

Abstract
We report a practical synthesis method of the reagent PhenoFluor on decagram scale, provide a new formulation of PhenoFluor as a toluene solution, which should decrease challenges associated with the moisture sensitivity of the reagent, and expand the substrate scope of deoxyfluorination with PhenoFluor to heteroaromatics.
Introduction
Over the past 5 years, a variety of new fluorination reactions of arenes have been reported,1−4 but reliable and practical methods that are general are still lacking. In our opinion, deoxyfluorination of phenols with commercially available PhenoFluor5 is currently the most versatile method for the synthesis of functionally complex small-molecule aryl fluorides in terms of substrate scope and operational simplicity. However, PhenoFluor is currently expensive ($0.80/mg),6 and its synthesis on larger scale has been challenging. Herein, we report an improved synthesis of PhenoFluor amenable to decagram scale, as well as a new formulation of PhenoFluor as a toluene solution. Combination of both should make access to and use of PhenoFluor more practical, more reliable, and less expensive.
Results and Discussion
The procedure described in our original report5a provides analytically pure PhenoFluor on gram scale, but it is not amenable to large-scale synthesis of the reagent. In the final step of the original PhenoFluor synthesis—nucleophilic fluorination of the chloroimidazolium salt 1(7) with CsF —acetonitrile is used as a solvent to ensure adequate solubility of CsF.8 It is crucial to maintain anhydrous conditions during the synthesis of PhenoFluor, which readily hydrolyses to the corresponding urea 2 (Scheme 1), the removal of which is problematic. Acetonitrile is notoriously difficult to dry, and as a consequence, the yield and purity of the final product are variable and difficult to control. To overcome the synthetic challenge and establish a robust synthetic method potentially applicable to the process-scale synthesis of PhenoFluor, we sought to avoid acetonitrile as a solvent and re-examine alternatives for the fluorination of chloroimidazolium salt 1.
Scheme 1. Hydrolysis of PhenoFluor to urea 2.

PhenoFluor can be prepared in two steps from the commercially available carbene 3, or in a five-step sequence from inexpensive 2,6-diisopropylaniline (Scheme 2). Condensation of 2,6-diisopropylaniline with 40% aqueous glyoxal to diimine 4 (crystalline yellow solid), followed by treatment of the diimine with paraformaldehyde in the presence of chlorotrimethylsilane in hot ethyl acetate afforded IPr-HCl (5) in 65% yield (2 steps). IPr-HCl (5) was deprotonated by potassium tert-butoxide and the resulting isolated IPr carbene (3) was treated with hexachloroethane in tetrahydrofuran to afford chloroimidazolium salt 1 in 81% yield over two steps. The four-step synthesis sequence to chloroimidazolium salt 1 does not require any special purification method, and we have prepared more than 40 g of solid, moisture-insensitive chloroimidazolium salt 1. In our original report, the carbene 3 was formed in situ before chlorination. However, carbene 3 is stable enough to be handled on the bench and was isolated under air through simple filtration and concentration.9 This two-step procedure was executed on 50 g scale. In addition, isolation of the carbene proved to be advantageous to obtain chloroimidazolium salt in higher purity, which facilitated isolation of pure PhenoFluor (vide infra).
Scheme 2. Preparation of chloroimidazolium salt 1.

We subsequently evaluated fluorination of the chloroimidazolium salt 1, and found that both solvent and fluoride source have a substantial impact on the effectiveness of PhenoFluor synthesis. We established that PhenoFluor is stable in hot, dry toluene, and no hydrolysis to the corresponding urea 2 was observed, in contrast to the observed hydrolysis upon heating of PhenoFluor in hot acetonitrile. We were pleased to find that toluene is a suitable solvent for the preparation of PhenoFluor: fluorination of 1 with CsF in a toluene suspension afforded PhenoFluor in high yield and purity on gram scale. The other alkali fluorides such as LiF, NaF, and KF were less reactive than CsF, and other fluoride sources (ZnF2, AgF) did not give the desired product. Furthermore, soluble fluoride sources, such as TMAF (tetramethylammonium fluoride) and TASF (tris(dimethylamino)sulfonium difluorotrimethylsilicate), were not effective for PhenoFluor formation.
The amounts of toluene and CsF, as well as the mesh size of the CsF and stirring rate were found to be important variables for efficient PhenoFluor synthesis. Both chloroimidazolium salt 1 and CsF should be finely ground prior to the reaction, likely due to their low solubility in toluene. We also found that larger amounts of toluene and CsF resulted in shorter reaction times. Completion of the reaction was judged by visual inspection: when no more solid chloroimidazolium salt was observed, the reaction mixtures were cooled. We ultimately found that for 25.0 g of 1, 10 equiv of CsF (82.6 g) and 181 mL of toluene were the most practical reaction parameters in a laboratory setting, which required heating at 100 °C for 96 h (Scheme 3). The reaction time can be shortened by more vigorous stirring, and larger quantities of toluene. Filtration to remove solids, followed by washing with acetonitrile (3 × 15 mL) afforded PhenoFluor (21.6 g, 98% purity) in 93% yield. We should note that no hydrolysis was observed on this scale and that the new procedure afforded more than 20 times the amount of PhenoFluor per batch compared to that from the original procedure.
Scheme 3. Large-scale synthesis of PhenoFluor.

The thermal stability and safety profile of PhenoFluor were evaluated by differential scanning calorimetry (DSC) (Figure 1). An exotherm of 0.15 kcal/g was observed at PhenoFluor’s decomposition temperature (213 °C). Compared to other deoxyfluorination reagents, PhenoFluor has a better safety profile due to its higher decomposition temperature and lack of a sharp, narrow exothermic peak in the DSC.5b
Figure 1.
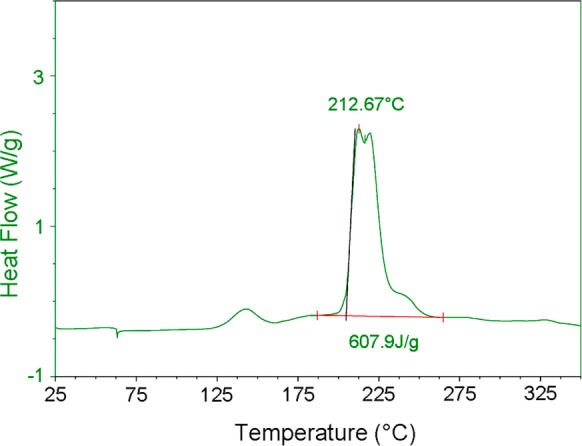
Differential scanning calorimetry of PhenoFluor.
Reactions with PhenoFluor must be carried out with dry solvents; wet solvents will result in the unproductive hydrolysis and formation of urea 2 (Scheme 1). PhenoFluor can be handled in ambient atmosphere for short periods of time; however, extended storage in ambient, moist atmosphere will result in partial or full hydrolysis. Additionally, it is important to note that the CsF used in deoxyfluorination reactions must also be carefully dried. CsF is hygroscopic, and wet CsF will hydrolyze PhenoFluor. We found it most convenient to dry CsF prior to use at 200 °C under vacuum for 24 h.
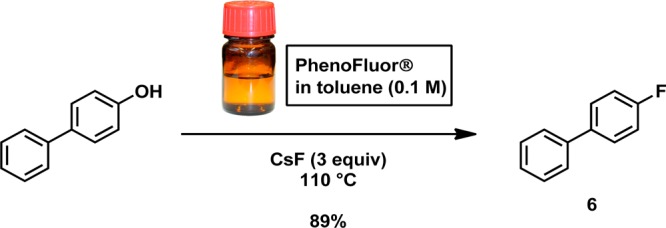
To increase the utility and straightforwardness of deoxyfluorination with PhenoFluor, we have prepared a 0.10 M solution of PhenoFluor in toluene. Deoxyfluorination reactions with the solution proceeded as smoothly as with solid PhenoFluor (Scheme 4). We have found the PhenoFluor solution is stable and storable in an inert atmosphere, such as a sure seal bottle. Dispensing from a sure seal bottle should further prevent adventitious hydrolysis, although CsF must still be used in dry form. Commercial availability of toluene solutions of PhenoFluor would facilitate PhenoFluor use.
Scheme 4. Deoxyfluorination reaction of PhenoFluor solution in toluene.
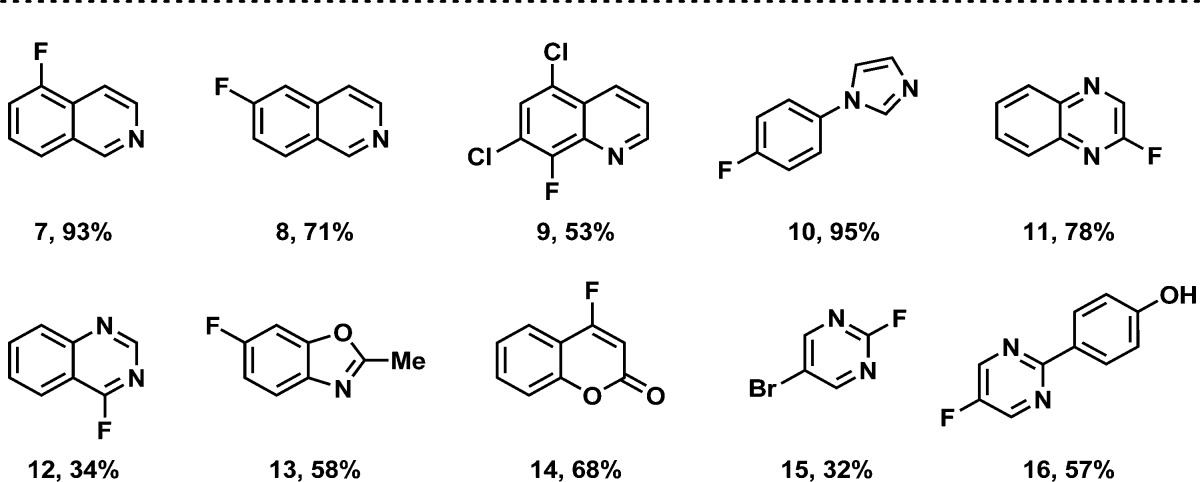
As demonstrated in our previous publications,5 the functional group tolerance of deoxyfluorination with PhenoFluor is high. Because PhenoFluor is currently being used in the pharmaceutical and agrochemical industries, we have evaluated a variety of heteroaromatic starting materials, and show those that give productive deoxyfluorination in Table 1. Basic nitrogen-containing heteroaromatics, such as isoquinolines, quinoline, and imidazole were well tolerated in deoxyfluorination reactions (7–10). Pyrimidine 15 was obtained in 32% yield, possibly due to its high volatility (yield determined by NMR using an internal standard is 62%). Selective deoxyfluorination occurs at the least electron-rich ring (16). We found that carboxamides with N–H bonds are not tolerated in PhenoFluor deoxyfluorination, although tertiary amides can be used.5a In addition, ortho substitution of phenols can result in inefficient or unproductive reactions, especially when the phenol is electron rich.
Table 1. Deoxyfluorination of heteroaromatics.
Conclusions
In conclusion, we have developed a robust synthetic method of PhenoFluor amenable to decagram scale. Furthermore, the synthetic utility is further increased by formulating PhenoFluor as a solution in toluene. PhenoFluor has a better safety profile than conventional deoxyfluorination reagents, but is more expensive. Our method shall give more facile access to PhenoFluor for more robust late-stage deoxyfluorination of alcohols and phenols.
Experimental Details
N,N′-1,3-Bis(2,6-diisopropylphenyl)imidazol-2-ylidene (3)
In an N2-filled glovebox, N,N′-1,3-bis(2,6-diisopropylphenyl)imidazolium chloride (5) (50.0 g, 118 mmol, 1.00 equiv) and t-BuOK (14.0 g, 125 mmol, 1.06 equiv) were placed in a round-bottom flask. THF (240 mL) was added, and the flask was capped with a rubber septum and then removed from the glovebox. The mixture was stirred for 3.5 h at 23 °C, then the solvent was evaporated in vacuo. The residue was dissolved in toluene (450 mL) with gentle heating (50–60 °C), and the hot solution was filtered through a pad of Celite eluting with toluene (50 mL). The filtrate was concentrated in vacuo to afford 39.1 g of the title compound as an off-white solid. The material 3 was used in the next step without any further purification.
N,N′-1,3-Bis(2,6-diisopropylphenyl)-2-chloroimidazolium chloride (1)
To a mixture of N,N′-1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene (3) (39.1 g) in THF (240 mL) was added 1,1,1,2,2,2-hexachloroethane (26.2 g, 111 mmol) at −45 °C. The mixture was warmed to 23 °C and stirred for 20 h. The reaction mixture was filtered, then the filter cake was washed with THF (3 × 100 mL) and toluene (2 × 100 mL) to afford 44.0 g of the title compound as a white solid (81% yield from 5). 1H NMR (600 MHz, CD2Cl2, 23 °C, δ): 8.87 (s, 2H), 7.67 (t, J = 7.8 Hz, 2H), 7.45 (d, J = 7.8 Hz, 4H), 2.36 (m, 4H), 1.32 (d, J = 7.0 Hz, 12H), 1.22 (d, J = 7.0 Hz, 12H). 13C NMR (125 MHz, CD2Cl2, 23 °C, δ): 145.5, 133.9, 133.1, 128.9, 128.4, 125.6, 29.7, 24.4, 23.5.
N,N′-1,3-Bis(2,6-diisopropylphenyl)-2,2-difluoroimidazolidene (PhenoFluor)
N,N′-1,3-Bis(2,6-diisopropylphenyl)-2-chloroimidazolium chloride (1) was finely ground using a mortar and dried at 80 °C under vacuum for 24 h. CsF was finely ground using a mortar in a glovebox then was removed from the glovebox and dried at 200 °C under vacuum for 24 h, prior to use. In a glovebox, N,N′-1,3-bis(2,6-diisopropylphenyl)-2-chloroimidazolium chloride (1) (25.0 g, 54.4 mmol, 1.00 equiv) and CsF (82.6 g, 544 mmol, 10.0 equiv) were placed in a pressure flask. Toluene (181 mL) was added and the flask was sealed, then removed from the glovebox. The flask was sonicated until the mixture appeared creamy (1 h), then stirred vigorously at 100 °C for 96 h. Completion of the reaction was judged by visual inspection: when stirring was stopped, chloroimidazolium salt was floating in the reaction mixture, while cesium salts dropped onto the bottom quickly. When no more chloroimidazolium salt was observed, the mixture was cooled to 23 °C. The flask was brought into a glovebox, and the mixture was filtered through a pad of Celite eluting with toluene (40 mL). The filtrate was concentrated and dried in vacuo (approximately 6–8 h). The residual solid was ground, washed with MeCN (3 × 15 mL) and dried on frit to afford 21.6 g (98% purity) of the title compound as an off-white solid (93% yield). PhenoFluor should be stored in an inert gas atmosphere. 1H NMR (600 MHz, C6D6, 23 °C, δ): 7.22 (t, J = 8.2 Hz, 2H), 7.12 (d, J = 8.2 Hz, 4H), 5.71 (s, 2H), 3.61 (m, 4H), 1.36 (d, J = 7.0 Hz, 12H), 1.18 (d, J = 7.0 Hz, 12H). 13C NMR (125 MHz, C6D6, 23 °C, δ): 150.9, 131.6, 129.9, 126.4 (t, J = 247.0 Hz), 124.5, 112.8, 28.9, 25.6, 24.2. 19F NMR (375 MHz, C6D6, 23 °C, δ): −34.0.
General Procedure for Deoxyfluorination with Solid PhenoFluor
PhenoFluor is moisture sensitive, and should be appropriately stored in an inert gas atmosphere. CsF was finely ground using a mortar in a glovebox, then was removed from the glovebox, and dried at 200 °C under vacuum for 24 h, prior to use. Reaction solvents and reagents must be dried for optimal results. In a glovebox, a heteroaromatic compound (0.500 mmol. 1.00 equiv), CsF (228 mg, 1.50 mmol, 3.00 equiv) and PhenoFluor (256–320 mg, 0.600–0.750 mmol, 1.20–1.50 equiv) were placed in a vial. Toluene or dioxane (5.0 mL) was added. The vial was sealed and removed from the glovebox. The mixture was stirred at 23 °C for 30 min and subsequently heated at 110 °C for 24 h. The mixture was cooled to 23 °C and then filtered through a pad of Celite eluting with CH2Cl2 (3 × 4 mL). The filtrate was concentrated in vacuo, and the residue was purified by flash silica gel column chromatography to afford the fluorinated compound.
General Procedure for Deoxyfluorination with PhenoFluor Solution in Toluene (c = 0.100 M)
Under air, a phenol (1.00 equiv) and previously dried CsF (3.00 equiv) were placed in a vial. The vial was evacuated and backfilled with N2 gas (3 times). A solution of PhenoFluor in toluene (0.100 M, 1.20 equiv) was added via syringe. The mixture was stirred at 23 °C for 30 min, then at 110 °C for 24 h. Once cooled to 23 °C, the mixture was filtered through a pad of Celite eluting with CH2Cl2 (3 × 3 mL). The filtrate was concentrated in vacuo and then purified by flash silica gel column chromatography.
Acknowledgments
We thank Filippo Sladojevich and Constanze N. Neumann for helpful discussions, and the NIH (NIH-NIGMS GM088237) as well as Daiichi-Sankyo Co., Ltd. for financial support.
Supporting Information Available
Detailed experimental procedures and spectroscopic characterization for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare the following competing financial interest(s): PhenoFluor is commercially available and licensed to SciFluor Life Sciences. T.R. may financially benefit from PhenoFluor sales.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- For recent reviews of fluorination reactions, see:; a Liang T.; Neumann C. N.; Ritter T. Angew. Chem., Int. Ed. 2013, 52, 8214–8264. [DOI] [PubMed] [Google Scholar]; b Ruzziconi R.; Buonerba F. J. Fluor. Chem. 2013, 152, 12–28. [Google Scholar]; c Hollingworth C.; Gouverneur V. Chem. Commun. 2012, 48, 2929–2942. [DOI] [PubMed] [Google Scholar]; d Jin Z.; Hammond G. B.; Xu B. Aldrichimica Acta 2012, 45, 67–83. [Google Scholar]; e Furuya T.; Klein J. E. M. N.; Ritter T. Synthesis 2010, 1804–1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For Pd-mediated aryl fluoride synthesis, see:; a Hull K. L.; Anani W. Q.; Sanford M. S. J. Am. Chem. Soc. 2006, 128, 7134–7135. [DOI] [PubMed] [Google Scholar]; b Ball N. D.; Sanford M. S. J. Am. Chem. Soc. 2009, 131, 3796–3797. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Furuya T.; Kaiser H. M.; Ritter T. Angew. Chem., Int. Ed. 2008, 47, 5993–5996. [DOI] [PubMed] [Google Scholar]; d Furuya T.; Ritter T. J. Am. Chem. Soc. 2008, 130, 10060–10061. [DOI] [PubMed] [Google Scholar]; e Furuya T.; Benitez D.; Tkatchouk E.; Strom A. E.; Tang P.; Goddard W. A. III; Ritter T. J. Am. Chem. Soc. 2010, 132, 3793–3807. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Mazzotti A. R.; Campbell M. G.; Tang P.; Murphy J. M.; Ritter T. J. Am. Chem. Soc. 2013, 135, 14012–14015. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Wang X.; Mei T.-S.; Yu J.-Q. J. Am. Chem. Soc. 2009, 131, 7520–7521. [DOI] [PubMed] [Google Scholar]; h Chan K. S. L.; Wasa M.; Wang X.; Yu J.-Q. Angew. Chem., Int. Ed. 2011, 50, 9081–9084. [DOI] [PubMed] [Google Scholar]; i Watson D. A.; Su M.; Teverovskiy G.; Zhang Y.; García-Fortanet J.; Kinzel T.; Buchwald S. L. Science 2009, 325, 1661–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]; j Noël T.; Maimone T. J.; Buchwald S. L. Angew. Chem., Int. Ed. 2011, 50, 8900–8903. [DOI] [PMC free article] [PubMed] [Google Scholar]; k Maimone T. J.; Milner P. J.; Kinzel T.; Zhang Y.; Takase M. K.; Buchwald S. L. J. Am. Chem. Soc. 2011, 133, 18106–18109. [DOI] [PMC free article] [PubMed] [Google Scholar]; l Lee H. G.; Milner P. J.; Buchwald S. L. Org. Lett. 2013, 15, 5602–5605. [DOI] [PMC free article] [PubMed] [Google Scholar]; m Lee H. G.; Milner P. J.; Buchwald S. L. J. Am. Chem. Soc. 2014, 136, 3792–3795. [DOI] [PMC free article] [PubMed] [Google Scholar]; n Wannberg J.; Wallinder C.; Ünlüsoy M.; Sköld C.; Larhed M. J. Org. Chem. 2013, 78, 4184–4189. [DOI] [PubMed] [Google Scholar]
- For other transition metal mediated aryl fluoride synthesis, see:; a Furuya T.; Strom A. E.; Ritter T. J. Am. Chem. Soc. 2009, 131, 1662–1663. [DOI] [PubMed] [Google Scholar]; b Furuya T.; Ritter T. Org. Lett. 2009, 11, 2860–2863. [DOI] [PubMed] [Google Scholar]; c Tang P.; Furuya T.; Ritter T. J. Am. Chem. Soc. 2010, 132, 12150–12154. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Tang P.; Ritter T. Tetrahedron 2011, 67, 4449–4454. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Fier P. S.; Hartwig J. F. Science 2013, 342, 956–960. [DOI] [PubMed] [Google Scholar]; f Casitas A.; Canta M.; Solà M.; Costas M.; Ribas X. J. Am. Chem. Soc. 2011, 133, 19386–19392. [DOI] [PubMed] [Google Scholar]; g Fier P. S.; Hartwig J. F. J. Am. Chem. Soc. 2012, 134, 10795–10798. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Fier P. S.; Luo J.; Hartwig J. F. J. Am. Chem. Soc. 2013, 135, 2552–2559. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Ye Y.; Sanford M. S. J. Am. Chem. Soc. 2013, 135, 4648–4651. [DOI] [PubMed] [Google Scholar]; j Ye Y.; Schimler S. D.; Hanley P. S.; Sanford M. S. J. Am. Chem. Soc. 2013, 135, 16292–16295. [DOI] [PubMed] [Google Scholar]; k Ichiishi N.; Canty A. J.; Yates B. F.; Sanford M. S. Org. Lett. 2013, 15, 5134–5137. [DOI] [PMC free article] [PubMed] [Google Scholar]; l Truong T.; Klimovica K.; Daugulis O. J. Am. Chem. Soc. 2013, 135, 9342–9345. [DOI] [PMC free article] [PubMed] [Google Scholar]; m Mu X.; Zhang H.; Chen P.; Liu G. Chem. Sci. 2014, 5, 275–280. [Google Scholar]; n Lee E.; Hooker J. M.; Ritter T. J. Am. Chem. Soc. 2012, 134, 17456–17458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For other recent aryl fluoride synthesis, see:; a Yamada S.; Gavryushin A.; Knochel P. Angew. Chem., Int. Ed. 2010, 49, 2215–2218. [DOI] [PubMed] [Google Scholar]; b Anbarasan P.; Neumann H.; Beller M. Angew. Chem., Int. Ed. 2010, 49, 2219–2222. [DOI] [PubMed] [Google Scholar]; c Wang B.; Qin L.; Neumann K. D.; Uppaluri S.; Cerny R. L.; DiMagno S. G. Org. Lett. 2010, 12, 3352–3355. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Vints I.; Gatenyo J.; Rozen S. J. Org. Chem. 2013, 78, 11794–11797. [DOI] [PubMed] [Google Scholar]
- a Tang P.; Wang W.; Ritter T. J. Am. Chem. Soc. 2011, 133, 11482–11484. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Sladojevich F.; Arlow S. I.; Tang P.; Ritter T. J. Am. Chem. Soc. 2013, 135, 2470–2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Available from Sigma-Aldrich and Strem Chemical Inc.
- Mendoza-Espinosa D.; Donnadieu B.; Bertrand G. J. Am. Chem. Soc. 2010, 132, 7264–7265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solubility of alkali metal fluorides, see:Wynn D. A.; Roth M. M.; Pollard B. D. Talanta 1984, 31, 1036–1040. [DOI] [PubMed] [Google Scholar]
- Arduengo A. J. III; Krafczyk R.; Schmutzler R. Tetrahedron 1999, 55, 14523–14534. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.