

Abstract
Age-related neurodegeneration has been studied extensively through the use of model organisms, including the genetically versatile Drosophila melanogaster. Various neurotoxic proteins have been expressed in fly eyes to approximate degeneration occurring in humans, and much has been learned from this heterologous system. Although Drosophila expedites scientific research through rapid generational times and relative inexpensiveness, one factor that can hinder analyses is the examination of milder forms of degeneration caused by some toxic proteins in fly eyes. Whereas several disease proteins cause massive degeneration that is easily observed by examining the external structure of the fly eye, others cause mild degeneration that is difficult to observe externally and requires laborious histological preparation to assess and monitor. Here, we describe a sensitive fluorescence-based method to observe, monitor, and quantify mild Drosophila eye degeneration caused by various proteins, including the polyglutamine disease proteins ataxin-3 (spinocerebellar ataxia type 3) and huntingtin (Huntington's disease), mutant a-synuclein (Parkinson's disease), and Ab42 (Alzheimer's disease). We show that membrane-targeted green fluorescent protein reports degeneration robustly and quantitatively. This simple yet powerful technique, which is amenable to large-scale screens, can help accelerate studies to understand age-related degeneration and to find factors that suppress it for therapeutic purposes.
Keywords: neurodegeneration, polyglutamine, spinocerebellar ataxia, Parkinson's, Alzheimer's, Huntington's
Age-dependent neurodegenerative diseases, including Alzheimer's and Parkinson's diseases, and polyglutamine (polyQ)-expansion disorders, such as Huntington's disease and several spinocerebellar ataxias, are health problems that will continue to worsen as human life expectancy increases worldwide. As these debilitating disorders remain poorly understood due to complex processes, strategies to treat them effectively remain elusive. Drosophila melanogaster, a powerful model organism with exceptionally flexible genetics, has been employed consistently and successfully to study the process of neurodegeneration and to identify genes that enhance or suppress degeneration from various toxic proteins (Jaiswal et al., 2012). By using the fruit fly, multiple groups have modeled human diseases and have identified key players in the pathogenesis of Alzheimer's and Parkinson's diseases, as well as polyQ-dependent disorders (Jackson et al., 1998; Warrick et al., 1998, 2005; Fernandez-Funez et al., 2000; Taylor et al., 2003; Pandey et al., 2007; Romero et al., 2008; Zhang et al., 2010; Casas-Tinto et al., 2011; Whitworth, 2011; Rincon-Limas et al., 2012; Choksi et al., 2013). Specifically, the Drosophila compound eye has emerged as a favorite heterologous system to study neurodegeneration and to find genes or molecules that modify it (Bonini and Fortini, 2003; Lenz et al., 2013).
The compound eye of Drosophila contains approximately 800 functional units called ommatidia, each containing eight photoreceptors (Paulk et al., 2013). The fly eye allows degeneration to be assessed quickly by observing the external eye structure through light microscopy in a live, anesthetized fly (Fig. 1A). However, this quick, simple, and informative method may not always adequately describe what is happening internally in milder cases of degeneration, thus requiring histology to assess disease (Fig. 1A, histological sections). Besides histology, ommatidial organization can also be studied through scanning and transmission electron microscopy in order to achieve greater detail (Taylor et al., 2003; Al-Ramahi et al., 2007; Lanson et al., 2011; Park et al., 2013). Histo-logical preparations and electron microscopic techniques provide a richly detailed view of structural abnormalities as a result of degenerative proteins expressed in fly eyes. However, they can be time consuming, neither always nor easily quantifiable, and not necessarily feasible to take advantage of one of the most appealing aspects of Drosophila studies: large-scale screens to identify disease modifiers for therapeutic purposes.
Fig. 1.
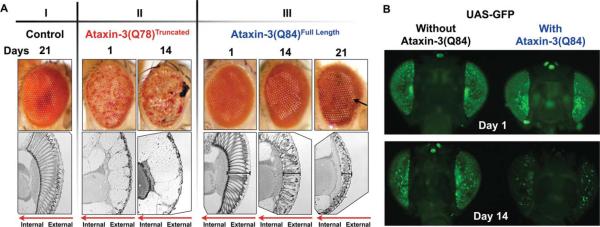
GFP reports degeneration caused by full-length pathogenic ataxin-3. A: Representative photos of external eye morphology and longitudinal histological sections of retinae from adult flies expressing the noted transgenes through the gmr-Gal4 driver. Flies were heterozygous for all transgenes. I: Control fly with one copy of the driver, gmr-Gal4. II: Note depigmentation and necrosis of external eyes when a truncated version of the polyQ-disease protein ataxin-3, ataxin-3(Q78)Truncated, is expressed, accompanied by nearly complete loss of the retina (histological sections). III: Expression of full-length pathogenic ataxin-3, ataxin-3(Q84), does not cause large anomalies to the external eye, but the retina is progressively disrupted with age. Bracketed lines, retinal depth; black arrow, depigmentation; red arrows in histological sections indicate directionality of eye structures. B: Flies expressing full-length pathogenic ataxin-3 alongside GFP show markedly reduced fluorescence by 14 days. Driver, gmr-Gal4. All flies were heterozygous for all constructs. Genotypes for flies in both panels are listed in Table I. Days denote time elapsed since adults eclosed from the pupal case.
A method that reports internal eye integrity and offers a sensitive, rapid, and quantitative assessment of neurodegeneration in intact flies would be beneficial for promptly and reliably assessing retinal degeneration. To this end, we examined whether we could use green fluorescent protein (GFP) expression to monitor internal retinal integrity without the need for histology. We found that membrane-targeted GFP is a sensitive reporter of internal retinal integrity. Membrane-GFP can be used to detect degeneration, and changes in it, caused by polyQ and non-polyQ proteins. This method also allows for quantitative analysis of degeneration in fly eyes.
MATERIALS AND METHODS
Unless otherwise specified, Drosophila lines were cultured and maintained in controlled environments at 25° C and 60% humidity (Tsou et al., 2012, 2013). For crosses containing nonpolyQ disease proteins (α-synucleinA30P and Aβ42), flies were raised at 29° C. Flies expressing wild-type ataxin-3 were created in our laboratory (Tsou et al., 2013) by utilizing the Gal4-UAS system (Brand and Perrimon, 1993). Publicly available stock lines, including gmr-Gal4, UAS-ataxin-3(Q78)Truncated (MJD.tr-Q78; Warrick et al., 1998), and UAS-ataxin-3(Q84)full-length (SCA3.fl-Q84.myc; Warrick et al., 2005), were obtained from the Bloomington Drosophila Stock Center (BDSC; stock Nos. 8121, 8605, 8150, and 33610; Department of Biology, Indiana University, Bloomington, IN). Other lines from BDSC include mCD8-GFP (stock Nos. 5130, 5137, here denoted as UAS-CD8-GFP; Lee and Luo, 2001), UAS-EGFP (stock No. 5431, here denoted as UAS-GFP), HTT.128Q.FL (stock No. 33808, here denoted as UAS-htt(Q128); Wu et al. 2011), Hsap-SNCA.A30P (stock No. 8147, here denoted as UAS-α-synucleinA30P; Auluck et al., 2002), Aβ1–42 (stock No. 32038, here denoted as UAS-Ab42; line generated by the Lawrence Goldstein laboratory). Genotypes of all of the flies shown in figures are listed in Table I.
TABLE I Genotype of Flies Used in Each Figure
| Figure | Genotype |
|---|---|
| 1 |
Panel A I w[*]; +; P{w[+mC]=longGMR-GAL4}3/ + II w[*]; P{w[+mC] = UAS-Hsap\MJD.tr-Q78}c211.2/+; P{w[+mC]=longGMR-GAL4}3/+ III w[*]; P{w[+mC]=longGMR-GAL4}2/+; P{w[+mC]=UAS-SCA3.fl-Q84.myc}7.2/ + Panel B w[*]; P{w[+mC]=longGMR-GAL4}2/+; P{w[+mC]=UAS-EGFP}34/+ w[*]; P{w[+mC]=longGMR-GAL4}2/+; P{w[+mC]=UAS-EGFP}34/P{w[+mC]=UAS-SCA3.fl-Q84.myc}7.2 |
| 2 |
Panel A w[*]; P{w[+mC]=longGMR-GAL4}2/P{w[+mC]=UAS-mCD8::GFP.L}LL5; + Panel C w[*]; P{w[+mC]=longGMR-GAL4}2/P{w[+mC]=UAS-mCD8::GFP.L}LL5; + w[*]; P{w[+mC]=longGMR-GAL4}2/P{w[+mC]=UAS-mCD8::GFP.L}LL5; P{w[+mC]=UAS-SCA3.fl-Q84.myc}7.2/+ Panel D w[*]; P{w[+mC]=longGMR-GAL4}2/+; P{w[+mC]=UAS-mCD8::GFP.L}LL6/+ w[*]; P{w[+mC]=longGMR-GAL4}2/P{w[+mC]=UAS-ataxin-3-WT-HA}E6; P{w[+mC]=UAS-mCD8::GFP.L}LL6/+ |
| 3 |
Panels A and B w[*]; P{w[+mC]=longGMR-GAL4}2/+; P{w[+mC]=UAS-SCA3.fl-Q84.myc}7.2/P{w[+mC]=UAS-mCD8::GFP.L}LL6 w[*]; P{w[+mC]=longGMR-GAL4}2/P{w[+mC]=UAS-ataxin-3-WT-HA}E6; P{w[+mC]=UAS-SCA3.fl-Q84.myc}7.2/P{w[+mC]=UAS-mCD8::GFP.L}LL6 |
| 4 |
Panel A w[*]; +; P{w[+mC]=longGMR-GAL4}3/ + w[*]; +; P{w[+mC]=longGMR-GAL4}3/P{w[+mC]=UAS-HTT.128Q.FL}f27b Panel B w[*]; P{w[+mC]=UAS-mCD8::GFP.L}LL5/ +; P{w[+mC]=longGMR-GAL4}3/ + w[*]; P{w[+mC]=UAS-mCD8::GFP.L}LL5/ +; P{w[+mC]=UAS-HTT.128Q.FL}f27b/P{w[+mC]=longGMR-GAL4}3 |
| 5 |
Panel A w[*]; P{w[+mC]=UAS-mCD8::GFP.L}LL5/ +; P{w[+mC]=UAS-Hsap\SNCA.A30P}40.1/P{w[+mC]=longGMR-GAL4}3 Panel B w[*]; P{w[+mC]=UAS-mCD8::GFP.L}LL5/ +; P{w[+mC]=longGMR-GAL4}3/ + w[*]; P{w[+mC] = UAS-mCD8::GFP.L}LL5/ +; P{w[+mC]=UAS-Hsap\SNCA.A30P}40.1/P{w[+mC]=longGMR-GAL4}3 |
| 6 | P{w[+mC]=UAS-Abeta1-42.G}1/w[*]; P{w[+mC]=UAS-mCD8::GFP.L}LL5/+; P{w[+mC]=longGMR-GAL4}3/+ w[*]; P{w[+mC] = UAS-mCD8::GFP.L}LL5/ +; P{w[+mC]=longGMR-GAL4}3/ + P{w[+mC]=UAS-Abeta1-42.G}1/w[*]; P{w[+mC]=UAS-mCD8::GFP.L}LL5/+; P{w[+mC]=longGMR-GAL4}3/+ |
Histology was performed by fixing whole flies overnight with 2% glutaraldehyde/2% paraformaldehyde in Tris-buffered saline. After fixation, flies were dehydrated using propylene oxide and 30%, 50%, 75%, and 100% ethanol. Flies were subsequently embedded in Poly/Bed812 (Polysciences, Warrington, PA), cut into 5-μM sections, and stained with toluidine blue as previously reported (Warrick et al., 1998, 2005; Tsou et al., 2013).
Images of the external Drosophila eyes were taken with an Olympus (Tokyo, Japan) SZ61 microscope and DP21 digital camera. Histological sections and fluorescence were imaged by using an Olympus BX53 microscope and DP72 digital camera. For examination with the fluorescent microscope, Drosophila heads were removed with spring scissors between the head and thorax, and heads were placed on microscope slides. Heads were arranged in pairs so that a single experimental fly would be imaged and directly compared to a single fly from its respective control group. Fluorescence was quantified by using the publicly available NIH ImageJ software. Areas of retinae measured with the Freeform tool in ImageJ were kept similar between control and experimental groups for image analysis. Mean fluorescence from the eyes of experimental flies was normalized to mean fluorescence from the eyes of their individual controls. P values were derived using two-tailed Student's t-tests comparing control and experimental groups, similar to analyses conducted by other groups (Ikegami et al., 2007; Di Cara et al., 2013; Stepp et al., 2013). For results summarized quantitatively in Figure 2B, we did not normalize the data to a specific control because we were determining variability among flies of the same genotype and age.
Fig. 2.

CD8-GFP is a uniform and sensitive reporter of retinal degeneration. A: Fluorescence images of intact fly heads expressing CD8-GFP driven by the gmr-Gal4 driver. Flies were heterozygous for both transgenes. Boxes denote the ocelli; dotted areas highlight surfaces used for quantification (as shown in B,E,F). B: Histogram summarizes arbitrary fluorescence units from fly eyes expressing membrane-bound GFP shown in A and other similar flies. Shown is mean fluorescence ± standard deviation. C: Fluorescence images of fly heads expressing the noted transgenes driven by gmr-Gal4. All flies were heterozygous for all transgenes. Boxes, ocelli display reduced GFP fluorescence over time in the presence of pathogenic ataxin-3. Magnifications of fly heads show fluorescence from individual ommatidia. D: Images of fly eyes expressing CD8-GFP in the absence or presence of wild-type ataxin-3(WT), driven by gmr-Gal4. All flies were heterozygous for all transgenes. E,F: Histograms show mean ± standard deviation of quantification of fluorescence from fly eyes shown in C,D and other images. P values are from Student's t-tests. The genotypes of flies in all panels are listed in Table I.
RESULTS AND DISCUSSION
GFP Reports Internal Retinal Degeneration Caused by a polyQ Protein
Spinocerebellar ataxia type 3 (SCA3, also known as Machado-Joseph disease) is considered to be the most common autosomal dominantly inherited ataxia and is one of at least 10 polyQ-related neurodegenerative diseases that include Huntington's disease and several SCAs (Todi et al., 2007; Williams and Paulson, 2008). SCA3 is an age-dependent disease that is caused by abnormal expansion in the polyQ tract of the deubiquitinating enzyme ataxin-3 (Costa Mdo and Paulson, 2012). This was one of the first neurodegenerative diseases to be modeled in vivo using the fruit fly in work done by the Bonini laboratory, where two different models were generated: 1) a truncated version of ataxin-3 that comprises the isolated 78-amino-acid polyQ stretch (Warrick et al., 1998) and 2) a full-length version of the human pathogenic ataxin-3 protein with 84 polyQ (Warrick et al., 2005).
As shown in Figure 1A, expression of the truncated version of pathogenic ataxin-3 protein in fly eyes caused robust degeneration of internal and external structures in adults (compare eye structures between Fig. 1AI and 1AII). However, expression of the full-length disease protein presented with a milder phenotype and did not severely impact the external fly eye (Fig. 1AIII; arrow highlights some depigmentation in adults 21 days after eclosing from the pupal case). In the presence of full-length pathogenic ataxin-3, the internal structures degenerated progressively over time, highlighted by ommatidial loss and contraction of the retinal depth. However, this marked loss of internal structure was not accompanied by large degeneration of the external eye (Fig. 1AIII).
We sought a method that would report rapidly and with high sensitivity milder forms of degeneration, as in the case of full-length pathogenic ataxin-3. This type of approach would be particularly important for large-scale genetic screens to identify modifiers of mild degeneration caused by neurotoxic proteins; such screens would be impractical if they required histology to assess the effect of thousands of genes. We investigated whether GFP expression in fly eyes can report retinal integrity, reasoning that ommatidial degeneration due to toxic proteins would lead to diminished GFP fluorescence because of fewer cells remaining in the retina.
To visualize neurodegeneration using a GFP-based method in vivo, we began by using a publicly available Drosophila line that expresses GFP by itself or that same line in combination with the full-length SCA3 disease peptide ataxin-3(Q84). As shown in Figure 1B, fly eyes expressing GFP showed nearly complete loss of fluorescence in the presence of ataxin-3(Q84) in 14-day-old flies. This feature was particularly striking in that we did not observe obvious external eye anomalies in the presence of ataxin-3(Q84) at 14 days, although internally the eyes were degenerating (Fig. 1AIII). Thus, GFP fluorescence mirrored internal degeneration. Despite this promising feature, flies that expressed UAS-GFP in the absence of the toxic protein displayed significant GFP mosaicism among different individuals, even within the same fly (Fig. 1B; other supportive data not shown). This variation could impede finer examination of degeneration caused by a specific protein. Therefore, we decided to examine other GFP variants to find better uniformity of expression.
Membrane-Targeted GFP Is a Uniform and Sensitive Reporter of Internal Retinal Organization in Drosophila
Work conducted in the Luo laboratory demonstrated the utility of membrane-targeted GFP to study the development and function of the Drosophila nervous system (Lee and Luo, 1999). These scientists generated a membrane-targeted GFP construct using the mouse CD8 protein for their mosaic analysis with a repressible cell marker system (Lee and Luo, 1999). CD8 is a transmembrane protein, therefore the cell surface is labeled with GFP. Although CD8-GFP has been used widely for various fly assays and models, to the best of our knowledge it has not been used to report adult retinal integrity.
We proceeded with using this system to examine retinal degeneration, because fly eyes expressing CD8-GFP in the absence of toxic proteins displayed little variability in fluorescence among individuals and over time (Fig. 2A,B). Drosophilae expressing the disease protein ataxin-3(Q84) in their eyes showed reduced GFP fluorescence that is particularly noticeable by day 7 and through day 14 (Fig. 2C, quantification in Fig. 2E), whereas eye-targeted expression of nonpathogenic ataxin-3(WT) in combination with membrane-targeted GFP did not lead to altered fluorescence (Fig. 2D, quantification in Fig. 2F). This finding indicates that reduced fluorescence and increased mosaicism in fly eyes expressing pathogenic ataxin-3 are not due to an inherent effect of ataxin-3 function on membrane-targeted GFP, but result from the degenerative property of this polyQ protein.
Especially notable is the fact that we observed perturbation in the consistency of GFP fluorescence in the presence of pathogenic ataxin-3 in 1-day-old adults. In Figure 2C, note how the uniformity of GFP fluorescence in eyes that did not express the toxic protein was compromised in eyes that expressed ataxin-3(Q84). Since we do not commonly observe obvious aberrations with histological sections at day 1 (Fig. 1AIII), this finding suggests that membrane-targeted GFP is a highly sensitive reporter of ommatidial degeneration. The sensitivity of CD8-GFP could stem from ommatidial loss at day 1 that is minor and not readily observed by individual longitudinal sections which, at 5 μm thickness, cover a small portion of the retinal array. Through fluorescence, we can instead examine a larger portion of the eye in one image and can readily detect disruptions in fluorescence uniformity resulting from ommatidial loss in various areas of the eye.
A useful aspect of this method is that it can be quantified (Fig. 2B,E,F). We used the NIH ImageJ software and measured overall GFP fluorescence in the compound eye. Coexpression of pathogenic ataxin-3 decreased GFP fluorescence by nearly 75% at day 14, although mean fluorescence was reduced in the presence of ataxin-3(Q84), even at day 1 (Fig. 2E).
One of our goals was to devise a method that would allow quantitative screens to discover modulators of retinal degeneration. A protein that is known to suppress polyQ-dependent degeneration in Drosophila is the normal form of ataxin-3. Wild-type ataxin-3 suppresses retinal degenerative phenotypes associated with several age-related disorders, including its own pathogenic version (SCA3), ataxin-1 (SCA1), and huntingtin (htt; Huntington's disease; Warrick et al., 2005; Tsou et al. 2013). Consequently, we examined whether coexpression of ataxin-3(WT) increases membrane-targeted GFP fluorescence in the presence of toxic ataxin-3 in the fly eye.
Based on histological examinations, ataxin-3(WT) suppressed retinal degeneration caused by a pathogenic version of itself. This suppressive effect was observable in 14-day-old adults, because on day 1 we did not observe clear anomalies in retinal organization as a result of the expression of ataxin-3(Q84) (Fig. 3A; see also Fig. 1AIII). The disease-causing version of ataxin-3 led to ommatidia loss and the presence of aggregates/inclusions (boxes in Fig. 3A) that contained the toxic protein (Warrick et al., 2005). Degeneration caused by ataxin-3(Q84) was suppressed by coexpressing normal ataxin-3; the ommatidia were present, and there were fewer aggregates/inclusions in adults aged 14 days. Still, eyes coexpressing ataxin-3(WT) and ataxin-3(Q84) were not completely normal; we observed some ommatidial loss at 14 days compared with 1-day-old adults of the same genotype (Fig. 3A; other supportive data not shown). Importantly, concomitant with improved histology, we observed enhanced GFP fluorescence when ataxin-3(Q84) was coexpressed with its wild-type version compared to when the toxic protein was expressed alone (Fig. 3B). These results highlight the feasibility of membrane-targeted GFP as a method that could be employed to discover modulators of age-related degeneration.
Fig. 3.

CD8-GFP reports suppression of polyQ-dependent degeneration in fly eyes. A: Histological sections of fly eyes expressing the noted transgenes. Driver, gmr-Gal4. The presence of normal ataxin-3 suppresses the loss of ommatidia caused by pathogenic ataxin-3(Q84) by day 14. Boxes, aggregates that contain the toxic protein. B: CD8-GFP reports suppression of degeneration when wild-type ataxin-3 is coexpressed alongside its pathogenic version. Driver, gmr-Gal4. Histograms summarize quantification of GFP fluorescence. Shown are mean ± standard deviation. P values are from Student's t-tests. All flies were heterozygous for all transgenes. The genotypes of flies are listed in Table I.
Membrane-Targeted GFP Can Reliably Report Retinal Degeneration Caused by Various Toxic Proteins
Next, we investigated whether the membrane-targeted GFP method can be applied widely to both polyQ-dependent and polyQ-independent disease models in Drosophila. First, we determined whether this method could reflect degeneration caused by another polyQ disease protein, htt, which causes Huntington's disease (Todi et al., 2007; Williams and Paulson, 2008). Expression of the wild-type form of htt in fly eyes does not cause anomalies, whereas expression of a disease-causing version with 128 polyQ leads to progressive loss of the ommatidia (Romero et al., 2008).
Similar to what we observed with ataxin-3-dependent degeneration (Fig. 1A), expression of full-length htt with a polyQ repeat of 128 residues, htt(Q128), did not cause noticeable external eye degeneration in 1-day-old adults; by day 14, the external eye showed mild roughness in the periphery (Fig. 4A, arrow). By histology, we noticed signs of degeneration on day 1, highlighted by loss of definition of the ommatidial array (Fig. 4A; although other adults appeared nearly normal structurally, data not shown). By day 14, there were marked signs of degeneration by histology when htt(Q128) was expressed in fly eyes (Fig. 4A).
Fig. 4.

CD8-GFP reports degeneration caused by full-length pathogenic huntingtin. A: Images and sections of fly eyes expressing the indicated transgene driven by gmr-Gal4. Expression of the Huntington's disease protein huntingtin with 128 polyQ, htt(Q128), does not detectably impact the external eye in 1-day-old adults. By 14 days, some roughness is observable in the periphery of the external eye (arrow). Internal sections show disruption of the ommatidial array in the presence of htt(Q128). All flies were heterozygous for all constructs. B: Membrane-targeted GFP reports degeneration caused by htt(Q128). On day 1, most fly eyes that express htt(Q128) have disrupted GFP uniformity, although a small minority does not show changes in fluorescence evenness compared with controls. By days 7 and 14, htt(Q128)-expressing eyes show markedly reduced fluorescence compared with controls. Driver, gmr-Gal4. Histograms show mean ± standard deviation of GFP fluorescence from the indicated lines. P values are from Student's t-tests. Flies were heterozygous for all transgenes. The genotypes of flies are listed in Table I.
With membrane-targeted GFP, we observed mosaicism at day 1 and a marked decrease in GFP intensity at days 7 and 14 in flies expressing htt(Q128) (Fig. 4B). In a minority of cases, htt(Q128)-expressing eyes did not present with clear perturbation in GFP uniformity on day 1 (Fig. 4B), similar to the variation in the phenotype that we observed through histology. Collectively, mean GFP fluorescence was significantly lower statistically in 1-dayold adult eyes expressing htt(Q128) compared with their controls (Fig. 4B, histograms). By days 7 and 14, htt(Q128)-expressing flies consistently demonstrated a marked reduction in fluorescence (Fig. 4B). From these results, we conclude that membrane-targeted GFP can report eye degeneration caused by different polyQ disease proteins.
Next, we examined whether membrane-targeted GFP can also report degeneration caused by non-polyQ disease proteins. We investigated degeneration caused by a mutant version of a-synuclein (mutation A30P), a protein that has been linked to Parkinson's disease (Kruger et al., 1998; Athanassiadou et al., 1999). Overexpression of normal or mutated forms of α-synuclein in fly eyes was previously found to cause mild retinal degeneration (Feany and Bender, 2000); similarly, targeted expression of normal or mutated versions of a-synuclein in dopaminergic neurons in flies was neurotoxic (Feany and Bender, 2000; Auluck et al., 2002).
External eye morphology remained mostly intact in flies expressing α-synucleinA30P, with only minor signs of depigmentation (Fig. 5A). Still, we found that, similar to polyQ-dependent degeneration, membrane-targeted GFP quantitatively reported toxicity from mutant α-synucleinA30P by days 14 and 28 (Fig. 5B). We did not notice differences in GFP fluorescence between α-synucleinA30P-expressing eyes and controls at day 1 or day 7 (data not shown).
Fig. 5.
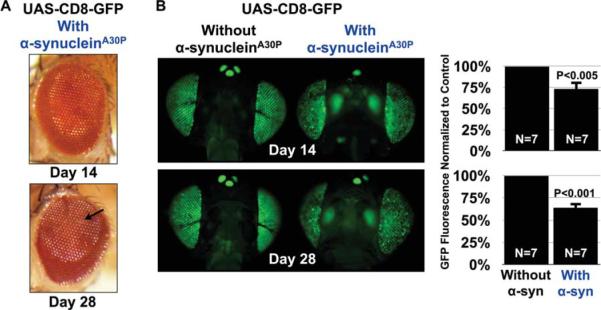
Reduced fluorescence from membrane-targeted GFP in the presence of α-synucleinA30P. A: Expression of a mutant form of asynuclein, alanine at position 30 mutated into proline, A30P, linked to familial Parkinson's disease (Kruger et al., 1998), does not greatly disrupt the external eye. Arrow, mild depigmentation. B: Reduced CD8-GFP fluorescence in the presence of α-synucleinA30P. Driver, gmr-Gal4. Shown in histograms are mean ± standard deviation of GFP fluorescence measured from flies with the indicated genotypes. P values are from Student's t-tests. All flies were heterozygous for all transgenes. The genotypes of flies are listed in Table I.
Lastly, we examined the ability of membrane-targeted GFP to report retinal degeneration caused by the expression of Aβ42. Aβ42 is a protein that is cleaved from the amyloid precursor protein (APP). This fragment of APP is found in plaques of Alzheimer's disease patients’ brains, and increased production of Ab42 is considered an etiological factor in this disease (Glenner and Wong, 1984; Kang et al., 1987; Tanzi and Bertram, 2005; Lans-bury and Lashuel, 2006; Thal et al., 2006).
Expression of Ab42 in fly eyes causes mild degeneration (Lenz et al., 2013). In our study, expression of this protein did not cause visible disruption of the external morphology of the compound eye at day 1 (Fig. 6). However, akin to the results from the proteins tested above (Figs. 2–5), eyes expressing membrane-targeted GFP and Aβ42 showed lower levels of fluorescence and increased mosaicism compared with controls not expressing this protein (Fig. 6).
Fig. 6.

Reduced fluorescence from CD8-GFP in the presence of Aβ42. Left: Expression of Aβ42 does not noticeably affect the external eye. Right: Reduced fluorescence from membrane-targeted GFP in the presence of Aβ42. Driver, gmr-Gal4. Histograms are from the quantification of GFP fluorescence from flies with the noted genotypes. Shown are mean ± standard deviation. P value is from Student's t-test. Flies were heterozygous for all transgenes. The genotypes are listed in Table I.
Overall, the findings shown in Figures 1–6 demonstrate the utility of membrane-targeted GFP as a sensitive, quantifiable, and rapid method to assess milder forms of protein-dependent degeneration in Drosophila eyes. The disease proteins that we tested led to reduced GFP fluorescence and progressive mosaicism, reflecting the loss of ommatidia. The exact mechanism of toxicity in fly eyes is probably different in each of these cases: toxicity from pathogenic ataxin-3 might be due to molecular sequelae unlike those linked to Aβ42-dependent degeneration. Also, the age of onset and rate of progression of toxicity from each protein can differ from one disease model to another, leading us to conclude that the precise ages of examination must be established on a case-by-case basis. In flies expressing htt(Q128), for example, we see clear signs of degeneration by day 7, whereas with a-synucleinA30P degeneration is observable at later time points. Nonetheless, the overall readout, i.e. loss of membrane-targeted GFP intensity and uniformity as a result of retinal degeneration, is consistent among the various toxic proteins we tested.
CONCLUSIONS
We reasoned that progressive loss of Drosophila retinal cells would be reflected by a concomitant decrease in fluorescence intensity and uniformity of GFP expressed in fly eyes. The data presented here demonstrate that membrane-targeted GFP is a sensitive reporter of Drosophila retinal degeneration caused by various human neuro-degenerative proteins. This technique is particularly useful in cases in which the external eye of the fly is not greatly or detectably impacted by a pathogenic protein, and could conceivably be used in lieu of time-consuming histology. An important benefit of this fluorescence-based method is that it is easily quantifiable, which may prove particularly valuable for undertakings that are based on numerical cutoffs for subsequent studies, as in the case of small-molecule, drug, or genetic screens.
ACKNOWLEDGMENTS
We thank Drs. Rodrigo Andrade and Michael Bannon (Wayne State University) for valuable discussions. The authors have no conflicts of interest.
Contract grant sponsor: NINDS, Contract grant number: R00NS064097 (to S.V.T.); Contract grant sponsor: NCI, Contract grant number: T32-CA009531 (to A.A.B.); Contract grant sponsor: National Ataxia Foundation (to S.V.T., W.-L.T.)
REFERENCES
- Al-Ramahi I, Perez AM, Lim J, Zhang M, Sorensen R, de Haro M, Branco J, Pulst SM, Zoghbi HY, Botas J. dAtaxin-2 mediates expanded Ataxin-1-induced neurodegeneration in a Drosophila model of SCA1. PLoS Genet. 2007;3:e234. doi: 10.1371/journal.pgen.0030234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Athanassiadou A, Voutsinas G, Psiouri L, Leroy E, Polymeropoulos MH, Ilias A, Maniatis GM, Papapetropoulos T. Genetic analysis of families with Parkinson disease that carry the Ala53Thr mutation in the gene encoding alpha-synuclein. Am J Hum Genet. 1999;65:555–558. doi: 10.1086/302486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auluck PK, Chan HY, Trojanowski JQ, Lee VM, Bonini NM. Chaperone suppression of alpha-synuclein toxicity in a Drosophila model for Parkinson's disease. Science. 2002;295:865–868. doi: 10.1126/science.1067389. [DOI] [PubMed] [Google Scholar]
- Bonini NM, Fortini ME. Human neurodegenerative disease modeling using Drosophila. Annu Rev Neurosci. 2003;26:627–656. doi: 10.1146/annurev.neuro.26.041002.131425. [DOI] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Casas-Tinto S, Zhang Y, Sanchez-Garcia J, Gomez-Velazquez M, Rincon-Limas DE, Fernandez-Funez P. The ER stress factor XBP1s prevents amyloid-beta neurotoxicity. Hum Mol Genet. 2011;20:2144–2160. doi: 10.1093/hmg/ddr100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choksi DK, Roy B, Chatterjee S, Yusuff T, Bakhoum MF, Sengupta U, Ambegaokar S, Kayed R, Jackson GR. TDP-43 Phosphorylation by casein kinase I (var epsilon) promotes oligomerization and enhances toxicity in vivo. Hum Mol Genet. 2013;23:1025–1035. doi: 10.1093/hmg/ddt498. [DOI] [PubMed] [Google Scholar]
- Costa Mdo C, Paulson HL. Toward understanding Machado-Joseph disease. Prog Neurobiol. 2012;97:239–257. doi: 10.1016/j.pneurobio.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Cara F, Duca E, Dunbar DR, Cagney G, Heck MM. Invadolysin, a conserved lipid-droplet-associated metalloproteinase, is required for mitochondrial function in Drosophila. J Cell Sci. 2013;126:4769–4781. doi: 10.1242/jcs.133306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feany MB, Bender WW. A Drosophila model of Parkinson's disease. Nature. 2000;404:394–398. doi: 10.1038/35006074. [DOI] [PubMed] [Google Scholar]
- Fernandez-Funez P, Nino-Rosales ML, de Gouyon B, She WC, Luchak JM, Martinez P, Turiegano E, Benito J, Capovilla M, Skinner PJ, McCall A, Canal I, Orr HT, Zoghbi HY, Botas J. Identification of genes that modify ataxin-1-induced neurodegeneration. Nature. 2000;408:101–106. doi: 10.1038/35040584. [DOI] [PubMed] [Google Scholar]
- Glenner GG, Wong CW. Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;120:885–890. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- Ikegami K, Heier RL, Taruishi M, Takagi H, Mukai M, Shimma S, Taira S, Hatanaka K, Morone N, Yao I, Campbell PK, Yuasa S, Janke C, Macgregor GR, Setou M. Loss of alpha-tubulin polyglutamylation in ROSA22 mice is associated with abnormal targeting of KIF1A and modulated synaptic function. Proc Natl Acad Sci U S A. 2007;104:3213–3218. doi: 10.1073/pnas.0611547104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson GR, Salecker I, Dong X, Yao X, Arnheim N, Faber PW, MacDonald ME, Zipursky SL. Polyglutamine-expanded human huntingtin transgenes induce degeneration of Drosophila photoreceptor neurons. Neuron. 1998;21:633–642. doi: 10.1016/s0896-6273(00)80573-5. [DOI] [PubMed] [Google Scholar]
- Jaiswal M, Sandoval H, Zhang K, Bayat V, Bellen HJ. Probing mechanisms that underlie human neurodegenerative diseases in Drosophila. Annu Rev Genet. 2012;46:371–396. doi: 10.1146/annurev-genet-110711-155456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang J, Lemaire HG, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH, Multhaup G, Beyreuther K, Muller-Hill B. The precursor of Alzheimer's disease amyloid A4 protein resembles a cell-surface receptor. Nature. 1987;325:733–736. doi: 10.1038/325733a0. [DOI] [PubMed] [Google Scholar]
- Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat Genet. 1998;18:106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- Lansbury PT, Lashuel HA. A century-old debate on protein aggregation and neurodegeneration enters the clinic. Nature. 2006;443:774–779. doi: 10.1038/nature05290. [DOI] [PubMed] [Google Scholar]
- Lanson NA, Jr, Maltare A, King H, Smith R, Kim JH, Taylor JP, Lloyd TE, Pandey UB. A Drosophila model of FUS-related neurodegeneration reveals genetic interaction between FUS and TDP-43. Hum Mol Genet. 2011;20:2510–2523. doi: 10.1093/hmg/ddr150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T, Luo L. Mosaic analysis with a repressible cell marker for studies of gene function in neuronal morphogenesis. Neuron. 1999;22:451–461. doi: 10.1016/s0896-6273(00)80701-1. [DOI] [PubMed] [Google Scholar]
- Lee T, Luo L. Mosaic analysis with a repressible cell marker (MARCM) for Drosophila neural development. Trends Neurosci. 2001;24:251–254. doi: 10.1016/s0166-2236(00)01791-4. [DOI] [PubMed] [Google Scholar]
- Lenz S, Karsten P, Schulz JB, Voigt A. Drosophila as a screening tool to study human neurodegenerative diseases. J Neurochem. 2013;127:453–460. doi: 10.1111/jnc.12446. [DOI] [PubMed] [Google Scholar]
- Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB, Schwartz SL, DiProspero NA, Knight MA, Schuldiner O, Padmanabhan R, Hild M, Berry DL, Garza D, Hubbert CC, Yao TP, Baehrecke EH, Taylor JP. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007;447:859–863. doi: 10.1038/nature05853. [DOI] [PubMed] [Google Scholar]
- Park J, Al-Ramahi I, Tan Q, Mollema N, Diaz-Garcia JR, Gallego-Flores T, Lu HC, Lagalwar S, Duvick L, Kang H, Lee Y, Jafar-Nejad P, Sayegh LS, Richman R, Liu X, Gao Y, Shaw CA, Arthur JS, Orr HT, Westbrook TF, Botas J, Zoghbi HY. RAS-MAPK-MSK1 pathway modulates ataxin 1 protein levels and toxicity in SCA1. Nature. 2013;498:325–331. doi: 10.1038/nature12204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulk A, Millard SS, van Swinderen B. Vision in Drosophila: seeing the world through a model's eyes. Annu Rev Entomol. 2013;58:313–332. doi: 10.1146/annurev-ento-120811-153715. [DOI] [PubMed] [Google Scholar]
- Rincon-Limas DE, Jensen K, Fernandez-Funez P. Drosophila models of proteinopathies: the little fly that could. Curr Pharm Des. 2012;18:1108–1122. doi: 10.2174/138161212799315894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero E, Cha GH, Verstreken P, Ly CV, Hughes RE, Bellen HJ, Botas J. Suppression of neurodegeneration and increased neuro-transmission caused by expanded full-length huntingtin accumulating in the cytoplasm. Neuron. 2008;57:27–40. doi: 10.1016/j.neuron.2007.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepp WH, Meyers JM, McBride AA. Sp100 provides intrinsic immunity against human papillomavirus infection. MBio. 2013;4:e00845–00813. doi: 10.1128/mBio.00845-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanzi RE, Bertram L. Twenty years of the Alzheimer's disease amyloid hypothesis: a genetic perspective. Cell. 2005;120:545–555. doi: 10.1016/j.cell.2005.02.008. [DOI] [PubMed] [Google Scholar]
- Taylor JP, Taye AA, Campbell C, Kazemi-Esfarjani P, Fischbeck KH, Min KT. Aberrant histone acetylation, altered transcription, and retinal degeneration in a Drosophila model of polyglutamine disease are rescued by CREB-binding protein. Genes Dev. 2003;17:1463–1468. doi: 10.1101/gad.1087503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thal DR, Capetillo-Zarate E, Del Tredici K, Braak H. The development of amyloid beta protein deposits in the aged brain. Sci Aging Knowledge Environ. 20062006:re1. doi: 10.1126/sageke.2006.6.re1. [DOI] [PubMed] [Google Scholar]
- Todi SV, Williams A, Paulson H. Polyglutamine repeat disorders, including Huntington's disease. In: Waxman SG, editor. Molecular neurology. 1st ed. Academic Press; London: 2007. pp. 257–276. [Google Scholar]
- Tsou WL, Sheedlo MJ, Morrow ME, Blount JR, McGregor KM, Das C, Todi SV. Systematic analysis of the physiological importance of deubiquitinating enzymes. PLoS One. 2012;7:e43112. doi: 10.1371/journal.pone.0043112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsou WL, Burr AA, Ouyang M, Blount JR, Scaglione KM, Todi SV. Ubiquitination regulates the neuroprotective function of the deubiquitinase ataxin-3 in vivo. J Biol Chem. 2013;288:34460–34469. doi: 10.1074/jbc.M113.513903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warrick JM, Paulson HL, Gray-Board GL, Bui QT, Fischbeck KH, Pittman RN, Bonini NM. Expanded polyglutamine protein forms nuclear inclusions and causes neural degeneration in Drosophila. Cell. 1998;93:939–949. doi: 10.1016/s0092-8674(00)81200-3. [DOI] [PubMed] [Google Scholar]
- Warrick JM, Morabito LM, Bilen J, Gordesky-Gold B, Faust LZ, Paulson HL, Bonini NM. Ataxin-3 suppresses polyglutamine neurodegeneration in Drosophila by a ubiquitin-associated mechanism. Mol Cell. 2005;18:37–48. doi: 10.1016/j.molcel.2005.02.030. [DOI] [PubMed] [Google Scholar]
- Whitworth AJ. Drosophila models of Parkinson's disease. Adv Genet. 2011;73:1–50. doi: 10.1016/B978-0-12-380860-8.00001-X. [DOI] [PubMed] [Google Scholar]
- Williams AJ, Paulson HL. Polyglutamine neurodegeneration: protein misfolding revisited. Trends Neurosci. 2008;31:521–528. doi: 10.1016/j.tins.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Shih HP, Vigont V, Hrdlicka L, Diggins L, Singh C, Mahoney M, Chesworth R, Shapiro G, Zimina O, Chen X, Wu Q, Glushankova L, Ahlijanian M, Koenig G, Mozhayeva GN, Kaznacheyeva E, Bezprozvanny I. Neuronal store-operated calcium entry pathway as a novel therapeutic target for Huntington's disease treatment. Chem Biol. 2011;18:777–793. doi: 10.1016/j.chembiol.2011.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Binari R, Zhou R, Perrimon N. A genomewide RNA interference screen for modifiers of aggregates formation by mutant Huntingtin in Drosophila. Genetics. 2010;184:1165–1179. doi: 10.1534/genetics.109.112516. [DOI] [PMC free article] [PubMed] [Google Scholar]