was monitored overlapping with the ER marker. No signal for VenusN-AtTPR7 and the negative control SCFPC could be observed.
In the case of Toc64 and HSP90 expression, as well as Toc64 and SCFPC, protoplasts were isolated from infiltrated tobacco leaves, since in microscopic pictures of the entire leaves the exact localization is difficult to determine, although fluorescence is already visible (Figures 4 and 5). A signal at 515 nm was restored expressing Toc64-VenusN together with SCFPC-HSP90 at the chloroplast envelope, which could be detected as ring shaped structures surrounding the chloroplasts. As above the control was photographed with identical microscope settings and did not show a fluorescence at 515 nm.
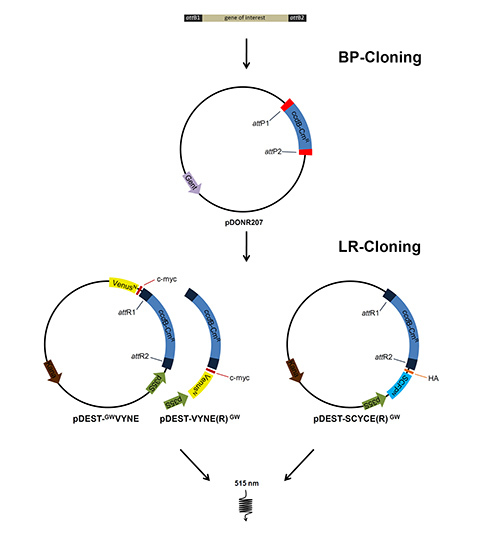 Figure 1. Cloning procedure of BiFC constructs. The genes of interest were amplified with oligonucleotides flanked by attB sites to allow BP recombination into an entry vector with attP sites, thus replacing the ccdB gene within the vector. Subsequently the entry vector was recombined with appropriate destination vectors using a LR recombinase. Transformation of these constructs into tobacco leaves allows expression of proteins fused to the split fluorescent proteins and to tags for antibody detection. Click here to view larger image.
Figure 1. Cloning procedure of BiFC constructs. The genes of interest were amplified with oligonucleotides flanked by attB sites to allow BP recombination into an entry vector with attP sites, thus replacing the ccdB gene within the vector. Subsequently the entry vector was recombined with appropriate destination vectors using a LR recombinase. Transformation of these constructs into tobacco leaves allows expression of proteins fused to the split fluorescent proteins and to tags for antibody detection. Click here to view larger image.
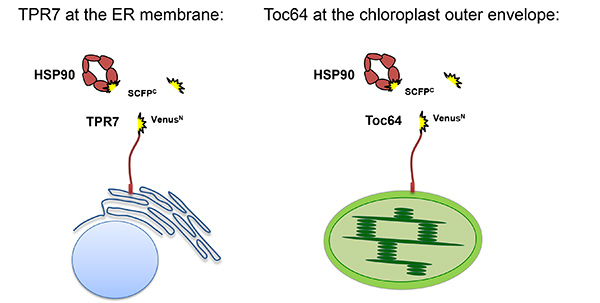 Figure 2. Schematic presentation of the proteins expressed in BiFC experiments. VenusN is coupled to the cytosolic parts of Toc64 or AtTPR7 residing in the chloroplast and ER, respectively. HSP90 is N-terminally fused to SCFPC, enabling interaction of the TPR domains of Toc64 and AtTPR7 with the HSP90 C-terminus. SCFPC alone is expressed in the cytosol as a control. Please click here to view a larger version of this figure.
Figure 2. Schematic presentation of the proteins expressed in BiFC experiments. VenusN is coupled to the cytosolic parts of Toc64 or AtTPR7 residing in the chloroplast and ER, respectively. HSP90 is N-terminally fused to SCFPC, enabling interaction of the TPR domains of Toc64 and AtTPR7 with the HSP90 C-terminus. SCFPC alone is expressed in the cytosol as a control. Please click here to view a larger version of this figure.
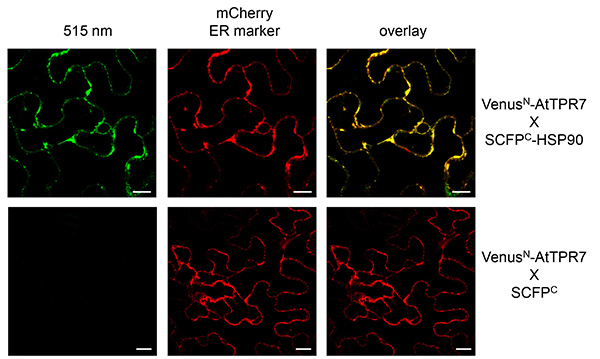 Figure 3. BiFC with AtTPR7 and HSP90 visualized in tobacco epidermal leaf cells. VenusN-AtTPR7 and SCFPC-HSP90 were cotransformed with the ER mCherry marker (middle panel) and transiently expressed in tobacco leaves. As a control VenusN-AtTPR7 was cotransformed with SCFPC alone and the ER mCherry marker (bottom panels). Reconstituted fluorescence was monitored at 515 nm (left panel). Overlay of the signal at 515 nm and the mCherry marker is shown (right panel). Scale bars: 10 µm. Please click here to view a larger version of this figure.
Figure 3. BiFC with AtTPR7 and HSP90 visualized in tobacco epidermal leaf cells. VenusN-AtTPR7 and SCFPC-HSP90 were cotransformed with the ER mCherry marker (middle panel) and transiently expressed in tobacco leaves. As a control VenusN-AtTPR7 was cotransformed with SCFPC alone and the ER mCherry marker (bottom panels). Reconstituted fluorescence was monitored at 515 nm (left panel). Overlay of the signal at 515 nm and the mCherry marker is shown (right panel). Scale bars: 10 µm. Please click here to view a larger version of this figure.
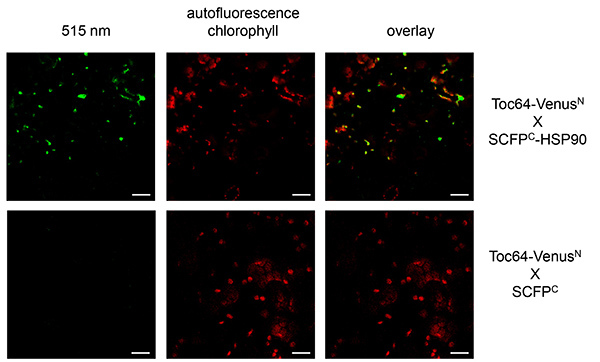 Figure 4. BiFC with Toc64 and HSP90 visualized in tobacco epidermal leaf cells. Toc64-VenusN and SCFPC-HSP90 were transiently expressed in tobacco leaves. As a control Toc64-VenusN was cotransformed with SCFPC alone (bottom panels). Reconstituted fluorescence was monitored at 515 nm (left panel). Overlay of the signal at 515 nm and the chlorophyll autofluorescence is shown (right panel). Chlorophyll autofluorescence is monitored at 480 nm. Scale bars: 10 µm. Please click here to view a larger version of this figure.
Figure 4. BiFC with Toc64 and HSP90 visualized in tobacco epidermal leaf cells. Toc64-VenusN and SCFPC-HSP90 were transiently expressed in tobacco leaves. As a control Toc64-VenusN was cotransformed with SCFPC alone (bottom panels). Reconstituted fluorescence was monitored at 515 nm (left panel). Overlay of the signal at 515 nm and the chlorophyll autofluorescence is shown (right panel). Chlorophyll autofluorescence is monitored at 480 nm. Scale bars: 10 µm. Please click here to view a larger version of this figure.
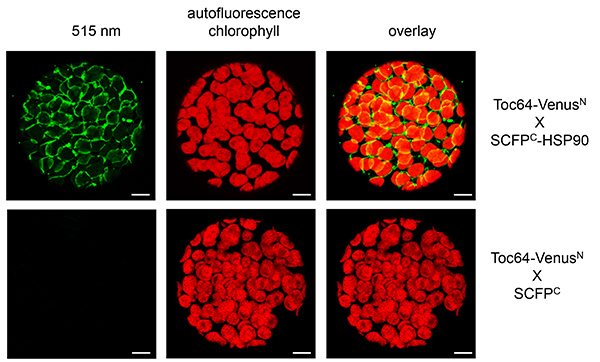 Figure 5. BiFC with Toc64 and HSP90 visualized in tobacco protoplasts. Toc64-VenusN and SCFPC-HSP90 were transiently expressed in tobacco leaves. As a control Toc64-VenusN was cotransformed with SCFPC alone (bottom panels). Reconstituted fluorescence was monitored at 515 nm (left panel) in isolated protoplasts. Overlay of the signal at 515 nm and the chlorophyll autofluorescence is shown (right panel). Chlorophyll autofluorescence is monitored at 480 nm. Scale bars: 10 µm. Please click here to view a larger version of this figure.
Figure 5. BiFC with Toc64 and HSP90 visualized in tobacco protoplasts. Toc64-VenusN and SCFPC-HSP90 were transiently expressed in tobacco leaves. As a control Toc64-VenusN was cotransformed with SCFPC alone (bottom panels). Reconstituted fluorescence was monitored at 515 nm (left panel) in isolated protoplasts. Overlay of the signal at 515 nm and the chlorophyll autofluorescence is shown (right panel). Chlorophyll autofluorescence is monitored at 480 nm. Scale bars: 10 µm. Please click here to view a larger version of this figure.
Discussion
Upon planning a BiFC experiment several points should be considered. Although no structural information about the proteins of interest is required, the topology has to be known when working with membrane spanning proteins. The fluorescent proteins have to reside in the same subcellular compartment or face the same side of a membrane to allow interaction. Naturally, when analyzing proteins which require an N-terminal targeting sequence, only a C-terminal tag can be considered. Since it is possible that the tag interferes with proper targeting or membrane insertion of the protein of interest it is advisable to test subcellular localization beforehand, for example, by expressing a GFP-tagged protein. Moreover, a negative control should always be included. In this example we generated a construct only expressing SCFPC in the cytosol. However, any protein that is not expected to interact can be used as a negative control. To verify proper expression of the constructs, especially if no fluorescence is visible, protein extracts of infiltrated leaves or protoplasts can be subjected to SDS-PAGE and protein expression can be verified with antibodies directed against the respective tags.
Fluorescent signals can be monitored either in intact leaves or isolated protoplasts. Although detection in entire leaves is faster, signals of more refined structures are better visualized in protoplasts. Moreover, mostly epidermal cells are monitored when looking at the entire leaf, which do not contain chloroplasts. Therefore, when analyzing chloroplast proteins, isolation of protoplasts is advisable.
The major advantage of the technique is the possibility to monitor protein-protein interactions in living plant cells. There is no need to break cells and to solubilize membrane protein complexes, as it is the case for example in coimmunoprecipitation experiments. Moreover, the application is simple since the only required materials are the vectors, agrobacteria and a standard fluorescence microscope (although higher quality images are achieved with a confocal laser scanning microscope). In contrast to in vitro pull-down assays with recombinant proteins, which only allow detection of an interaction if both proteins are interacting directly, BiFC can also detect protein complexes which require additional, endogenous proteins present in the cell. However, this also means that BiFC provides no prove of a direct protein-protein interaction, which always has to be verified by other techniques. Moreover, due to overexpression by strong promoters unspecific interactions might occur, which have to be ruled out by appropriate negative controls. To this end a protein not predicted to interact with the protein of interest, but residing in the same compartment, or constructs lacking the protein-protein interaction domains should be used. In addition, a dilution series of the prey cDNA with a noninteracting cDNA as well as observation of the fluorescence in a time dependent manner after transformation can help to validate the results. To ensure discrimination of true fluorescent signals and artifacts the BiFC signals should be quantified and set into relation to another expressed fluorescent protein, for example a marker protein7,8. Another drawback of the BiFC method is, that interactions of the proteins may also be hindered sterically by the relatively large fluorescent tags.
Application of agrobacterium-mediated transformation in other plants (for example, Arabidopsis) is limited, however, it is possible to transform the plasmid DNA directly either into isolated Arabidopsis protoplasts or to transform cells using a particle gun. However, plasmid DNA should be isolated using a MAXI Kit, since it should be highly concentrated and as pure as possible for protoplast transformation. Another problem we observed due to high expression of the target proteins was unspecific aggregation in the cytosol, especially when working with mitochondrial membrane proteins. This problem can be overcome by biolistic transformation of onion cells.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We would like to thank Jürgen Soll for helpful discussions and Chris Carrie for critical reading of the manuscript. This project was funded by the DFG and Fonds der chemischen Industrie (grants numbers SFB 1035, project A04 to S.S. and Do 187/22 to R.S.).
References
- Citovsky V, et al. Subcellular localization of interacting proteins by bimolecular fluorescence complementation in planta. J. Mol. Biol. 2006;362:1120–1131. doi: 10.1016/j.jmb.2006.08.017. [DOI] [PubMed] [Google Scholar]
- Schutze K, Harter K, Chaban C. Bimolecular fluorescence complementation (BiFC) to study protein-protein interactions in living plant cells. Methods Mol. Biol. 2009;479:189–202. doi: 10.1007/978-1-59745-289-2_12. [DOI] [PubMed] [Google Scholar]
- Weinthal D, Tzfira T. Imaging protein-protein interactions in plant cells by bimolecular fluorescence complementation assay. Trends Plant Sci. 2009;14:59–63. doi: 10.1016/j.tplants.2008.11.002. [DOI] [PubMed] [Google Scholar]
- Ohad N, Shichrur K, Yalovsky S. The analysis of protein-protein interactions in plants by bimolecular fluorescence complementation. Plant Physiol. 2007;145:1090–1099. doi: 10.1104/pp.107.107284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohad N, Yalovsky S. Utilizing bimolecular fluorescence complementation (BiFC) to assay protein-protein interaction in plants. Methods Mol. Biol. 2010;655:347–358. doi: 10.1007/978-1-60761-765-5_23. [DOI] [PubMed] [Google Scholar]
- Tsien RY. The green fluorescent protein. Annu. Rev. Biochem. 1998;67:509–544. doi: 10.1146/annurev.biochem.67.1.509. [DOI] [PubMed] [Google Scholar]
- Lee LY, et al. Screening a cDNA library for protein-protein interactions directly in planta. Plant Cell. 2012;24:1746–1759. doi: 10.1105/tpc.112.097998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarlane HE, Shin JJ, Bird DA, Samuels AL. Arabidopsis ABCG transporters, which are required for export of diverse cuticular lipids, dimerize in different combinations. Plant Cell. 2010;22:3066–3075. doi: 10.1105/tpc.110.077974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunschede B, Bals T, Funke S, Schunemann D. Interaction studies between the chloroplast signal recognition particle subunit cpSRP43 and the full-length translocase Alb3 reveal a membrane-embedded binding region in Alb3 protein. J. Biol. Chem. 2011;286:35187–35195. doi: 10.1074/jbc.M111.250746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehl C, Waadt R, Kudla J, Mendel RR, Hansch R. New GATEWAY vectors for high throughput analyses of protein-protein interactions by bimolecular fluorescence complementation. Mol. Plant. 2009;2:1051–1058. doi: 10.1093/mp/ssp040. [DOI] [PubMed] [Google Scholar]
- Qbadou S, et al. The molecular chaperone Hsp90 delivers precursor proteins to the chloroplast import receptor Toc64. EMBO J. 2006;25:1836–1847. doi: 10.1038/sj.emboj.7601091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweiger R, Muller NC, Schmitt MJ, Soll J, Schwenkert S. AtTPR7 is a chaperone docking protein of the Sec translocon in Arabidopsis. J. Cell Sci. 2012. [DOI] [PubMed]
- Schweiger R, S S. AtTPR7 as part of the Arabidopsis Sec post-translocon. Plant Signal Behav. 2013;8 doi: 10.4161/psb.25286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson BK, Cai X, Nebenfuhr A. A multicolored set of in vivo organelle markers for co-localization studies in Arabidopsis and other plants. Plant J. 2007;51:1126–1136. doi: 10.1111/j.1365-313X.2007.03212.x. [DOI] [PubMed] [Google Scholar]
- Kerppola TK. Bimolecular fluorescence complementation (BiFC) analysis as a probe of protein interactions in living cells. Annu. Rev. Biophys. 2008;37:465–487. doi: 10.1146/annurev.biophys.37.032807.125842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koop HU, et al. Integration of foreign sequences into the tobacco plastome via polyethylene glycol-mediated protoplast transformation. Planta. 1996;199:193–201. doi: 10.1007/BF00196559. [DOI] [PubMed] [Google Scholar]