

Abstract
Probe mapping is a common approach for identifying potential binding sites in structure-based drug design; however, it typically relies on energy minimizations of probes in the gas phase and a static protein structure. The mixed-solvent molecular dynamics (MixMD) approach was recently developed to account for full protein flexibility and solvation effects in hot-spot mapping. Our first study used only acetonitrile as a probe, and here, we have augmented the set of functional group probes through careful testing and parameter validation. A diverse range of probes are needed in order to map complex binding interactions. A small variation in probe parameters can adversely effect mixed-solvent behavior, which we highlight with isopropanol. We tested 11 solvents to identify six with appropriate behavior in TIP3P water to use as organic probes in the MixMD method. In addition to acetonitrile and isopropanol, we have identified acetone, N-methylacetamide, imidazole, and pyrimidine. These probe solvents will enable MixMD studies to recover hydrogen-bonding sites, hydrophobic pockets, protein–protein interactions, and aromatic hotspots. Also, we show that ternary-solvent systems can be incorporated within a single simulation. Importantly, these binary and ternary solvents do not require artificial repulsion terms like other methods. Within merely 5 ns, layered solvent boxes become evenly mixed for soluble probes. We used radial distribution functions to evaluate solvent behavior, determine adequate mixing, and confirm the absence of phase separation. We recommend that radial distribution functions should be used to assess adequate sampling in all mixed-solvent techniques rather than the current practice of examining the solvent ratios at the edges of the solvent box.
Introduction
Computational probe mapping is frequently applied in structure-based drug design (SBDD) to identify potential binding pockets along a protein surface (i.e., hot spots).1−4 However, mapping is usually limited by the implementation of gas-phase minimizations, wherein protein flexibility and solvent competition are ignored. This leads to a rugged potential surface and many local energy minima, where druggable sites are indistinguishable from irrelevant minima. Furthermore, predictions based on a static receptor structure often neglect important binding-site features, such as the presence of transient cavities5 or bridging water molecules.6 As a result, appropriately modeling the binding potential continues to be a challenge for SBDD.
Several computational and experimental models have emerged that address the limitations of traditional solvent mapping. The multiple protein structure (MPS) method addressed the issue of protein flexibility by creating pharmacophore models from consensus mapping of conformational ensembles with solvent probes.7−10 While this technique has demonstrated success in mapping the binding sites of pharmaceutically relevant targets,8,11,12 it cannot account for desolvation penalties or water-bridging contacts, which may be essential to accurate prediction. The multiple solvent crystal structure (MSCS)13,14 technique uses X-ray crystallography to determine receptor structures in the presence of competing water and organic solvent. This paved the way for fragment-based methods by providing the first experimental identification of preferred sites for probe binding. MSCS results have demonstrated high potential for use in drug development and have inspired several new computational approaches.
Simultaneous protein flexibility and solvent mapping was first implemented in 2009 when molecular dynamics (MD) simulations were used as a tool to map hot spots.15 Seco et al. performed MD simulations with seven proteins that were solvated by a 20% volume/volume (v/v) solution of water and isopropanol (IPA). Although their method sought to detect binding sites and predict druggability, it did not address aromatic hotspots and was unable to reproduce some of the experimental binding sites. Yang and Wang have conducted similar studies that included phenol with the alcohol/water mix to address aromatic interactions.16,17 Another MD-based approach, site identification by ligand competitive saturation (SILCS), was implemented with a ternary-solvent box composed of 1 M benzene (BNZ), 1 M propane, and water to locate hot spots and to reproduce experimental binding sites.18,19 The authors introduced a dummy atom with a virtual repulsion term for both BNZ and propane in order to solubilize the hydrophobic solvents and prevent probe–probe aggregation. However, this repulsive term may alter proper mapping. Their fragment maps for BNZ along the trypsin surface found abundant local minima that were mapped equally or better than the valid binding site.19 In addition, Guvench and MacKerell noted that the repulsive term may also prevent the mapping of secondary binding sites due to the unnatural physical interactions that are enforced between the hydrophobic probes.15 The most recent technique was developed by Bakan et al.,20 which uses mixtures of water with multiple probes simultaneously (IPA, acetamide, acetic acid, isopropylamine). Although each of these MD-based techniques have shown some success in identifying binding sites, they were limited in their ability to selectively map hot spots without the use of either a weighting or artificial repulsion term. Many spurious minima were also identified.
Over the past few years, we have worked to develop mixed-solvent molecular dynamics (MixMD) using AMBER, with an aim of solvent mapping across a wide range of targets.21,22 Instead of simply showing that probes could occupy regions of known binding sites, we sought to establish a stronger foundation that would allow for application of our method to target systems without requiring a priori knowledge of the binding sites. Our initial study with MixMD used a 50% w/w solvent box composed of acetonitrile (ACN) and water, and the results showed that MixMD reproduced binding sites from MSCS studies with excellent convergence to the true hot spots and no spurious minima.21,22
We were interested in extending the probe set for MixMD to fully enable identification of hydrogen-bonding sites, hydrophobic contacts, protein–protein interactions, and aromatic pockets. Here, we show what happens when poor parameters are used in mapping a protein. We then use MD simulations of water-probe boxes to examine which remain evenly mixed. It is important to identify additional functional sites, particularly aromatic sites, for MixMD to be most useful for SBDD. We explain our choice of using OPLS parameters23 for IPA in MixMD.21 We also identify additional probe solvents that expand the ability of MixMD to locate important interaction profiles in protein–ligand binding: acetone (ACE), N-methylacetamide (NMA), imidazole (IMI), and pyrimidine (1P3). We highlight the importance of developing a solid foundation for mapping with MD, particularly as it relates to the validation of parameters through detailed evaluation of metrics for probe–probe dispersion.
Methods
Probe Parameters
Solvent probes were selected to represent specific interaction types for use in MixMD (Table 1). Parameters for ACE,24 ACN,25 and NMA26 were obtained from studies of liquid solvent with AMBER. AMBER parameters have not been specifically developed for IPA or aromatic heterocycles. Therefore, we explored the suitability of OPLS parameters, which were developed to work with TIP3P water27 and to reproduce the empirical data for pure liquids.28 The adaptation of nonbonded parameters from OPLS for use in AMBER simply required the conversion of σ to rmin, where 21/6 × σ/2 = rmin. This is necessary because BOSS29 and AMBER30 use different combining rules for van der Waals (vdw) parameters. A comprehensive list of the parameters used in our MixMD simulations is given in Table S1 and Figure S1 of the Supporting Information.
Table 1. List of Solvent Probes Included in Development of MixMD Functional Group Types and Their Interaction Profile.
| solvent | mapping role |
|---|---|
| isopropanol (IPA) | hydrogen bond donor/acceptor, small hydrophobic |
| acetonitrile (ACN) | polar, amphipathic |
| acetone (ACE) | polar, hydrogen bond acceptor only |
| N-methylacetamide (NMA) | protein backbone |
| imidazole (IMI) | 5-membered polar aromatic |
| pyridine (PYR) | 6-membered aromatic (more soluble) |
| pyridazine (1P2) | 6-membered aromatic (more soluble) |
| pyrimidine (1P3) | 6-membered aromatic (more soluble) |
| pyrazine (1P4) | 6-membered aromatic (more soluble) |
| benzene (BEN) | 6-membered aromatic (less soluble) |
| phenol (IPH) | 6-membered aromatic (less soluble) |
MD Simulations
Pure solvent boxes of no fewer than 200 probe molecules were built using the tleap module in AMBERTOOLS. MD simulations were performed in AMBER1031 through the sander module with SHAKE32 and a 1 fs time step.27 Each solvent box was subjected to 1000 steps of steepest descent followed by 49,000 cycles of conjugate gradient minimization and then heated over 20 ps from 10 to 300 K at constant volume. Two nanoseconds of equilibration were followed by a 5 ns simulation at constant pressure with the temperature held at 300 K by a weak-coupling algorithm.33 The Berendsen coupling method was chosen because it was used in the original publication of AMBER parameters for the solvent molecules used in this study. Particle mesh Ewald34 was implemented with a vdw cutoff of 8 Å. The behavior of the pure-probe boxes was used to confirm proper behavior in our setup. Detailed results are in Table S2 and the text of the Supporting Information. The final boxes of organic solvent were also used in the setup of the mixed boxes.
Mixed-solvent boxes were created in tleap by solvating a single probe molecule with a layer of TIP3P water27 and then a layer of the equilibrated box of pure, organic solvent. The size of the outer box of probes is adjusted to achieve the desired solvent ratio. At least 2500 water molecules and the equivalent mass of probe molecules necessary to achieve a 50% w/w solution were used in order to ensure that realistic solvent behavior would be observed (Table S3, Supporting Information). Each layered mixed-solvent box underwent the same procedure for minimization, heating, equilibration, and production simulation as described for the pure-solvent boxes above.
It should be noted that Berendsen’s coupling method33 can sometimes be problematic for simulations of mixtures because differential heating can occur for different components of the system.35 Our use of the method with the small boxes could be considered a worst-case scenario, where probes may (or may not) have a small bias to aggregate. Any mixture of water and probe solvent that displays homogeneous behavior under these circumstances is likely to be robust to a wide range of applications. In our examination of thermolysin with IPA+water, we chose the Andersen coupling method to be consistent with our other published applications of MixMD.21,22 There is no bias from the temperature coupling that can cause the differences seen in the two parameter choices for IPA when used in MixMD of thermolysin.
We characterized the distribution of probe density by computing atom–atom radial distribution functions (RDFs) with the radial command in ptraj. The final 1 ns of production time was used to calculate the RDF with a bin size of 0.1 Å. When the solvent probes are fully solubilized into water, they are well dispersed throughout the solvent box, and the RDF converges toward unity at the vdw cutoff. When phase separation occurs in simulations of mixed solvent, too many probes and too little water occupy the local microenvironment around each probe. This makes the local environment too dense with organic probes, and the RDF will not converge to unity within the vdw cutoff. Accordingly, we used these RDF descriptions of mixed vs aggregated solvent systems to evaluate the structure of each mixed-solvent box.
The carbon–carbon (rCC), carbon–oxygen (rCO), carbon–nitrogen (rCN), nitrogen–nitrogen (rNN), nitrogen–oxygen (rNO), and oxygen–oxygen (rOO) distributions were assessed between probe–probe, water–water, and probe–water as was relevant for each probe molecule. For brevity, only representative RDFs are shown for each case below. Cumulative density distributions of rOO for water–water distances in mixed solvent were also examined to determine the number density of water within the volume sphere. For the case of a 50% w/w mixed-solvent box, equitable distribution of solvent probes and water within the system will give a cumulative plot for rOO of water–water distances that differs from the rOO of pure water. The distribution of water molecules relative to one another is less than in pure solvent because obviously the presence of a miscible probe reduces the number of water molecules that can occupy the local microenvironment around each water.
Results and Discussion
Earlier papers—ours included—have used the behavior at the edges of the simulation box to prove even mixing of probes and water. The reasoning was that the box edges are far from the protein, outside the vdw cutoff, and should have minimal bias in the ratio of probe to water. If the edges of the box had the same ratio as the setup solvent box, then there was even mixing. However, taking a step back to these basic simulations of solvent boxes provided a better means of measuring mixing: the use of a RDF. RDFs of the solvents relative to one another can be calculated for simulations of proteins, and we recommend they be used in all mixed-solvent simulations to judge whether the behavior of the probes and waters are reasonable.
The RDF, or pair correlation function g(r), can be used to describe the structure of the solvent based on atom/molecule pairs in a system. The function g(r) enables a precise description of how likely a probe molecule will have another probe present at a separation distance of r. This function may be simplified for a liquid system in a solvent box, where g(r) is proportional to the observed number density (ρo) divided by the expected number density (ρe). The value ρo is the total number of atoms at a given distance, and ρe is the number expected at that same distance if the solvent were uniformly distributed. If the probe solvent and water are evenly mixed, the RDF will converge to 1.0 at long ranges. If it converges to a larger number, this indicates phase separation (further discussion below). The values of g(r) at 8 Å are given for the mixed water–solvent boxes in Table 2.
Table 2. Proper Mixing of 50/50 Solvent Boxes, Including the Pair Correlation Function g(r) at a Distance of 8 Å.
| probe | mixed, g(r) | probe | mixed, g(r) |
|---|---|---|---|
| isopropanol (IPAOPLS) | yes, 1.06 | imidazole (IMI)a | yes, 1.06 |
| isopropanol (IPASeco) | no, 1.51 | pyridine (PYR)a | no, 1.63 |
| acetonitrile (ACN)b | yes, 1.02 | pyridazine (1P2)a | no, 1.30 |
| acetone (ACE)a | yes, 1.12 | pyrimidine (1P3)a | yes, 1.01 |
| acetone (ACE)b | yes, 1.03 | pyrazine (1P4)a | no, 1.57 |
| N-methylacetamide (NMA)a | yes, 1.10 | benzene (BNZ)a | no, 1.79 |
| N-methylacetamide (NMA)b | yes, 1.01 | phenol (IPH)a | no, 1.48 |
OPLS parameters.
AMBER parameters.
Previously, MixMD was validated with ACN as a probe, which specifies hot spots for amphipathic nitrogen-containing ligands.21 We applied AMBER parameters for ACN from Grabuleda et al.,25 and they accurately reproduce experimental data from MSCS. For completeness, we ran simulations of ACN+H2O boxes to confirm that the RDF demonstrated appropriate convergence to unity (Figure S2, Supporting Information).
Importance of Realistic Parameters: The Case of Isopropanol
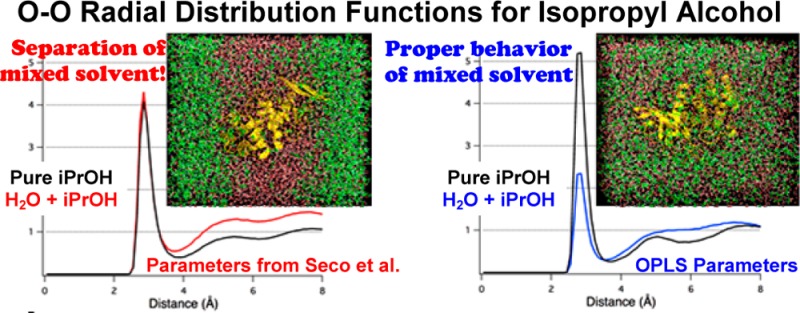
In trying to reproduce simulations of thermolysin in IPA+H2O published by Seco et al.,15 we found that the IPA phase-separated from water (Figure 1A). It is unclear whether the authors observed this same behavior, and they do not note which temperature-coupling method was used. They noted “partial phase separation” in the paper, and they rescaled the reference values for probe density to account for the solvent behavior. However, the bulk phase separation we observed signifies the implementation of inadequate parameters for liquid IPA, which were derived from the AMBER parameter files for threonine with charges assigned after RESP calculations (IPASeco).
Figure 1.

Snapshots of MixMD simulations of thermolysin in IPA+H2O executed using the protocol outlined in Seco et al.15 (A) Fully flexible simulations of thermolysin were performed with a mixed box of 50% w/w IPASeco+H2O. Five independent runs were calculated, and every simulation resulted in the solvent layers separating between 4 and 5 ns and remaining separated when simulations were extended to 10 ns. This solvent behavior was unrealistic because IPA and water are completely miscible. (B) The same simulation using IPAOPLS23 resulted in solvent remaining well distributed for the entire equilibration and 10 ns of production in all five independent simulations.
We turned to the work of Jorgensen et al. for alcohol parameters because they were developed to specifically reproduce a variety of pure-solvent behaviors and interactions with TIP3P water.28 For small boxes without protein, we compared our simulations of IPAOPLS+H2O to experiments by Langdon and Keyes that determined the density for IPA+H2O systems at different temperatures.36 We found that our mixed box of IPAOPLS+H2O reproduced the experimental density at 308 K (0.830g/mL) to within 0.06%. Our ability to replicate this fundamental data confirmed the use of good parameters for our IPA+H2O systems. Indeed, MixMD simulations of thermolysin resulted in proper mixing using the IPA parameters from Jorgensen et al. (IPAOPLS, Figure 1B).
The RDFs for the IPA probes (rOO and rCC) and water (rOO) in mixed-solvent boxes further justified the use of OPLS parameters. Figure 2 compares the O–O RDF for both mixed-solvent and pure-solvent boxes of IPA. The rOO for (IPA-IPA)OPLS converged to unity with an appropriately shorter peak than that of pure liquid alcohol. However, the rOO for (IPA-IPA)Seco remained well above 1.0 at 8 Å and featured a large peak for the first solvent shell, the same as seen in the simulation of a pure IPA box. All simulations with IPASeco feature RDFs that indicate aggregation of the solvent molecules. For this first example, the count data behind the RDFs will be discussed in detail to further explain our interpretation, which is supported by viewing the boxes.
Figure 2.
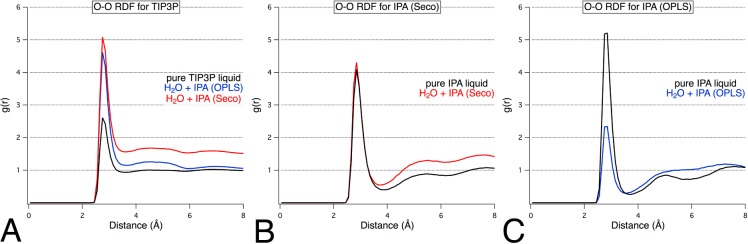
O–O radial distribution functions for (A) water–water and (B,C) probe–probe in a mixed-solvent environment with IPA. The impact of different parameters for IPA is demonstrated by the poor convergence to unity for O–O in simulations of 50% w/w IPASeco (B) when compared to the appropriate convergence obtained for the same mixed solvent with IPAOPLS parameters (C).
IPA and the great majority of organic solvents chosen for this study are miscible with water (Table 3). The definition of miscibility is that the two solutions are homogeneously distributed at all ratios of the two solvents. It might be reasonable to find some bias or local structure in the first and second solvent shells, but bulk separation should not be seen at long ranges. This has critical correspondence with one of the basic underlying assumptions of RDFs. RDFs are founded on a uniform distribution of solvent to describe ρe (solvent count/box volume); any deviations from uniformity (1.0) reflects the local structure of the solvent.
Table 3. List of Probes for MixMD and Experimental Observations from Two Different Sources.
| solvent | MSDS | CRC handbook |
|---|---|---|
| acetone (ACE) | soluble | miscible |
| acetonitrile (ACN) | soluble | miscible |
| benzene (BNZ) | no data | slightly soluble |
| imidazole (IMI) | soluble at about 50g/L | very soluble |
| isopropanol (IPA) | soluble | miscible |
| N-methylacetamide (NMA) | soluble | no data |
| phenol (PHO) | no data | soluble |
| pyrazine (1P4) | no data | soluble |
| pyridazine (1P2) | no data | miscible |
| pyridine (PYR) | soluble | miscible |
| pyrimidine (1P3) | no data | miscible |
In all the RDFs in Figure 2, water has its maximum g(r) at 2.75 Å, and the IPA maxima are at 2.85 Å. For pure water at 2.75 Å, the maximum g(r) is 2.6, and the cumulative count of water is 1.4 molecules (Table 4). If we define the first solvent shell between 2.45 and 3.15 Å, it contains 3.9 neighboring water molecules. When simulated in a mixture with IPAOPLS, there are 1.1 waters within 2.75 Å and 2.9 waters within 3.15 Å (Table 4). Of course, both counts are reduced because some of the interactions are occasionally fulfilled by IPA. This is also incorporated into ρe, where the larger box volume reduces the expected density. It is that reduced expectation (actually 0.244 at 2.75 Å) that results in a g(r)of 4.6. This is higher than the g(r) maximum from the pure water simulation, but it does not reflect more interactions with water molecules. Within a 7.95 Å radius sphere, the observed and expected counts are nearly equal and g(r) approaches 1.0, showing that the molecules have a uniform distribution over larger scales.
Table 4. Detailed Values Behind the RDFs in Figure 2a.
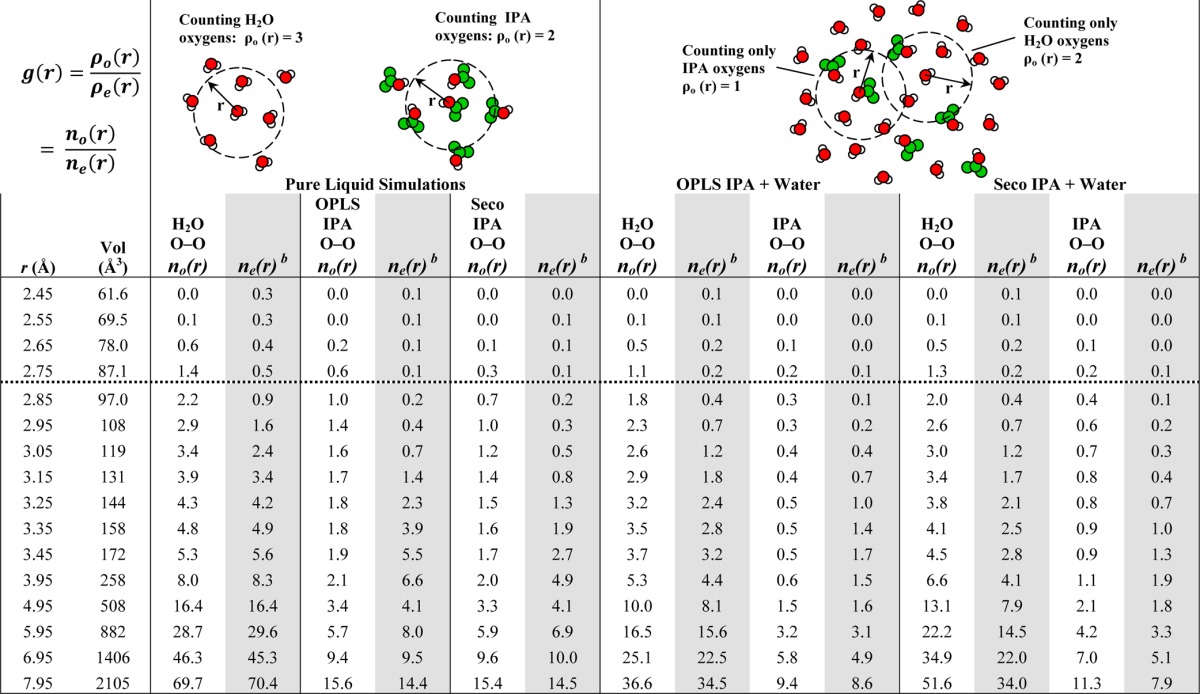
Both ρo and ρe depend on the volume of the simulation box; this drops out and makes each g(r) = no(r)/ne(r). The values in this table have been rounded; the g(r) in the figures are the exact values. Dashed horizontal line indicates the maxima in g(r).
Values for ne(r) are based on the volume of the count sphere, the number of solvent in the whole box, and size of the whole box during the simulation.
For the mixture of water and IPASeco, a dramatically different picture emerges. Within a 7.95 Å radius sphere, the expected count of water was 34.0, but the observed count was 51.6 water molecules (Table 4). Clearly, there are too many waters close to one another! Furthermore, g(r) asymptotically approaches 1.5 at 7.95 Å. Phase separation (as in Figure 1A) is best quantified by high g(r) at long r, but even the local changes reflected the phenomenon. Within 2.75 and 3.15 Å, there are 1.3 and 3.4 waters (Table 4), respectively, which is much closer to the values shown in the pure-water boxes. The mixed box equilibrated to a slightly larger volume (1% change), which is reflected in the slight differences in the expected counts between IPASeco+water and IPAOPLS+water. However, the 1% change does not explain the maximum g(r) of 5.1, which is based on the increase in number of water molecules.
The same patterns are seen in the RDFs of the IPA oxygens (Table 4). Pure IPA simulations show appropriate behavior for both sets of parameters (black lines in Figure 2BC, Table S2 and text in Supporting Information). Though the RDFs may imply IPAOPLS has slightly tighter solvent shells, they both sum to the same number of IPA molecules in the first solvent shell (2.1 for IPAOPLS and 2.0 for IPASeco). For mixed simulations, both IPASeco and IPAOPLS show lower counts than pure-solvent boxes at their g(r) maxima (2.85 Å) and within their solvent shells (3.95 Å). However, g(r) of IPASeco unmistakably approaches 1.4 at 7.95 Å.
Additional Probes for Hydrogen-Bonding and Protein–Protein Interactions
Influenced by the discrepancies in solvent behavior observed for IPASeco and IPAOPLS, we carefully examined the behavior of existing parameters for additional solvent probes to determine their applicability in MixMD. The inclusion of various probe types, including complicated molecular fragments, would facilitate the development of accurate pharmacophores for druggable hot spots, ligand binding sites, and protein–protein interactions. The selection of additional probes for expanding the functional groups represented in MixMD was based on existing MSCS data and common interaction types found in protein–ligand systems. IPA maps both hydrogen bond-accepting and -donating locations, but additional hydrogen-bonding probes are needed to develop robust pharmacophore maps of binding sites. ACE and NMA were selected because each molecule represents a different hydrogen-bonding profile. ACE locates where the protein donates hydrogen bonds, and NMA represents protein–protein or peptide binding.
Specific parameters for ACE and NMA had been developed for use with AMBER24,26 and OPLS.28,37 Simulations of ACE+H2O and NMA+H2O were performed using each parameter set to assess their suitability. The g(r) was calculated for probe–probe, probe–water, and water–water interactions within the binary-solvent boxes. Analysis of the resulting RDF plots indicated that in each case all g(r) converged to unity near 8 Å. We found that both the AMBER and OPLS parameters for liquid ACE and NMA demonstrated correct behavior according to RDF plots and visual inspection (Figure 3). The O–O RDF for water from the mixed-solvent simulations also illustrate that solvent mixing has occurred. Analysis of the cumulative plots for the rOO of water in solution with ACE or NMA showed that the number density of water within the local volume sphere was half the number density observed in pure water simulations, which is the expected result for a well-distributed system of 50% w/w probe and water. Visual analysis of the trajectory snapshots served to corroborate the occurrence of proper solvent mixing. However, the RDF’s deviation from unity at 8 Å was slightly higher for the OPLS descriptions of ACE and NMA (Table 2). For this reason, we would recommend that the AMBER parameters be used for ACE and NMA in our MixMD method, especially when the AMBER parameters are also used to describe the protein.
Figure 3.

(A,D) O–O and (B,E) N–N radial distribution functions for probe–probe and O–O RDFs for water–water (C,F) in a mixed-solvent environment with ACE and NMA. The impact of optimized parameters from AMBER versus general solvent parameters from OPLS is shown in the slightly better convergence to unity for simulations using AMBER parameters (D–F) compared to OPLS (A–C) parameters.
Search for an Aromatic Probe
In particular, we wanted to find parameters for soluble heterocycles that could map aromatic hot spots without requiring an artificial repulsive term. No other MD approach for probe mapping has successfully integrated aromatic probes without an artificial interaction term. Futhermore, we were particularly interested in developing probes that matched common heterocycles used in modern pharmaceuticals. As a result, our initial investigation into an appropriate aromatic probe led to IMI.
A highly polar diazole, IMI, is miscible with water and is present as a functional group in a wide range of biologically active molecules, including mercaptopurine (anticancer), ketoconazole (antifungal), and moxonidine (antihypertensive). IMI parameters were derived from Jorgensen and McDonald,38 and then simulations of pure IMI and IMI+H2O were conducted to assess the potential of IMI as a probe for MixMD. The proper convergence of the RDF plots to 1.0 showed that IMI was properly solubilized and well distributed within the binary-solvent system (Figure 4). Our pure-solvent RDFs were consistent with the results of Jorgensen and co-workers38,39 and further validated our parameter choice and the accuracy of our protocol. Also, visualization of simulation snapshots confirmed that the IMI probe was well dispersed in the aqueous solution.
Figure 4.
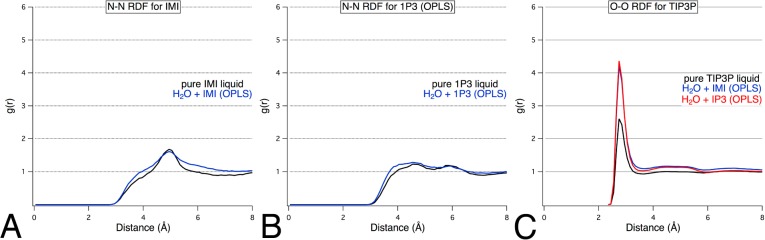
N–N radial distribution functions for (A,B) probe–probe and (C) water–water in a mixed-solvent environment with (A) IMI and (B) 1P3. Appropriate convergence was obtained for both probe types.
Many other nitrogen-containing heterocycles were examined. OPLS parameters for PYR, 1P2, 1P3, and 1P4 were each simulated in a binary water–probe solution. Visualization of the simulations of PYR+H2O, 1P2+H2O, and 1P4+H2O clearly showed that they underwent phase separation. This was proved by RDFs that converged to values well above 1.0 at 8 Å (Figure 5). However, 1P3+H2O yielded a well-dispersed system with well-behaved RDF plots (Figure 4).
Figure 5.

Probe–probe radial distribution functions for mixed solvent environments composed of (A) BNZ+H2O, (B) PYR+H2O, (C) IPH+H2O, (D) 1P2+H2O, and (E) 1P4+H2O clearly showed phase separation, with g(r) values well above 1.0 at 8 Å.
We also examined two nonpolar aromatic probes, BNZ and IPH, simply to confirm that they are not appropriate probes when an artificial repulsive term is not used. Comparison of the resultant RDF plots to the reference RDF plot of pure water showed that the g(r) values for BNZ+H2O and IPH+H2O do not converge to unity within 8 Å (Figure 5). In fact, the g(r) for BNZ+H2O indicated convergence to a value of 2. The simulations with IPH as a probe molecule showed a converged g(r) of approximately 1.45 at 8 Å. Visualization of snapshots from these simulations showed significant aggregation of the probe molecules, verifying that these probes are not be soluble enough for use with MixMD without inclusion of a repulsive term.
Because BNZ is one of the solvent probes used in SILCS, we were able to compare the RDF results for our MixMD simulations of BNZ+H2O to the published results from SILCS mapping studies.18 The BNZ probe used in SILCS was parametrized based on the CHARMM force field and contained a repulsive term to correct for probe–probe aggregation. Although a nonbonded cutoff of 8 Å with a 5–8 Å switching term was applied in SILCS, the probe–probe distance for BNZ converged to 1.0 at ∼13 Å (solvent box size was 72 Å × 58 Å × 43 Å). In comparison, the soluble aromatic probes applied in MixMD converged at approximately 6–7 Å. The first solvent shell of BNZ in SILCS was observed at 9 Å, while we observed the first peak at 4–5 Å. The presence of the second hydrophobic probe (propane) in SILCS simulations may have influenced these differences in RDF. However, the use of a repulsive term to prevent probe–probe aggregation does not correspond with the experimental properties of BNZ, suggesting that SILCS simulations with high concentrations of BNZ may yield incorrect results.
To determine whether BNZ and IPH would disperse more readily in the presence of an “intermediary” probe, ternary mixed-solvent boxes were constructed to contain 1 M aromatic probe and 1 M IPA in water. We hypothesized that the introduction of the IPA molecule would enhance available interactions and aid in the dispersion of the aromatic probes into water. However, this was not observed in simulations of the ternary-solvent boxes. At 5 ns of production time, the BNZ molecules were predominantly clustered along one side of the solvent box, while the IPA molecules were dispersed throughout the water. An additional 20 ns of production time did not change the observed aggregation (Figure 6). Ternary-solvent simulations with IPH indicated a similar result; after 5 ns of production time, IPH was separated with some dispersion toward the interior. Elongating the run time out to 25 ns showed no significant improvement (Figure 6).
Figure 6.
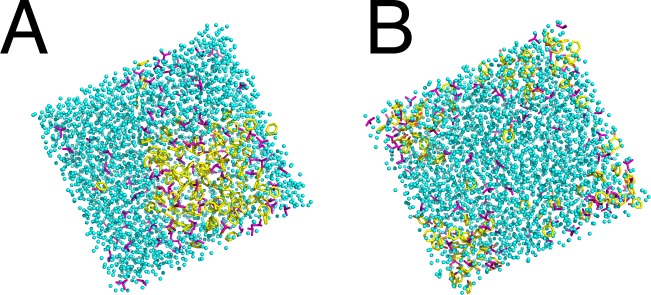
Representative snapshots of MD trajectory for solvent boxes of 1 M aromatic probe (yellow) and 1 M IPA (purple) in water (cyan) for (a) BNZ+IPA+H2O at 25 ns and (b) IPH+IPA+H2O at 25 ns.
Solubility
Although all of the probes selected for use in our validation study had experimental data establishing their solubility in water, not all of these probes were soluble in simulation (Table 3). The solvation conditions applied in a MixMD simulation included high probe concentrations, which may have affected probe solubility. To determine whether the outcomes we observed were justified based on experimental data, we considered their miscibility. The terms soluble and miscible are frequently used interchangeably; however, they are two distinct properties. Solubility is dependent upon temperature and pressure and refers to the ability of a solute (solid, liquid, or gas) to dissolve into a solvent (solid, liquid, or gas), thereby forming a homogeneous mixture. Miscibility refers to the ability of two liquids to form a homogeneous solution, independent of proportion.
All probes that have been experimentally categorized as miscible were well dispersed in our computational simulations, with two exceptions (Table 3). Pyridine is a weak base with a nontrivial concentration of protonated pyridinium ions (pKa of 5.3) following solubilization into water. This effect was not simulated in our studies, and we suggest that the charged species may be responsible for solubilizing the uncharged pyridine molecules into water. A similar explanation may also be extended to pyridazine.
Ternary-Solvent System: Toward Full Pharmacophore Modeling using MixMD
One advantage of using SILCS as a tool for solvent mapping has been that the ternary-solvent box can enable the development of a complete pharmacophore model from a single MD run because the three probe types identify aromatic, hydrophobic, and hydrogen-bonding sites.18,19 Using three of the probes with parameter sets we have validated for MixMD, we constructed a ternary system with IMI (aromatic probe), IPA (hydrophobic and hydrogen-bonding probe), and water at a concentration of 33% w/w by probe. Visualization of the trajectory data indicated appropriate solvent mixing, and our RDF results showed that both organic probes were fully dispersed into solution (Figure 7). Total run time required a comparable amount of simulation time to a binary MixMD simulation; the total cost was an additional 4 or 8 h of production time on an 8-core CPU as compared to a binary IPA+H2O or IMI+H2O simulation, respectively.
Figure 7.

(A) Final snapshot from the production simulation of a mixed-solvent box containing IMI+IPA+H2O illustrates that the probes are well mixed within the system. (B–D) Probe–probe and water–water radial distribution functions for IMI and IPA establish convergence to unity within the trisolvent environment.
We suggest that the use of this ternary MixMD model may offer several advantages over the SILCS approach. We have shown that MixMD can be used to identify maximally occupied sites without recovering additional spurious minima, which is a feature that was not consistently seen in the SILCS studies.19 In addition, instead of requiring water to act as a hydrogen-bonding probe, in MixMD, the druggable hydrogen-bonding sites can be identified in competition with water using IPA as the probe. This is important for ascertaining whether a drug-like molecule that contains a similar function group could displace water molecules at the proposed hydrogen-bonding site. A further advantage of using MixMD is the array of available probes, which can be tailored to an investigator’s mapping needs. For example, either IMI or 1P3 could be used in the ternary box to locate aromatic hotspots that are specific to the size of the ring. The ternary mixture of 33% w/w ACE+ACN+H2O was also simulated, and the results depicted a well-distributed solvent system for use in hot spot mapping with MixMD (Figure S3, Supporting Information).
Conclusion
We have examined solvent parameters for neat liquid from the literature for use in hot spot mapping through MixMD. Our work highlights the importance of parameter validation and analysis of simulation data when performing computational solvent mapping in order to obtain appropriate behavior. Influenced by the poor results obtained using a standard set of parameters used in the field for liquid isopropanol in water, we identified a set of functional group probes with specific parameters available for liquid simulation. We pursued validation of these parameters for neat and mixed-solvent simulations and identified six probes for use with TIP3P in MixMD: acetonitrile, acetone, imidazole, isopropanol, N-methylacetamide, and pyrimidine. Of course, these probes should be used with more proteins beyond thermolysin (Figure 1) to prove their applicability. We have previously applied OPLS IPA parameters to also examine elastase, HEWL, HEWL, p53 core, and RNase A.22 Furthermore, we are currently using all of these probes to map 10 additional protein systems, but these are separate studies in their own right.
The extent of ideal dispersion is most easily quantified by using the cumulative RDF of rOO for water. Although published results for pyridine and pyridazine indicated that they are miscible with water, these two probes were observed to separate from the aqueous phase during our simulations, rendering them unsuitable for solvent mapping in our context. We hypothesized that this disparity between experiment and computation was caused by the presence of the corresponding ionized species, which were not simulated. In order to avoid unphysical mapping or reduced accuracy, our simulations did not employ the use of an artificial repulsion or weighted density term. We have successfully expanded the range of probes that can be incorporated into MixMD studies to allow for the mapping of druggable sites, including hydrogen-bonding regions, aromatic pockets, and protein–protein interfaces. With the proposed ternary-solvent systems, a full pharmacophore model could be created from the simulation of a single protein+probes+water system, which would greatly reduce the computational costs of MixMD studies. This is a clear advantage of the SILCS method that we would like to incorporate. Ultimately, investigators could “mix and match” the probes used in MixMD to identify hotspots with a greater degree of specificity.
Acknowledgments
This work has been supported by the National Institutes of Health (GM65372). K.W.L. thanks the Rackham Graduate School, Pharmacological Sciences Training Program (GM07767), and the American Foundation for Pharmaceutical Education for funding.
Supporting Information Available
Full description of the structures and parameters of all probe solvents, results of the pure-solvent simulations, listing of the exact number of water and probe solvents for each mixed-box simulation, and RDFs for ACN+H2O and ACE+ACN+H2O simulations. This material is available free of charge via the Internet at http://pubs.acs.org..
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Dennis S.; Camacho C. J.; Vajda S. Continuum electrostatic analysis of preferred solvation sites around proteins in solution. Proteins 2000, 38, 176–188. [PubMed] [Google Scholar]
- Goodford P. J. A computational procedure for determining energetically favorable binding sites on biologically important macromolecules. J. Med. Chem. 1985, 28, 849–857. [DOI] [PubMed] [Google Scholar]
- Guarnieri F.; Mezei M. Simulated annealing of chemical potential: A general procedure for locating bound waters. Application to the study of the differential hydration propensities of the major and minor grooves of DNA. J. Am. Chem. Soc. 1996, 118, 8493–8494. [Google Scholar]
- Stultz C. M.; Karplus M. MCSS functionality maps for a flexible protein. Proteins 1999, 37, 512–529. [PubMed] [Google Scholar]
- Wassman C. D.; Baronio R.; Demir O.; Wallentine B. D.; Chen C. K.; Hall L. V.; Salehi F.; Lin D. W.; Chung B. P.; Hatfield G. W.; Richard Chamberlin A.; Luecke H.; Lathrop R. H.; Kaiser P.; Amaro R. E. Computational identification of a transiently open L1/S3 pocket for reactivation of mutant p53. Nat. Commun. 2013, 4, 1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mobley D. L.; Dill K. A. Binding of small-molecule ligands to proteins: “What you see” is not always “what you get”. Structure 2009, 17, 489–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson H. A.; Masukawa K. M.; McCammon J. A. Method for including the dynamic fluctuations of a protein in computer-aided drug design. J. Phys. Chem. A 1999, 103, 10213–10219. [Google Scholar]
- Carlson H. A.; Masukawa K. M.; Rubins K.; Bushman F. D.; Jorgensen W. L.; Lins R. D.; Briggs J. M.; McCammon J. A. Developing a dynamic pharmacophore model for HIV-1 integrase. J. Med. Chem. 2000, 43, 2100–2014. [DOI] [PubMed] [Google Scholar]
- Damm K. L.; Carlson H. A. Exploring experimental sources of multiple protein conformations in structure-based drug design. J. Am. Chem. Soc. 2007, 129, 8225–8235. [DOI] [PubMed] [Google Scholar]
- Meagher K. L.; Lerner M. G.; Carlson H. A. Refining the multiple protein structure pharmacophore method: Consistency across three independent HIV-1 protease models. J. Med. Chem. 2006, 49, 3478–3484. [DOI] [PubMed] [Google Scholar]
- Bowman A. L.; Nikolovska-Coleska Z.; Zhong H.; Wang S.; Carlson H. A. Small molecule inhibitors of the MDM2-p53 interaction discovered by ensemble-based receptor models. J. Am. Chem. Soc. 2007, 129, 12809–12814. [DOI] [PubMed] [Google Scholar]
- Damm K. L.; Ung P. M.; Quintero J. J.; Gestwicki J. E.; Carlson H. A. A poke in the eye: inhibiting HIV-1 protease through its flap-recognition pocket. Biopolymers 2008, 89, 643–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen K. N.; Bellamacina C. R.; Ding X.; Jeffery C. J.; Mattos C.; Petsko G. A.; Ringe D. An experimental approach to mapping the binding surfaces of crystalline proteins. J. Phys. Chem. B 1996, 100, 2605–2611. [Google Scholar]
- Mattos C.; Ringe D. Locating and characterizing binding sites on proteins. Nat. Biotechnol. 1996, 14, 595–599. [DOI] [PubMed] [Google Scholar]
- Seco J.; Luque F. J.; Barril X. Binding site detection and druggability index from first principles. J. Med. Chem. 2009, 52, 2363–2371. [DOI] [PubMed] [Google Scholar]
- Yang C. Y.; Wang S. M. Analysis of flexibility and hotspots in Bcl-xL and Mcl-1proteins for the design of selective small-molecule inhibitors. ACS Med. Chem. Lett. 2012, 3, 308–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C. Y.; Wang S. M. Computational analysis of protein hotspots. ACS Med. Chem. Lett. 2010, 1, 125–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guvench O.; MacKerell A. D. Jr. Computational fragment-based binding site identification by ligand competitive saturation. PLoS Comput. Biol. 2009, 5, e1000435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman E. P.; Yu W.; Guvench O.; Mackerell A. D. Reproducing crystal binding modes of ligand functional groups using site-identification by ligand competitive saturation (SILCS) simulations. J. Chem. Inf. Model. 2011, 51, 877–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakan A.; Nevins N.; Lakdawala A. S.; Bahar I. Druggability assessment of allosteric proteins by dynamics simulations in the presence of probe molecules. J. Chem. Theory Comput. 2012, 8, 2435–2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lexa K. W.; Carlson H. A. Full protein flexibility is essential for proper hot-spot mapping. J. Am. Chem. Soc. 2011, 133, 200–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lexa K. W.; Carlson H. A. Improving protocols for protein mapping through proper comparison to crystallography data. J. Chem. Inf. Model. 2013, 53, 391–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen W. L. Optimized intermolecular potential functions for liquid alcohols. J. Phys. Chem. 1986, 90, 1276–1284. [Google Scholar]
- Pavone M.; Brancato G.; Morelli G.; Barone V. Spectroscopic properties in the liquid phase: combining high-level ab initio calculations and classical molecular dynamics. ChemPhysChem 2006, 7, 148–156. [DOI] [PubMed] [Google Scholar]
- Grabuleda X.; Jaime C.; Kollman P. A. Molecular dynamics simulation studies of liquid acetonitrile: New six-site model. J. Comput. Chem. 2000, 21, 901–908. [Google Scholar]
- Caldwell J. W.; Kollman P. A. Structure and properties of neat liquids using nonadditive molecular dynamics: Water, methanol, and N-methylacetamide. J. Phys. Chem. B 1995, 99, 6208–6219. [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar]
- Jorgensen W. L.; Maxwell D. S.; Tirado-Rives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar]
- Jorgensen W. L.; Tirado-Rives J. Molecular modeling of organic and biomolecular systems using BOSS and MCPRO. J. Comput. Chem. 2005, 26, 1689–1700. [DOI] [PubMed] [Google Scholar]
- Cornell W. D.; Cieplak P.; Bayly C. I.; Gould I. R.; Merz K. M.; Ferguson D. M.; Spellmeyer D. C.; Fox T.; Caldwell J. W.; Kollman P. A. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. [Google Scholar]
- Case D. A.; Darden T. A.; Cheatham I., T E. ; Simmerling C. L.; Wang J.; Duke R. E.; Luo R.; Crowley M.; Walker R. C.; Zhang W.; Merz K. M.; Wang B.; Hayik S.; Roitberg A.; Seabra G.; Kolossváry I.; Wong K. F.; Paesani F.; Vanicek J.; Wu X.; Brozell S. R.; Steinbrecher T.; Gohlke H.; Yang L.; Tan C.; Mongan J.; Hornak V.; Cui G.; Mathews D. H.; Seetin M. G.; Sagui C.; Babin V.; Kollman P. A.. AMBER 10; University of California: San Francisco, 2008. [Google Scholar]
- Ryckaert J. P.; Ciccotti G.; Berendsen H. J. C. Numerical-integration of cartesian equations of motion of a system with constraints: Molecular-dynamics of N-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar]
- Berendsen H. J. C.; Postma J. P. M.; Vangunsteren W. F.; Dinola A.; Haak J. R. Molecular-dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar]
- Darden T.; York D.; Pedersen L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar]
- Feller S. E.; Zhang Y.; Pastor R. W.; Brooks B. R. Constant pressure molecular dynamics simulation: The Langevin piston method. J. Chem. Phys. 1995, 103, 4613–4621. [Google Scholar]
- Langdon W. M.; Keyes D. B. Isopropyl alcohol–water system: Density-composition data and pycnometric technique. Ind. Eng. Chem. 1943, 35, 459–464. [Google Scholar]
- Kaminski G.; Jorgensen W. L. Performance of the AMBER94, MMFF94, and OPLS-AA force fields for modeling organic liquids. J. Phys. Chem. 1996, 100, 18010–18013. [Google Scholar]
- Jorgensen W. L.; McDonald N. A. Development of an all-atom force field for heterocycles. Properties of liquid pyridine and diazenes. J. Mol. Struct.: THEOCHEM 1998, 424, 145–155. [Google Scholar]
- McDonald N. A.; Jorgensen W. L. Development of an all-atom force field for heterocycles. properties of liquid pyrrole, furan, diazoles, and oxazoles. J. Phys. Chem. B 1998, 102, 8049–8059. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.