

Abstract
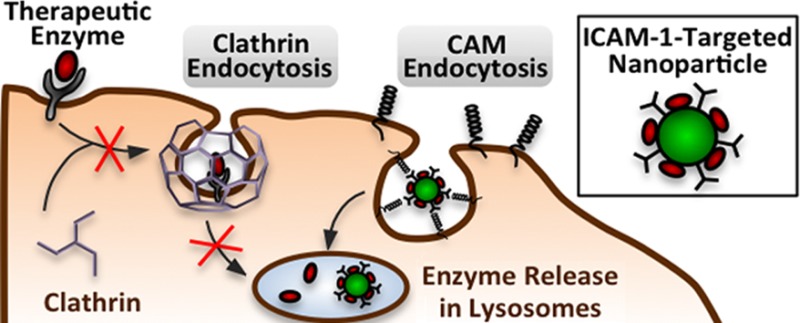
Drugs often use endocytosis to achieve intracellular delivery, either by passive uptake from the extracellular fluid or by active targeting of cell surface features such as endocytic receptors. An example is enzyme replacement therapy, a clinically practiced treatment for several lysosomal storage diseases where glycosylated recombinant enzymes naturally target the mannose-6-phosphate receptor and are internalized by clathrin mediated endocytosis (CME). However, lysosomal substrate accumulation, a hallmark of these diseases, has been indirectly linked to aberrant endocytic activity. These effects are poorly understood, creating an obstacle to therapeutic efficiency. Here we explored endocytic activity in fibroblasts from patients with type A Niemann–Pick disease, a lysosomal storage disease characterized by acid sphingomyelinase (ASM) deficiency. The uptake of fluid phase markers and clathrin-associated ligands, formation of endocytic structures, and recruitment of intracellular clathrin to ligand binding sites were all altered, demonstrating aberrant CME in these cells. Model polymer nanocarriers targeted to intercellular adhesion molecule-1 (ICAM-1), which are internalized by a clathrin-independent route, enhanced the intracellular delivery of recombinant ASM more than 10-fold compared to free enzyme. This strategy reduced substrate accumulation and restored clathrin endocytic activity to wild-type levels. There appears to be a relationship between lysosomal storage and diminished CME, and bypassing this pathway by targeting ICAM-1 may enhance future therapies for lysosomal storage diseases.
Keywords: type A−B Niemann−Pick disease, clathrin endocytosis, CAM-mediated endocytosis, ICAM-1-targeted nanocarriers, enzyme replacement therapy
Introduction
Drug delivery strategies commonly use endocytosis to access the cell interior; however, the potential for disease to affect endocytic behavior remains relatively unexplored, hindering the design of robust therapeutics. Endocytosis encompasses several energy-dependent and highly regulated processes through which cells internalize substances and objects from their surroundings in membrane-bound vesicles, permitting essential functions such as nutrient uptake, signal transmission, plasmalemma recycling, and pathogen defense.1−3 Many endocytic pathways have been described, each distinguished by its molecular players, vesicle size, and cargo.2 Perhaps the most studied and most widespread mechanism in eukaryotic cells is clathrin-mediated endocytosis (CME), recognizable via electron microscopy by the distinct lattice of clathrin proteins that enclose newly formed vesicles.3−5 Clathrin-coated vesicle formation begins at plasma membrane invagination sites called pits and ultimately involves as many as 30 proteins (clathrin, adaptor protein-2, epsin, dynamin, and others) in a multistep process of recruitment, budding, and vesicle release into the cytosol.5,6 As clathrin-coated vesicles form, adaptor protein-2 and other specific adaptor proteins recruit transmembrane receptors into the fledgling vesicle by binding to their cytosolic domains and the clathrin lattice simultaneously.5,6 Receptors of transferrin, insulin, low-density lipoprotein, growth factors, and numerous other ligands have been shown to internalize in this fashion.5−7 Receptor recruitment mechanisms vary, and internalization can be independent of ligand attachment (constitutive) or specific to the conformation of a ligand–receptor complex.5 As a result, the clathrin pathway provides flexibility for drug design, enabling continuous uptake of compounds in the fluid phase and selective uptake of drugs conjugated to antibodies, peptides, and natural ligands that target receptors in clathrin-coated vesicles.7
Lysosomal storage diseases offer an example where CME is key to intracellular drug delivery. They include around 50 different inherited disorders where a dysfunctional lysosomal protein (hydrolase, transmembrane protein, chaperone, etc.) leads to aberrant accumulation of nondegraded substrates in this compartment.8 Patients present with varying, but often devastating symptoms including abnormal tissue development, peripheral organ enlargement, seizures, and neurodegeneration.8 Although treatment options remain limited, intravenous infusion of recombinant enzymes has proven therapeutic in the clinics for a few lysosomal storage diseases (Gaucher, Fabry, Pompe, and mucopolysaccharidosis types I, II, and VI).9,10 Known as enzyme replacement therapy, this treatment takes advantage of exposed mannose-6-phosphate residues on lysosomal hydrolases, which target cell surface mannose-6-phosphate receptors and internalize in clathrin-coated vesicles.11,12 Upon trafficking to endosomal compartments, the enzyme–receptor complex dissociates, the receptor recycles to the cell surface, and the enzyme is delivered to the lysosome.11,12
Several obstacles have hindered enzyme replacement therapy, including enzyme clearance from the bloodstream, immune reactions, and difficulty traversing cellular barriers.12 Another confounding variable may be aberrant cell behavior owing to substrate storage. Several studies have described intracellular “traffic jams”, including altered transport of endocytic vesicles, impaired recycling of membrane proteins to the cell surface, cytoskeletal rearrangement, altered signaling, and diminished biosynthesis.13−16 Lipidoses in particular (lysosomal storage diseases characterized by lipid accumulation) have been associated with abnormal trafficking of plasma membrane materials (e.g., sphingolipids and the mannose-6-phosphate receptor) to subcellular compartments.17−20 These findings offer indirect evidence of endocytic deficits in these diseases, potentially reducing the efficiency of current therapies; yet, the internalization of other receptors amenable for drug targeting warrants further investigation.
We have previously reported lysosomal enzyme delivery (acid sphingomyelinase, α-galactosidase, and α-glucosidase; deficient in type A–B Niemann–Pick, Fabry, and Pompe diseases, respectively) by targeting intercellular adhesion molecule-1 (ICAM-1).21−26 This receptor is highly expressed in inflammatory conditions, including lysosomal storage disease, and mediates a nonclassical endocytic mechanism independent of clathrin, caveolin, and other known pathways.23,27 ICAM-1 engagement induces nano- and micrometer-scale vesicle formation, permitting targeting and internalization of nanocarriers of a variety of dimensions.28 Cell culture and mouse models have demonstrated that polymer nanocarriers targeted to ICAM-1 (anti-ICAM NCs) markedly increase accumulation of enzymes in lysosomes and relieve lysosomal storage.21,22,24,25,28 Importantly, anti-ICAM NC uptake seems to surpass that of otherwise equivalent nanocarriers targeted to the transferrin receptor, a receptor associated with clathrin-coated vesicles.26 Furthermore, these nanocarriers readily internalize even when classical endocytic pathways are pharmacologically inhibited, suggesting a useful treatment modality for pathological endocytic dysfunction.23,29
Here we have examined clathrin-mediated endocytic activity in wild-type fibroblasts and fibroblasts from patients with type A Niemann–Pick disease (NPD). In these cells, the absence of acid sphingomyelinase (ASM) leads to marked accumulation of sphingomyelin in intracellular compartments, which has been linked to aberrant mannose-6-phosphate receptor internalization.19,30 Whether this represents an isolated endocytic deficit or a symptom of broader dysfunction remains an open question. We therefore examined the uptake of other clathrin-associated ligands, the formation of clathrin-coated vesicles, and the advantage of targeting ICAM-1 as an alternative enzyme delivery strategy.
Experimental Section
Antibodies and Reagents
The murine monoclonal antibody against human ICAM-1 (anti-ICAM) was R6.5 from the American Type Culture Collection, as in our previous studies.22 Alexa Fluor 594 transferrin, BODIPY-FL C12–Sphingomyelin, 10000 MW Texas Red dextran, and fluorescent secondary antibodies were from Molecular Probes (Eugene, OR). Anti-transferrin and anti-clathrin heavy chain were from Calbiochem (La Jolla, CA). Green Fluoresbrite 100 nm polystyrene beads were from Polysciences (Warrington, PA). Recombinant human ASM was a gift from Dr. Edward Schuchman (Department of Genetics and Genomics Sciences, Mount Sinai School of Medicine, New York, NY).31 Unless otherwise noted, all other reagents were from Sigma-Aldrich (St. Louis, MO).
Cell Cultures
Wild-type and ASM-deficient skin fibroblasts from type A NPD patients (homozygous for the R496L mutation) were kindly provided by Dr. Edward Schuchman. Cells were seeded on glass coverslips and cultured in Dulbecco’s Modified Eagle Medium (Gibco BRL, Grand Island, NY) supplemented with 10% fetal bovine serum, 2 mM glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin. Cells were incubated at 37 °C, 5% CO2, and 95% relative humidity and, where indicated, stimulated with 10 ng/mL tumor necrosis factor-α (TNFα) overnight prior to the assay to upregulate ICAM-1 expression.22
Preparation of ASM-Loaded Nanocarriers Targeted to ICAM-1
Model polymer nanocarriers were prepared by coating 100 nm diameter polystyrene beads by surface absorption with a mix of anti-ICAM and ASM (anti-ICAM/ASM NCs) at a 50:50 mass ratio as described.22 Unbound molecules were separated by centrifugation at 13000g for 3 min and removed. Coated carriers were resuspended in a solution of phosphate buffered saline supplemented with 1% bovine serum albumin and then sonicated at low power to separate aggregates. This protocol has been shown to produce carriers with active ASM capable of degrading accumulated lysosomal substrates.22 Where indicated, either anti-ICAM or the enzyme cargo were labeled with 125I, and the amount of radiolabeled antibody or enzyme per carrier was determined with a gamma counter. Final carrier diameter (190 ± 7 nm) and polydispersity index (0.16 ± 0.02) were determined by dynamic light scattering as described previously.23 Enzyme loading efficiency was 80% with a content of 230 ± 24 ASM molecules and 135 ± 17 anti-ICAM molecules per carrier. Lysosomal enzymes have been shown to remain steadily attached to these nanocarrier formulations, with only minimal release by mechanical stress (∼10% release by pipetting, centrifugation, or sonication), storage (<5% release after 3 days in saline at 4 °C), or physiological fluid (∼10% release after 5 h in serum at 37 °C).21,25 While these prototype carriers are not intended for clinical use, they are a valid model with coating efficacy, targeting, and intracellular transport comparable to anti-ICAM NCs made of poly(lactic-co-glycolic acid), a material approved by the US Food and Drug Administration.24,32
Transferrin Uptake, Clathrin Distribution, and Vesicle Formation
Wild-type and NPD fibroblasts were incubated with medium containing 200 μg/mL Alexa Fluor-594 transferrin for 1 h at 37 °C to measure uptake by CME.23 Cells were then washed and fixed with 2% paraformaldehyde at room temperature. To distinguish the surface-bound fraction of transferrin, fixed cells were stained with goat anti-transferrin then fluorescein isothiocyanate (FITC)-labeled rat anti-goat IgG. This protocol renders double-labeled surface transferrin (green + red) vs single-labeled internal transferrin. Alternatively, to measure the colocalization of clathrin and transferrin, fixed cells were permeabilized with 0.2% Triton X-100 and stained with mouse anti-human clathrin heavy chain, then FITC goat anti-mouse IgG. The samples were washed and analyzed by fluorescence microscopy. Vesicle formation was confirmed by transmission and scanning electron microscopy (TEM and SEM) in cells treated with transferrin for 30 min. For TEM or SEM, ∼10 different frames from two independent cell samples were examined. Pits were defined as invaginations of the plasmalemma ≥50 nm in diameter with a visible electron-dense protein coat under the cytosolic leaflet of the plasmalemma in the case of TEM, or an electron-light halo surrounding the opening of the invagination for SEM. Semiquantitve analysis was only obtained from SEM, where each frame encompassed ∼5 pits (≥50 pits analyzed per condition).
Fluorescent samples were observed with an Eclipse TE2000-U microscope, using 60× PlanApo objectives, and filters optimized for Texas Red and FITC fluorescence (Nikon, Melville, NY). Color channels were imaged separately with an Orca-ICCD camera (Hamamatsu, Bridgewater, NJ), merged, and analyzed with ImagePro 3.0 software (Media Cybernetics, Silver Spring, MD). The transferrin channel was pseudocolored green and the secondary stains red. As described previously, a custom ImagePro macro was used to quantify all surface (yellow) vs internalized (green) material.23
Bulk Endocytosis and Endocytic Pathway Inhibition
Wild-type and NPD fibroblasts were pretreated for 30 min at 37 °C with control media or media supplemented with one or a combination of the following pharmacological inhibitors: 50 μM monodansylcadaverine (MDC; inhibits clathrin endocytosis), 1 μg/mL filipin (inhibits caveolar endocytosis), and 3 mM amiloride (inhibits CAM endocytosis).23 Cells were then incubated for 1 h with inhibitor-supplemented media containing 1 mg/mL Texas Red dextran (a nondegradable fluid phase marker for endocytosis). Cells were fixed and the number of fluorescent dextran-filled compartments were quantified by fluorescence microscopy using an algorithm that quantifies fluorescent objects whose intensity is above a threshold background level.21
Delivery and Functional Activity of ASM
Following overnight treatment with TNFα to mimic inflammation in many lysosomal storage diseases,12 NPD fibroblasts were incubated at 37 °C with a similar concentration of free 125I-ASM or 125I-ASM on anti-ICAM NCs for 1 h (∼2.3 μg/mL). Cells were then washed to remove unbound ASM and scraped from the well in a lysis buffer (2% Triton X-100). The mass of ASM present in cell lysates was determined in a gamma counter as described.22
ASM activity was measured with an AmplexRed Sphingomyelinase Assay Kit (Molecular Probes, Eugene, OR). TNFα-stimulated NPD fibroblasts were incubated with free ASM and anti-ICAM/ASM NCs for 1 h at 37 °C, washed, and lysed as described above. ASM in the cell lysate hydrolyzes sphingomyelin in the assay kit, ultimately reacting to form fluorescent resorufin (excitation and emission maxima of 571 and 585 nm, respectively). Fluorescence was measured using an F2500 fluorescent spectrophotometer (Hitachi, Schaumburg, IL). To determine intracellular ASM activity, surface-bound ASM was removed with an acid glycine wash prior to collecting cell lysates, a technique that has been shown to detach many types of ligands (including antibody conjugates) bound to their respective cell-surface receptors.33
Reduction of Lipid Storage
To verify ASM activity within the lysosome, fibroblasts were incubated overnight with TNFα and fluorescent BODIPY-FL-C12-sphingomyelin, which integrates with cellular sphingomyelin stores and fluoresces red (620 nm) at high concentrations and green (530 nm) at physiological concentrations.22 Cells were incubated for 1 h with free ASM or anti-ICAM/ASM NCs, then washed and fixed 3 h later, permitting degradation of intracellular sphingomyelin stores. Samples were analyzed by fluorescence microscopy for reversal of the lysosomal storage phenotype.
Results
Reduced Formation of Clathrin-Coated Pits and Endocytosis of Clathrin Ligands by NPD Fibroblasts
The clathrin-mediated uptake of glycosylated enzymes via the mannose-6-phosphate receptor represents the basis of current lysosomal enzyme replacement therapies, yet the diminished uptake of this receptor in type A–B Niemann–Pick disease poses an obstacle for therapeutic efficiency.19 Internalization parameters (binding efficiency, uptake kinetics, intracellular routing, etc.) vary widely between ligands, even those utilizing the same endocytic pathway.7 Hence, we examined the uptake of fluorescent transferrin, a ligand with a well-described clathrin-mediated internalization pathway.34 Wild-type fibroblasts internalized transferrin within widespread intracellular compartments within 1 h, unlike NPD fibroblasts (Figure 1A). We counted less than half as much internalized transferrin in NPD cells as wild-type cells (32 ± 4 vs 88 ± 28); however, there was a comparable amount of transferrin bound to the surface of the plasma membrane in both cell types (Figure 1B). These findings suggest that the transferrin receptor may still be available on the cell surface for binding, but NPD fibroblasts less readily internalize the ligand–receptor complex.
Figure 1.
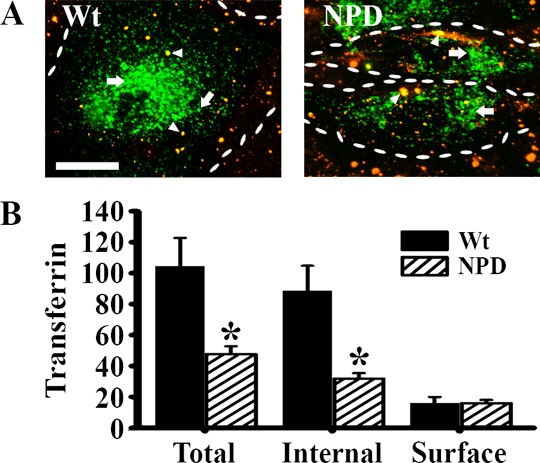
Binding and internalization of transferrin in NPD fibroblasts. (A) Fluorescence microscopy images of wild-type (Wt) and NPD fibroblasts incubated with transferrin (green) for 1 h at 37 °C. Unbound transferrin was washed away, cells were fixed, and surface transferrin was immunostained (red + green = yellow; arrowheads) to distinguish from the internalized ligand (green; arrows). Dashed lines = cell borders. Scale bar = 10 μm. (B) The fractions of internal and surface transferrin were quantified. Data are the mean ± SEM. *Comparison with Wt cells (p < 0.05 by Student’s t test).
Electron microscopy enabled visual verification of CME dysfunction. TEM of wild-type and NPD fibroblasts exposed to transferrin revealed far fewer membrane invaginations and clathrin-coated pits on the surface of NPD cells (Figure 2A). In parallel, SEM enabled visualization of the cell surface and quantification of endocytic invaginations (Figure 2B). In addition to dramatic bulges underneath the cell surface (asterisks, presumably engorged storage compartments), NPD fibroblasts exhibited smaller pit diameters (107 ± 8 vs 140 ± 6 nm in wild-type cells) and fewer pits over 100 nm (6 ± 3 vs 18 ± 5 pits per μm2; Figure 2C), verifying diminished CME capability.
Figure 2.

Electron microscopy of clathrin-coated pits in NPD fibroblasts. Wt and NPD fibroblasts were incubated with transferrin for 30 min at 37 °C, washed, and fixed. (A) Nascent clathrin vesicles and membrane invaginations were imaged by transmission electron microscopy (TEM; arrowheads point to only some, not all, examples). Scale bar = 200 nm. (B) Endocytic pits on the cell surface (arrowheads show only some, not all, examples) and bulges beneath the membrane (asterisks) were imaged by scanning electron microscopy (SEM). Scale bar = 1 μm. (C) The number and size of the pits in (B) were quantified. Data are the mean ± SEM. *Comparison with Wt cells (p < 0.05 by Student’s t test).
Abnormal Distribution and Ligand-Mediated Recruitment of Clathrin in NPD Fibroblasts
Clathrin recruitment to ligand binding sites represents a key step in clathrin-coated vesicle formation, so we examined the intracellular distribution of clathrin heavy chain. Without transferrin added to the cell media, clathrin occupied a central area within the wild-type cell body (possibly associating with the Golgi and assisting with transport through the biosynthetic route), but a perinuclear area within the NPD cells, potentially indicating sequestration around lysosomes (Figure 3A). With the addition of transferrin to the cell media, the distribution broadened in wild-type cells (Figure 3B), where it largely colocalized with transferrin (63% ± 6%; Figure 3C). On the contrary, clathrin remained largely within 3 μm of the nucleus in NPD cells (85% ± 4% vs 36% ± 11% perinuclear localization in wild-type cells), resulting in low colocalization with transferrin (17% ± 4%). This is consistent with reduced transferrin uptake and endocytic pit formation.
Figure 3.
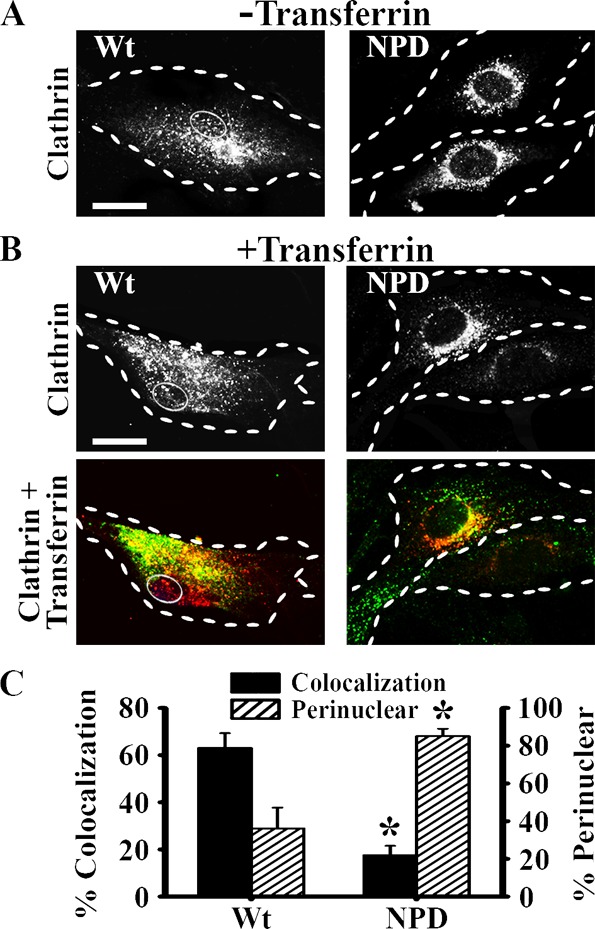
Colocalization of transferrin with clathrin in NPD fibroblasts. (A) Fluorescence microscopy images of Wt and NPD fibroblasts showing the distribution of clathrin heavy chain in the absence of ligands added in the cell media. (B) Cells were incubated with transferrin for 1 h at 37 °C, washed, fixed, and clathrin heavy chain was immunostained red. Top panels illustrate the distribution of clathrin. Bottom panels show colocalization (yellow) of clathrin (red) and transferrin (green) in the same cells. (A,B) Dashed lines = cell borders. Thin solid line = nuclei in Wt cells. Scale bar =10 μm. (C) Quantification of the percent of bound transferrin that colocalized with clathrin, and the percent of clathrin distributed within ∼3 μm of the cell nucleus (perinuclear). Data are the mean ± SEM. *Comparison with Wt cells (p < 0.05 by Student’s t test).
Reduced Pinocytosis via the Clathrin-Mediated Route in NPD Fibroblasts
Diminished clathrin recruitment to ligand binding sites suggests a mechanism for diminished CME, which can potentially affect not only ligand internalization but also constitutive pinocytosis. We examined the uptake of Texas Red dextran (a nondegradable fluid phase marker for endocytosis). By fluorescence microscopy, we observed a more widespread distribution of dextran-filled endocytic vesicles throughout the cytosol in NPD fibroblasts compared to the wild-type (Figure 4A), consistent with aberrant intracellular trafficking observed in some lipidoses.13,20 Indeed, we observed only ∼60% as much dextran uptake in NPD fibroblasts as wild-type cells, indicative of diminished endocytosis overall (Figure 4B). To evaluate whether the reduction was due to impaired CME as observed above, we treated cells with MDC, an inhibitor of clathrin-mediated uptake. In wild-type cells, MDC decreased pinocytosis of dextran by 48%, indicating that this pathway accounts for dextran uptake. Contrarily, applying MDC to NPD cells had little effect, confirming that the disease disrupted the clathrin pathway.
Figure 4.
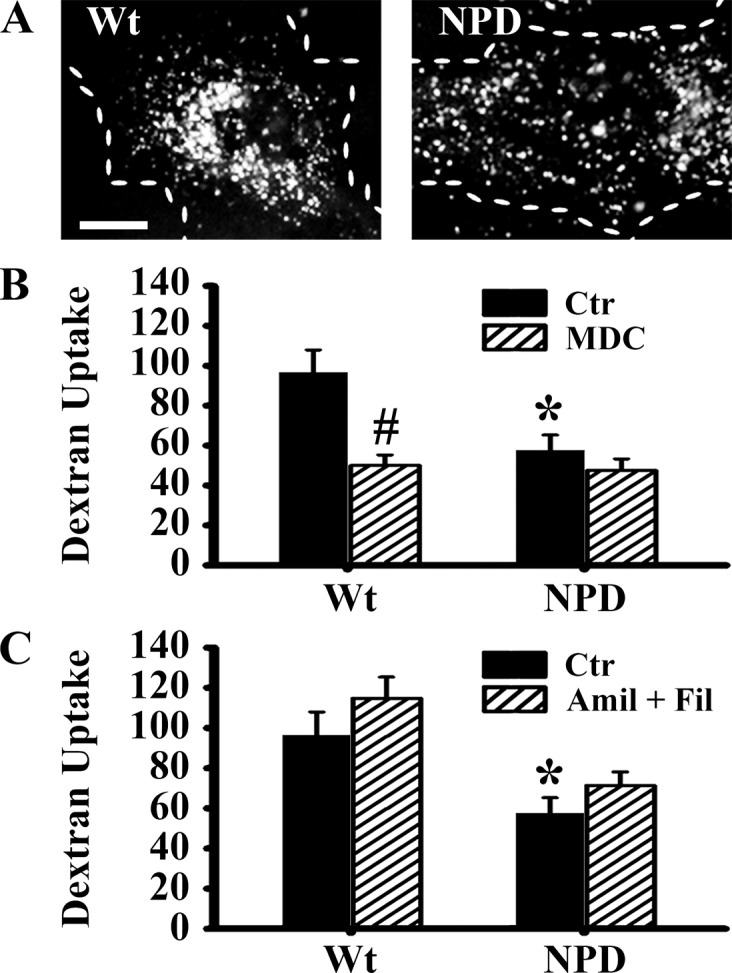
Clathrin-mediatd pinocytosis in NPD fibroblasts. (A) Fluorescence microscopy images of Wt and NPD fibroblasts incubated for 1 h at 37 °C in media containing Texas Red dextran. Dashed lines = cell borders. Scale bar = 10 μm. The number of dextran-positive vesicles was quantified by fluorescence microscopy in cells treated with or without (B) 50 μM monodansylcadaverine (MDC, an inhibitor of clathrin endocytosis) and (C) a cocktail of 1 μg/mL flipin (inhibits caveolar endocytosis) and 3 mM amiloride (inhibits macropinocytosis and CAM-mediated endocytosis). (B,C) Data are the mean ± SEM. *Comparison between NPD and Wt cells within each condition. #Comparison between control and inhibitor within each cell type group (p < 0.05 by Student’s t test).
Next we evaluated whether CME can compensate for uptake when other pathways are inhibited. Wild-type cells treated with a cocktail of filipin and amiloride (inhibitors of the caveolar, and CAM + macropinocytosis pathways) maintained basal levels of bulk uptake (115 ± 11 vesicles, Figure 4C). Others have reported compensation between clathrin/caveolar pathways in response to inhibition, which may be the case here.35 However, such compensation was not observed in NPD cells treated with the same inhibitor cocktail, further validating that CME is impaired in these cells. Taken together, these data demonstrate that not only CME of ligands, but also clathrin-mediated pinocytosis is affected in NPD fibroblasts.
Enhanced Enzyme Uptake and Intracellular Activity by ICAM-1-Targeted Carriers vs Clathrin-Mediated ASM Delivery
Because several enzyme replacement therapies depend on CME via the mannose-6-phosphate receptor,22 deficient receptor-mediated and pinocytic CME in NPD cells may affect the efficacy of this strategy, calling for other therapeutic approaches. We have previously reported ASM delivery to NPD fibroblasts using nanocarriers targeted to ICAM-1 on the cell surface, inducing endocytosis and lysosomal delivery via a well-studied pathway distinct from clathrin, caveolin, and other known pathways.21−25 Here we compared the uptake of ASM carried by anti-ICAM NCs to the uptake of free ASM. Over the course of 1 h, NPD fibroblasts internalized greater amounts of enzyme (>10 fold) when presented on anti-ICAM NCs (Figure 5A). As a result, NPD cells treated with anti-ICAM/ASM NCs recovered more basal ASM activity than those treated with free enzyme (∼30% vs ∼3% of the ASM activity of wild-type cells; Figure 5B). Furthermore, activity of anti-ICAM/ASM NCs was localized to the intracellular space (∼57% intracellular enzyme activity), whereas most free ASM activity was localized to the cell surface (∼10% intracellular enzyme activity), as evidenced after washing the cells with acid glycine (Figure 5C). Therefore, alternative targeting strategies, such as targeting the CAM pathway, offer a means to bypass dysfunctional clathrin endocytosis and deliver enzymes to intracellular targets.
Figure 5.

ASM delivery efficiency and functional activity. (A) TNFα-stimulated NPD fibroblasts were incubated for 1 h at 37 °C with free 125I-ASM or anti-ICAM/125I-ASM NCs. The amount of cell-associated ASM was determined in a gamma counter from the cell lysates. (B) Additionally, ASM activity was measured with a sphingomyelinase kit (Molecular Probes) to detect the conversion of sphingomyelin into fluorescent Resorufin in cell lysates. Data is presented as a percentage of Wt ASM activity. (C) Intracellular enzyme activity after removing the surface-bound ASM fraction with an acid glycine solution. Data are a percentage of total ASM activity. (A–C) Data are the mean ± SEM. *Comparison with cells treated with free ASM (p < 0.05 by Student’s t test).
Substrate Reduction by Exogenous ASM Restores Clathrin Mediated Endocytosis
Given the enhanced intracellular ASM delivery by anti-ICAM NCs, we tested their potential to attenuate lysosomal storage, the central goal of enzyme replacement therapy. NPD and wild-type fibroblasts were incubated overnight with fluorescent sphingomyelin to permit visualization of lipid stores, and NPD fibroblasts were treated with free or carrier-bound ASM as before (Figure 6A). Untreated NPD fibroblasts accumulated ∼5-fold more sphingomyelin in compartments around the nucleus than wild-type cells (Figure 6B). Treatment with anti-ICAM/ASM NCs reduced ∼89% of storage, whereas treatment with the same amount of enzyme free in solution reduced only ∼35% of storage, consistent with impaired CME.
Figure 6.
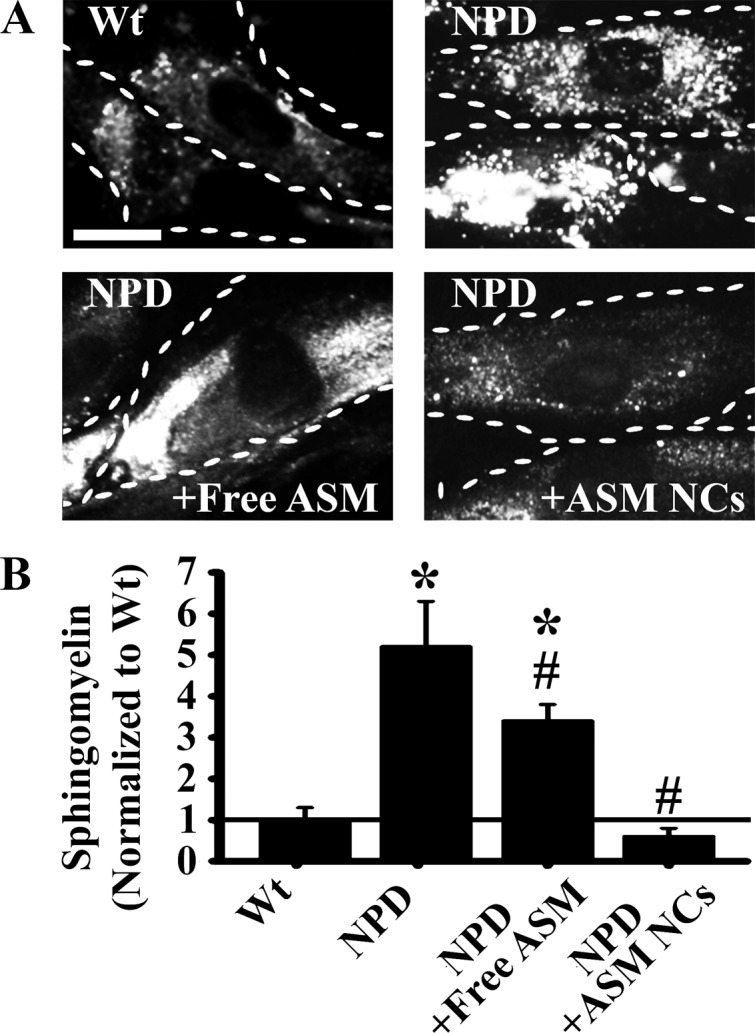
Reduction of sphingomyelin accumulation in NPD fibroblasts by ASM delivery. TNF-stimulated Wt and NPD fibroblasts were incubated overnight with fluorescent BODIPY-FL-C12-sphingomyelin to permit visualization of lipid accumulation. (A) Fluorescence microscopy images of cells receiving no treatment, free ASM, or anti-ICAM/ASM NCs for 1 h at 37 °C. Cells were washed and fixed 3 h later to permit sphingomyelin degradation. Dashed lines = cell borders. Scale bar = 10 μm. (B) Quantification of sphingomyelin fluorescence, normalized to the Wt level. Data are the mean ± SEM. *Comparison with Wt. #Comparison with untreated NPD control (p < 0.05 by Student’s t test).
Finally, the reduction of lysosomal storage by anti-ICAM NCs correlated with restoration of CME. NPD fibroblasts treated with anti-ICAM/ASM NCs demonstrated a more widespread distribution of clathrin heavy chain and a 4-fold improvement of clathrin–transferrin colocalization, reaching levels comparable to that of the wild-type cells (Figure 7C). Furthermore, treated cells exhibited recovery of transferrin uptake, from ∼36% to ∼71% of the wild-type levels (Figure 7A), suggesting a relationship between lysosomal storage and endocytic efficiency.
Figure 7.
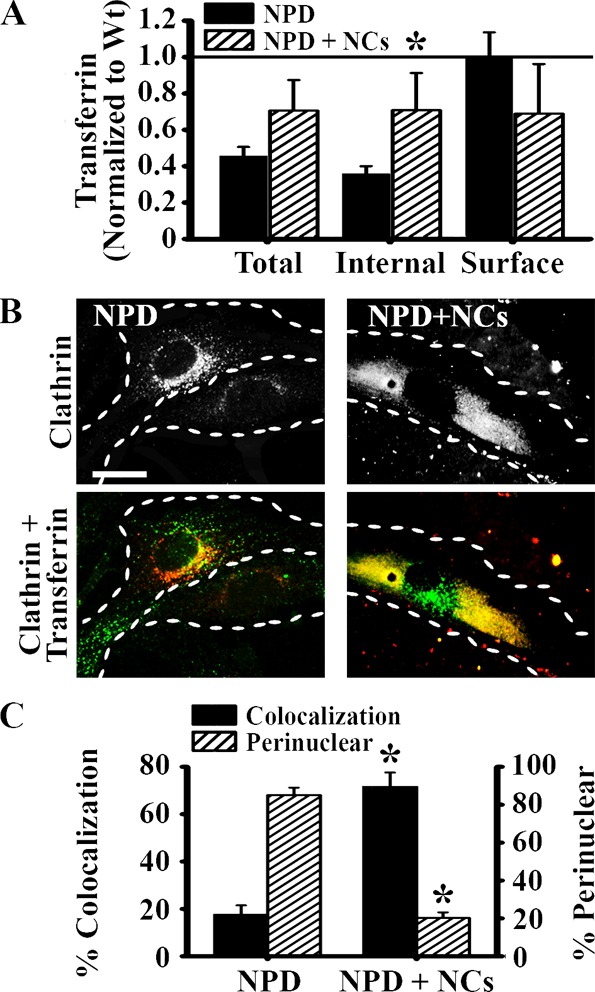
Recovery of clathrin distribution, recruitment, and endocytosis in NPD fibroblasts treated with anti-ICAM/ASM NCs. (A) Untreated NPD fibroblasts vs NPD fibroblasts treated with anti-ICAM/ASM NCs were incubated with fluorescent transferrin as in Figure 1. Data are normalized to the Wt level. (B) Fluorescence microscopy images of untreated and treated NPD fibroblasts incubated for 1 h at 37 °C with transferrin, and then immunostained for clathrin heavy chain. Top panels illustrate the distribution of clathrin. Bottom panels show the colocalization (yellow) of clathrin (red) and transferrin (green) in the same cells. Dashed lines = cell borders. Scale bar = 10 μm. (C) Quantification of the percent of bound transferrin that colocalized with clathrin, and the percent of clathrin distributed within ∼3 μm of the cell nucleus. (A,C) Data are the mean ± SEM. *Comparison with untreated NPD cells (p < 0.05 by Student’s t test).
Discussion
As one of the predominant endocytic mechanisms in most mammalian cells, CME represents a valuable target for intracellular drug delivery, either through passive association with the fluid phase of clathrin-coated vesicles or active targeting of the cargo receptors that regulate this pathway.34 In the case of enzyme replacement therapy for lysosomal storage disorders, where therapeutic recombinant enzymes target the mannose-6-phosphate receptor, clathrin endocytic activity is key to efficient enzyme uptake.9 Using a patient fibroblast model, we have examined the effect of type A NPD on CME, the implications for enzyme delivery, and the potential role for ICAM-1-targeted nanocarriers to enhance enzyme delivery by bypassing the clathrin pathway.
The lysosomal storage phenotype at the cellular level appears to correlate with diminished uptake of clathrin-associated cargo. We observed diminished internalization of fluorescent transferrin in ASM-deficient fibroblasts as compared to the wild-type (Figure 1). The quantity of transferrin associated with the cell surface remained the same, suggesting that the ligand–receptor interactions may remain intact at the cell surface, while the internalization machinery or the recycling kinetics may be impaired. Several groups have also observed impaired receptor mobilization in cells with lysosomal storage disorders. For example, atypical mannose-6-phosphate receptor internalization and trafficking has been observed in aveolar macrophages from ASM-knockout mice, in fibroblasts with I-cell disease, and in fibroblasts with Pompe disease.19,36,37 Interestingly, previous studies have also described either enhancement or reduction of mannose-6-phosphate receptor expression in different lysosomal storage disorders,19,36,37 while our work did not identify changes in this parameter with regard to the transferrin receptor. Because the mannose-6-phosphate receptor is directly involved in transporting lysosomal enzymes in the biosynthetic route,11 it is possible that the receptor accumulates at this stage as an effect of lysosomal storage disease, thereby altering expression on the cell surface and explaining differences with our study. It is also possible that our experimental set up may not detect a reduction of transferrin receptor expression on the cell surface because we do not directly evaluate this, but rather experiments involve concomitant endocytosis, which may in itself modify the amount of receptor exposed at the plasmalemma. Importantly, transferrin is often an advantageous targeting moiety that can be chemically conjugated to drugs or nanoscale drug carriers, and the mannose-6-phosphate receptor is also a key target for current enzyme replacement therapies. Yet, results shown here indicate that these may not represent the most efficient targeting schemes for lysosomal storage diseases.12,35,38
We examined endocytosis in these cells and found that dysfunction was not limited to specific receptors but also affected clathrin-mediated pinocytosis, posing significant implications to current therapies. Upon examination with TEM, NPD fibroblasts exposed to transferrin formed fewer clathrin pits (observable by the characteristic cytosolic lattices) and exhibited generally less invaginations and furrows in their membrane (Figure 2). These observations correlate well with the smaller size and number of endocytic pits observed by SEM (Figure 2) and with the diminished clathrin-mediated uptake of extracellular fluid (Figure 4). Given established diameters for traditional pinocytic vesicles formed by macropinocytic, clathrin-, and caveolin-mediated pathways (>1 μm, ∼120 nm, ∼60 nm, respectively), the pits we observed were most consistent with CME in size.2 Aberrant pit formation in NPD cells is a relevant parameter for therapeutic design, restricting the amount of material and the size of objects these cells internalize. For example, diminished fluid phase uptake may impair substrate reduction therapy, a treatment strategy where small molecules remove excess storage or inhibit metabolite biosynthesis in order to attenuate storage.12 And while smaller pit sizes may still accommodate entry of small molecules, macromolecules and drug conjugates larger than 100 nm in diameter (such as enzyme-loaded nanoparticles or certain viral gene delivery vectors) may be excluded.
Diminished pit formation can be explained by poor trafficking of endocytic machinery (e.g., clathrin) to the pit site. The diseased cells exhibited perinuclear clathrin localization that was poorly recruited to transferrin binding sites (Figure 3B). Pits recruit clathrin directly from the cytosol,5 and cytosolic diffusion may be impaired due to the storage phenotype. Clathrin may also become entrapped in disrupted cellular processes. For example, clathrin coats assemble on endosomes/lysosomes and the trans-Golgi network during the formation of transport vesicles and on autophagosomes during lysosome reformation.39,40 Aberrant membrane fusion due to elevated cholesterol (a feature of several lipidoses, including type A–B NPD30) has been linked to vesicle and autophagosome accumulation.14,16,41 The delayed turnover of these intermediate compartments may lead to sequestration of a fraction of the clathrin pool, consistent with the observed perinuclear clathrin localization (Figure 3) and limiting clathrin availability at the plasma membrane.
Impaired CME in NPD cells may be responsible for poor internalization of free ASM. Inefficient enzyme uptake has long been an obstacle to enzyme replacement therapy; the aforementioned aberrant uptake of mannose-6-phosphate receptors has been linked to poor internalization of ASM in a murine NPD model and α-glucosidase in Pompe fibroblasts.9,19,37 It is worth noting, however, that this is not a universal finding. Renal tubular cells with metachromatic leukodystrophy (a lysosomal storage disease characterized by sulfatide accumulation) showed a 2-fold increase in enzyme uptake via this receptor in one study.42 This is a key aspect to consider: given that different cell types and tissues in the body are differently affected by NPD (as for other lysosomal storage disorders), it is likely that the manner and severity of endocytic dysfunction will vary (e.g., neurons are severely affected in type A NPD, vs fibroblasts used here as an available model). Therefore, a thorough and comparative investigation of endocytic patterns is necessary in other cells types. Furthermore, systematic examination of these patterns in different lysosomal storage diseases may help identify efficient routes of internalization. For example, the present study demonstrated the utility of nanocarriers that target the CAM pathway (a clathrin- and caveolin- independent pathway), offering a well-studied means to bypass aberrant CME and more efficiently deliver enzyme cargo.22,24,43 Therapeutic effect only requires 10–20% of wild-type enzyme activity within the lysosome,12 which was achieved with anti-ICAM/ASM NCs after only 1 h. The nanocarriers more efficiently reduced sphingomyelin storage (Figure 6), and the reduction of the storage phenotype led to a recovery of clathrin-mediated endocytosis (Figure 7).
There is likely a correlation between lysosomal storage and clathrin endocytic activity. The restorative effect could be due to relief of the intracellular traffic jam, permitting increased cytosolic diffusion of endocytic machinery and active transport of trafficking compartments along opened cytoskeletal pathways. Alternatively, several lipidoses (such as NPD) are characterized by increased cholesterol storage, which has been implicated in aberrant suppression of lipid synthesis, switching between lipid biosynthetic and degredative pathways, and abnormal lipid trafficking between the membrane and intracellular compartments.20,44,45 Atypical membrane composition resulting from these disrupted processes may play a role in endocytic dysfunction and could be revived upon normalizing stored lipids to a basal level. It would be interesting to test if a similar restoration of CME could be observed in NPD cells following substrate reduction therapy, using compounds that have been demonstrated to reduce lipid accumulation, such as cyclodextrin or Miglustat.12
For drugs, drug conjugates, and drug carriers that target endocytosis for cellular uptake, the relationship between disease and endocytic behavior represents a crucial and often overlooked design parameter. For example, this work examined for the first time a broader relationship between lysosomal storage and CME. Impairment of clathrin-associated ligand uptake in ASM-deficient fibroblasts poses a challenge to current enzyme replacement therapies dependent on this pathway, as well as novel therapies that may seek to use clathrin-targeting agents. Targeting alternate mechanisms of endocytosis, such as the CAM pathway, may improve therapeutic delivery to desired intracellular compartments, whether for small molecule, gene delivery, or enzyme replacement platforms. Recognizing the relative activity of different endocytic pathways in diseased cells may help identify appropriate drug targets, and whether by ligand-mediated or pinocytic uptake, improve the internalization efficiency of drug delivery systems overall. Additionally, by analogy to endocytic defects observed in lysosomal storage diseases, it becomes vital to examine whether accumulation within endolysosomal compartments of foreign materials employed in drug delivery could impart similar endocytic alterations. Because most drug carrier strategies converge at the endolysosomal route, this knowledge will be key in designing strategies for intracellular delivery built of materials whose degradation does not cause secondary endocytic defects.
Acknowledgments
We thank Dr. Edward Schuchmann (Mount Sinai School of Medicine, New York, NY) for providing recombinant human ASM and NPD fibroblasts. This study was funded by NIH and NSF grants R01-HL098416 and CBET-1402756, respectively, to S. Muro and by a fellowship to the University of Maryland from the Howard Hughes Medical Institute Undergraduate Science Education Program for J. Rappaport.
Glossary
Abbreviations Used
- ASM
acid sphingomyelinase
- CME
clathrin-mediated endocytosis
- ICAM-1
intercellular adhesion molecule-1
- anti-ICAM NCs
nanocarriers targeted to ICAM-1
- NPD
type A–B Niemann–Pick disease
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Mellman I. Endocytosis and Molecular Sorting. Annu. Rev. Cell. Dev. Biol. 1996, 12, 575–625. [DOI] [PubMed] [Google Scholar]
- Conner S. D.; Schmid S. L. Regulated Portals of Entry into the Cell. Nature 2003, 422, 37–44. [DOI] [PubMed] [Google Scholar]
- Marsh M.; McMahon H. T. The Structural Era of Endocytosis. Science 1999, 285, 215–220. [DOI] [PubMed] [Google Scholar]
- Parkar N. S.; Akpa B. S.; Nitsche L. C.; Wedgewood L. E.; Place A. T.; Sverdlov M. S.; Chaga O.; Minshall R. D. Vesicle Formation and Endocytosis: Function, Machinery, Mechanisms, and Modeling. Antioxid. Redox Signaling 2009, 11, 1301–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon H. T.; Boucrot E. Molecular Mechanism and Physiological Functions of Clathrin-Mediated Endocytosis. Nature Rev. Mol. Cell. Biol. 2011, 12, 517–533. [DOI] [PubMed] [Google Scholar]
- Mousavi S. A.; Malerod L.; Berg T.; Kjeken R. Clathrin-Dependent Endocytosis. Biochem. J. 2004, 377, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muro S. Challenges in Design and Characterization of Ligand-Targeted Drug Delivery Systems. J. Controlled Release 2012, 164, 125–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futerman A. H.; van Meer G. The Cell Biology of Lysosomal Storage Disorders. Nature Rev. Mol. Cell. Biol. 2004, 5, 554–565. [DOI] [PubMed] [Google Scholar]
- Desnick R. J.; Schuchman E. H. Enzyme Replacement and Enhancement Therapies: Lessons from Lysosomal Disorders. Nature Rev. Genet. 2002, 3, 954–966. [DOI] [PubMed] [Google Scholar]
- Beck M. New Therapeutic Options for Lysosomal Storage Disorders: Enzyme Replacement, Small Molecules and Gene Therapy. Hum. Genet. 2007, 121, 1–22. [DOI] [PubMed] [Google Scholar]
- Neufeld E. F. The Uptake of Enzymes into Lysosomes: an Overview. Birth Defects Orig. Artic. Ser. 1980, 16, 77–84. [PubMed] [Google Scholar]
- Muro S. Strategies for Delivery of Therapeutics Into the Central Nervous System for Treatment of Lysosomal Storage Disorders. Drug Delivery Transl. Res. 2012, 2, 169–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons K.; Gruenberg J. Jamming the Endosomal System: Lipid Rafts and Lysosomal Storage Diseases. Trends Cell. Biol. 2000, 10, 459–462. [DOI] [PubMed] [Google Scholar]
- Fraldi A.; Annunziata F.; Lombardi A.; Kaiser H. J.; Medina D. L.; Spampanato C.; Fedele A. O.; Polishchuk R.; Sorrentino N. C.; Simons K.; Ballabio A. Lysosomal Fusion and SNARE Function are Impaired by Cholesterol Accumulation in Lysosomal Storage Disorders. EMBO J. 2010, 29, 3607–20. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Ballabio A.; Gieselmann V. Lysosomal Disorders: from Storage to Cellular Damage. Biochim. Biophys. Acta 2009, 1793, 684–696. [DOI] [PubMed] [Google Scholar]
- Fukuda T.; Ewan L.; Bauer M.; Mattaliano R. J.; Zaal K.; Ralston E.; Plotz P. H.; Raben N. Dysfunction of Endocytic and Autophagic Pathways in a Lysosomal Storage Disease. Ann. Neurol. 2006, 59, 700–708. [DOI] [PubMed] [Google Scholar]
- Marks D. L.; Pagano R. E. Endocytosis and Sorting of Glycosphingolipids in Sphingolipid Storage Disease. Trends Cell. Biol. 2002, 12, 605–613. [DOI] [PubMed] [Google Scholar]
- Takahashi M.; Kobayashi T. Cholesterol Regulation of Rab-Mediated Sphingolipid Endocytosis. Glycoconjugate J. 2009, 26, 705–710. [DOI] [PubMed] [Google Scholar]
- Dhami R.; Schuchman E. H. Mannose 6-Phosphate Receptor-Mediated Uptake is Defective in Acid Sphingomyelinase-Deficient Macrophages: Implications for Niemann–Pick Disease Enzyme Replacement Therapy. J. Biol. Chem. 2004, 279, 1526–1532. [DOI] [PubMed] [Google Scholar]
- Puri V.; Watanabe R.; Dominguez M.; Sun X.; Wheatley C. L.; Marks D. L.; Pagano R. E. Cholesterol Modulates Membrane Traffic Along the Endocytic Pathway in Sphingolipid-Storage Diseases. Nature Cell Biol. 1999, 1, 386–388. [DOI] [PubMed] [Google Scholar]
- Hsu J.; Northrup L.; Bhowmick T.; Muro S. Enhanced Delivery of α-Glucosidase for Pompe Disease by ICAM-1-Targeted Nanocarriers: Comparative Performance of a Strategy for Three Distinct Lysosomal Storage Disorders. Nanomedicine 2012, 8, 731–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muro S.; Schuchman E. H.; Muzykantov V. R. Lysosomal Enzyme Delivery by ICAM-1-Targeted Nanocarriers Bypassing Glycosylation- and Clathrin-Dependent Endocytosis. Mol. Ther. 2006, 13, 135–341. [DOI] [PubMed] [Google Scholar]
- Muro S.; Wiewrodt R.; Thomas A.; Koniaris L.; Albelda S. M.; Muzykantov V. R.; Koval M. A Novel Endocytic Pathway Induced by Clustering Endothelial ICAM-1 or PECAM-1. J. Cell Sci. 2003, 116, 1599–1609. [DOI] [PubMed] [Google Scholar]
- Garnacho C.; Dhami R.; Simone E.; Dziubla T.; Leferovich J.; Schuchman E. H.; Muzykantov V.; Muro S. Delivery of Acid Sphingomyelinase in Normal and Niemann–Pick Disease Mice Using Intercellular Adhesion Molecule-1-Targeted Polymer Nanocarriers. J. Pharmacol. Exp. Ther. 2008, 325, 400–408. [DOI] [PubMed] [Google Scholar]
- Hsu J.; Serrano D.; Bhowmick T.; Kumar K.; Shen Y.; Kuo Y. C.; Garnacho C.; Muro S. Enhanced Endothelial Delivery and Biochemical Effects of α-Galactosidase by ICAM-1-Targeted Nanocarriers for Fabry Disease. J. Controlled Release 2011, 149, 323–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papademetriou J.; Garnacho C.; Serrano D.; Bhowmick T.; Schuchman E. H.; Muro S. Comparative Binding, Endocytosis, and Biodistribution of Antibodies and Antibody-Coated Carriers for Targeted Delivery of Lysosomal Enzymes to ICAM-1 versus Transferrin Receptor. J. Inherit. Metab. Dis. 2013, 36, 467–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muro S., Intercellular Adhesion Molecule-1 and Vascular Cell Adhesion Molecule-1. In Endothelial Biomedicine, 1st ed.; Aird W., Ed.; Cambridge University Press: New York, 2007; pp 1058–1070. [Google Scholar]
- Muro S.; Garnacho C.; Champion J. A.; Leferovich J.; Gajewski C.; Schuchman E. H.; Mitragotri S.; Muzykantov V. R. Control of Endothelial Targeting and Intracellular Delivery of Therapeutic Enzymes by Modulating the Size and Shape of ICAM-1-Targeted Carriers. Mol. Ther. 2008, 16, 1450–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu J.; Rappaport J.; Muro S. Specific Binding, Uptake, and Transport of ICAM-1-Targeted Nanocarriers Across Endothelial and Subendothelial Components of the Blood–Brain Barrier. Pharm. Res. 2014, 31, 1855–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuchman E.; Desnick R., Niemann–Pick disease types A and B: Acid Sphingomyelinase Deficiencies. In The Metabolic and Molecular Bases of Inherited Disease, 8th ed.; Scriver C., Beaudet A., Sly W., Valle D., Childs B., Kinzler K., Vogelstein B., Eds.; McGraw-Hill: New York, 2000; pp 3589–3610. [Google Scholar]
- He X.; Miranda S. R.; Xiong X.; Dagan A.; Gatt S.; Schuchman E. H. Characterization of Human Acid Sphingomyelinase Purified from the Media of Overexpressing Chinese Hamster Ovary Cells. Biochim. Biophys. Acta 1999, 1432, 251–264. [DOI] [PubMed] [Google Scholar]
- Muro S.; Dziubla T.; Qiu W.; Leferovich J.; Cui X.; Berk E.; Muzykantov V. R. Endothelial Targeting of High-Affinity Multivalent Polymer Nanocarriers Directed to Intercellular Adhesion Molecule 1. J. Pharmacol. Exp. Ther. 2006, 317, 1161–1169. [DOI] [PubMed] [Google Scholar]
- Muro S.; Muzykantov V.; Murciano J-C.. Characterization of Endothelial Internalization and Targeting of Antibody–Enzyme Conjugates in Cell Cultures and in Laboratory Animals. In Methods in Molecular Biology; Bioconjugation Protocols Series, 1st ed.; Niemeyer C. M., Ed.; Humana Press: Totowa, NJ, 2004; pp 21–36. [DOI] [PubMed] [Google Scholar]
- Bareford L. M.; Swaan P. W. Endocytic Mechanisms for Targeted Drug Delivery. Adv. Drug Delivery Rev. 2007, 59, 748–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty G. J.; McMahon H. T. Mechanisms of Endocytosis. Annu. Rev. Biochem. 2009, 78, 857–902. [DOI] [PubMed] [Google Scholar]
- Otomo T.; Higaki K.; Nanba E.; Ozono K.; Sakai N. Lysosomal Storage Causes Cellular Dysfunction in Mucolipidosis II Skin Fibroblasts. J. Biol. Chem. 2011, 286, 35283–35290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardone M.; Porto C.; Tarallo A.; Vicinanza M.; Rossi B.; Polishchuk E.; Donaudy F.; Andria G.; De Matteis M. A.; Parenti G. Abnormal Mannose-6-Phosphate Receptor Trafficking Impairs Recombinant alpha-Glucosidase Uptake in Pompe Disease Fibroblasts. Pathogenetics 2008, 1, 6–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Z. M.; Li H.; Sun H.; Ho K. Targeted Drug Delivery via the Transferrin Receptor-Mediated Endocytosis Pathway. Pharmacol. Rev. 2002, 54, 561–587. [DOI] [PubMed] [Google Scholar]
- Traub L. M.; Bannykh S. I.; Rodel J. E.; Aridor M.; Balch W. E.; Kornfeld S. AP-2-Containing Clathrin Coats Assemble on Mature Lysosomes. J. Cell Biol. 1996, 135, 1801–1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong Y.; Liu M.; Ma L.; Du W.; Zhang H.; Tian Y.; Cao Z.; Li Y.; Ren H.; Zhang C.; Li L.; Chen S.; Xi J.; Yu L. Clathrin and Phosphatidylinositol-4,5-bisphosphate Regulate Autophagic Lysosome Reformation. Nature Cell Biol. 2012, 14, 924–934. [DOI] [PubMed] [Google Scholar]
- Lieberman A. P.; Puertollano R.; Raben N.; Slaugenhaupt S.; Walkley S. U.; Ballabio A. Autophagy in Lysosomal Storage Disorders. Autophagy 2012, 8, 719–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein D.; Yaghootfam A.; Matzner U.; Koch B.; Braulke T.; Gieselmann V. Mannose 6-Phosphate Receptor-Dependent Endocytosis of Lysosomal Enzymes is Increased in Sulfatide-Storing Kidney Cells. Biol. Chem. 2009, 390, 41–48. [DOI] [PubMed] [Google Scholar]
- Muro S.; Cui X.; Gajewski C.; Murciano J. C.; Muzykantov V. R.; Koval M. Slow Intracellular Trafficking of Catalase Nanoparticles Targeted to ICAM-1 Protects Endothelial Cells from Oxidative Stress. Am. J. Physiol.: Cell. Physiol. 2003, 285, C1339–C1347. [DOI] [PubMed] [Google Scholar]
- Liscum L.; Faust J. R. Low Density Lipoprotein (LDL)-Mediated Suppression of Cholesterol Synthesis and LDL Uptake is Defective in Niemann–Pick type C Fibroblasts. J. Biol. Chem. 1987, 262, 17002–17008. [PubMed] [Google Scholar]
- Hortsch R.; Lee E.; Erathodiyil N.; Hebbar S.; Steinert S.; Lee J. Y.; Chua D. S.; Kraut R. Glycolipid Trafficking in Drosophila Undergoes Pathway Switching in Response to Aberrant Cholesterol Levels. Mol. Biol. Cell 2010, 21, 778–790. [DOI] [PMC free article] [PubMed] [Google Scholar]