

Summary
The commensal microbiota impacts specific immune cell populations and their functions at peripheral sites, such as gut mucosal tissues. However, it remains unknown whether gut microbiota control immunity through regulation of hematopoiesis at primary immune sites. We reveal that germ-free mice display reduced proportions and differentiation potential ofspecific myeloid cellprogenitors of both yolk sac and bone marrow origin. Homeostaticinnate immune defects may lead to impaired early responses to pathogens. Indeed, following systemic infection with Listeria monocytogenes, germ-free and oral antibiotic treated mice display increased pathogen burden and acute death. Re-colonization of germ-free mice with a complex microbiota restores defects inmyelopoiesis and resistance to Listeria. Thesefindingsreveal that gut bacteriadirect innate immune cell development via promoting hematopoiesis, contributing to our appreciation of the deep evolutionary connection between mammals and their microbiota.
Introduction
The vast majority of our interactions with bacteriaare symbiotic in nature, consisting of colonization by a complex and diverse microbiota that inhabit humans for life.Rather than causing inflammation, commensal microbes largely direct beneficial immune functions and often engender health. In particular, the microbiota shapesglobalimmune cell repertoires, thereby altering host susceptibility to inflammation and infection at sites of colonization(Hill et al., 2012; Kamada et al., 2012; Mazmanian et al., 2008; Naik et al., 2012). Specific gut bacteria or bacterial products have been shown to suppressintestinal inflammation(e.g., colitis) in micethrough a variety of immune mechanisms (Atarashi et al., 2013; Round and Mazmanian, 2010; Smith et al., 2013).Furthermore, the impact of commensal microbes on host immune responses is not limited to mucosal interfaces, but extends tosystemic compartments; gut microbes regulate immune responses that influence organ-specific autoimmunity in animal models of multiple sclerosis, rheumatoid arthritis and type 1 diabetes(Lee et al., 2011; Markle et al., 2013; Wu et al., 2010). While numerous examples illustrate how the microbiota contributes to immune function at mucosal and systemic sites, little is known about the influencesofgut bacteria on cellular development within primary immune tissues.
The immune system begins to develop in utero, but full maturation requires both genetic and environmental signals that further shape immunity after birth. Lymphoid and myeloid cells develop largely from hematopoietic stem cells (HSCs) within primary tissues, where molecular cues orchestrate immune cell differentiation from uncommitted HSCs and progenitor cells via regulation of transcription factors and epigenetic modifications (Weissman, 1994). Additionally, certain phagocyte populations (including Langerhans cells and microglia), derived from embryonic precursors, are maintained independently of HSCs(Sieweke and Allen, 2013). Genetic contributions (i.e., molecular cues encoded by the host genome) to lineage commitment pathways that control the myeloid repertoire are well studied (Georgopoulos, 2002). However, environmental factors that influence hematopoiesis have not been extensively defined. Based on emerging data that the microbiota represents an integral environmental factor in shaping numerous features of the immune system, we reasoned that gut bacteria may be controlling central immunity. We report herein that commensal microbes promote the maintenance of both HSC and embryonic-derived myeloid cells during steady-state conditions. The absence of commensal microbes leads to defects in severalinnate immune cell populations (including neutrophils, monocytes and macrophages) within systemic sites. By controlling the differentiation of innate immunity, the gut microbiota prepares the host to rapidly mount immune responses upon pathogen encounter, as germ-free and antibiotic treated mice are impaired in clearance of systemic bacterial infection. Our study reveals that gut microbesevolved to actively shape immunity at its core—via regulation of hematopoiesis.
Results
Germ-Free Animals Display Global Defects in Innate Immune Cells
The commensal gut microbiota profoundly influencescellular proportions, migration and functions of various immune cell subsets. Recent studies have provided numerous examples illustrating how gut bacteria modulate innate and adaptive immune responses at mucosal surfaces during infection, inflammation and autoimmunity(Kamada et al., 2013; Round and Mazmanian, 2009). With such pervasive effects, we reasoned that the microbiota may regulate hematopoiesis—the developmental programming of the immune system. Initially, to determine if the microbiota has global effects on systemic immune cell populations, we profiled myeloid cells in the spleen of colonized (SPF; specific pathogen free) and germ-free (GF) mice. Indeed, GF animals display reduced proportions and total numbers of F4/80hi and F4/80locells compared to SPF mice (Figures 1A-C).F4/80hi cells are mainly macrophages, while F4/80lo splenocytes are a heterogeneous population of macrophages, monocytes and neutrophils (Schulz et al., 2012). Intriguingly, all three cell subsets are reduced in GF mice(Figure S1A). Furthermore, treatment of SPF mice with antibiotics also results in diminished myeloid cell populations in the spleen (Figure S1B). Thus, gut bacteria dynamically influence innate immune cell proportions at secondary immune sites in the periphery.
Figure 1. GF Mice Are Deficient in Resident Myeloid Cell Populations in the Spleen and Bone Marrow.
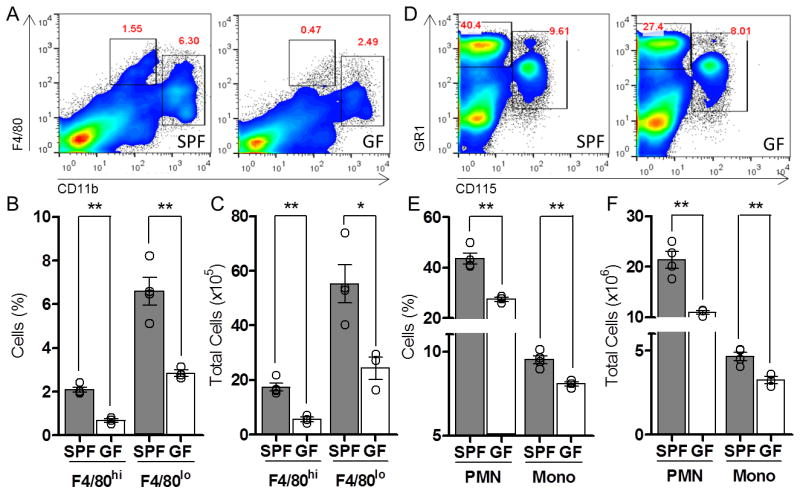
(A-C) Splenic phagocyte profile among SPF and GF mice. Representative flow cytometry plots (A), cell proportions (B), and total cell number (C) of CD11blo F4/80hi and CD11bhi F4/80lo splenic cells in SPF and GF mice. (D-F) Bone marrow populations of neutrophils (Gr1hi CD115neg) and monocytes (Gr1hi CD115hi) among SPF and GF mice. Representative flow cytometry plots (D), cell proportions (E) and total cell number (F) within the bone marrow of SPF and GF mice. For all panels, data are representative of at least 3 independent trials with n≥ 4 mice / group. Each symbol represents data from a single animal. Error bars represent standard error of mean (SEM). *p<0.05, **p<0.01. PMN: polymorphonuclear cells; Mono: monocytes. See also Figure S1.
Myeloid cell precursors differentiate into various phagocyte lineages that are stored in the bone marrow, which are a major source of cells that populate peripheral tissues(Geissmann et al., 2010). The reduction of splenic macrophages, monocytes and neutrophils in GF mice suggests that defects in host immunity may include compromiseddevelopment in primary immune sites. Accordingly, we observed a reduction of myeloid cells within the bone marrow of GF mice (Figures 1D-F). A similar decrease was observed in the liver, a site of alternative immune cell development (Figure S1C). A global defect in myeloid cell populations in primary immune sites of GF mice demonstrates that gut bacteria shape the architecture of the immune system early in cellular development.
Commensal Microbes Enhance Myelopoiesis
We reasoned that reductions in several phagocytic cell subsets in GF mice may reflect a primary defect in the maintenance of myeloid cell populations. To test if commensal microbes promote myelopoiesis, we pulsed SPF and GF mice with 5-Ethynyl-2′-deoxyuridine (EdU), a thymidine analog, to compare the percentage of dividing leukocytes. Both F4/80hi and F4/80lo phagocytes from GF mice showed reduced EdU incorporation compared to SPF animals (Figure 2A,B). F4/80hi macrophages are largely derived from embryonic yolk sac progenitors and are maintained independently of HSCs (Schulz et al., 2012; Sieweke and Allen, 2013). F4/80lo leukocytes, however, are of hematopoietic origin and reduced EdU incorporation by these cells in GF mice indicates defects in the expansion and/or differentiation of bone marrow progenitor cells (Schulz et al., 2012). These studies uncover a role for commensal microbes in promoting the maintenance of both splenic yolk sac-derived and HSC-derived myeloid cells.
Figure 2. The Microbiota Directs Myelopoiesis.
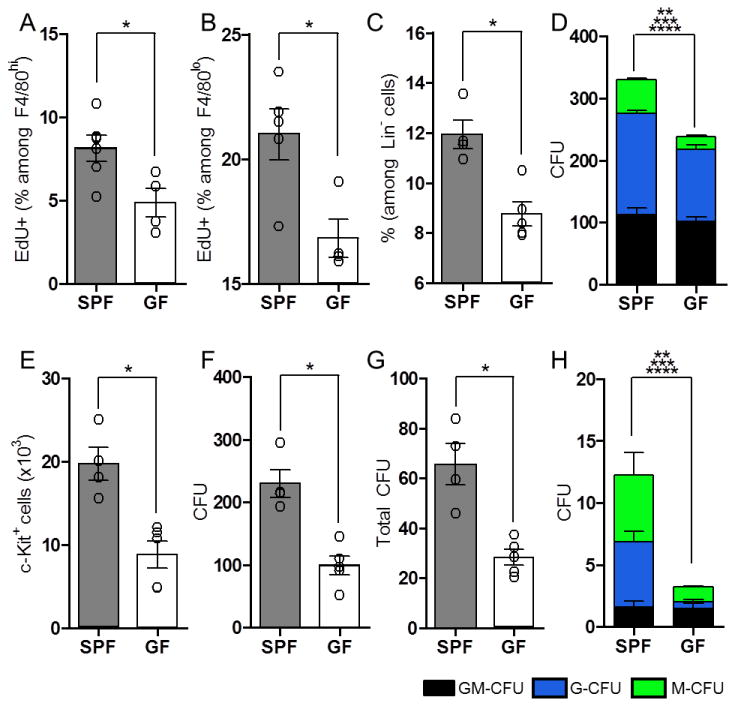
The percentage of F4/80hi CD11b+ (A) and F4/80lo CD11b+ (B) splenocytes with incorporated EdU, following single dose administration. (C) The frequency of LKS- CD34+ Fc γ Rhi granulocyte and/or monocyte progenitors (GMPs) among lineage negative (Lin-) progenitors from bone marrow of SPF and GF mice, as assessed by flow cytometry. (D) Distribution of cell types following purified LKS- CD34+ Fc γ Rhi cell culture in methylcellulose medium. Colonies were identified and counted to assess the proportion of granulocyte-monocytes (GM-CFU; black), granulocytes (G-CFU; blue) and monocytes (M-CFU; green). (E) Total numbers of c-Kit+ CD11b- progenitors from methylcellulose cultures of LKS- CD34+ Fc γ Rhi progenitors, as assessed by flow cytometry. (F) Cells harvested from methylcellulose cultures of LKS- CD34+ Fc γ Rhi progenitors were re-plated at equal numbers in fresh methylcellulose, and cultured to assess their colony forming capacity. (G and H) Splenic cells isolated from SPF and GF mice were cultured in methylcellulose to assess the colony forming capacity of progenitors from SPF and GF mice. Total CFUs (G), and GM-CFUs, G-CFUs and M-CFUs (H) are shown. For each panel, data are representative of at least 2-3 independent trials with n≥ 4/ group. Each symbol represents data from a single animal. Error bars represent SEM. *p<0.05 for all panels. **p<0.05 (comparing total CFU between SPF and GF for (D) and (H)), ***p<0.05 (comparing G-CFU between SPF and GF for (D) and (H)), ****p<0.05 (comparing M-CFU between SPF and GF for (D) and (H)). CFU: colony forming units. See also Figure S2.
The reduction of F4/80lo cells in GF mice led us to further investigate the contribution of commensal microbes on HSCs and myeloid progenitor cells in the bone marrow. No differences were detected in the proportion or differentiation potential of LKS+ cells (HSCs and multipotent progenitors; MPPs), LKS- cells (total lineage-restricted progenitors), or common myeloid progenitor cells (CMPs) between SPF and GF mice (Figure S2A-F). Remarkably, GF miceare significant reduced in the proportion of bone marrow granulocyte and/or monocyte progenitors (GMPs), identified as LKS- CD34+ FcγRhi cells (Figure 2C). GMPs consist of progenitor cells, downstream of HSCs and CMPs during hematopoiesis, with restricted myeloid differentiation potential (Akashi et al., 2000). To further examine the effects of gut microbiota on innate immune cells, we tested if commensal microbes affect the differentiation potential and self-maintenance capacity of GMPs. Methylcellulose culture of LKS- CD34+ FcγRhi cells from GF mice displayed reduced granulocyte (G-CFU) and monocyte (M-CFU) colony formation compared to cells from SPF mice (Figure 2D). Furthermore, passage of LKS- CD34+ FcγRhi cells isolated from GF mice in primary methylcellulose culture yielded reduced recovery of c-Kit+ CD11b- progenitor cells compared to SPF GMPs (Figure 2E). This suggeststhe ability of GMPs to maintain cells with progenitor potential isdefective in the absence of commensal microbes(Rodrigues et al., 2008). Consistent with this notion, secondary cultures of unfractionated cells derived from GF GMPs generated fewer colonies compared to cells isolated from SPF mice (Figure 2F). The commensal microbiota therefore promotes steady-state myelopoiesis by specifically maintaining GMP proportions and enhancing their differentiation to mature myeloid cells in the bone marrow.
Extramedullary hematopoiesis (outside the bone marrow) further contributes to the maintenance and inflammatory responses of tissue-resident phagocytic cells (Jenkins et al., 2011; Massberg et al., 2007; Robbins et al., 2012; Swirski et al., 2009). We therefore investigated whether commensal microbes influence the hematopoietic potential of progenitors located in the spleen. Similar to GMPs from the bone marrow, splenocytes isolated from GF mice displayed reduced colony formation in methylcellulose, compared to SPF mice, with significant reductions in both neutrophil and monocyte production (Figures 2G, H). Overall, we conclude that the microbiota shapes innate immune profiles by promoting myeloid progenitor development and differentiation in the bone marrow and extramedullary sites, revealing that gut bacteria control immunity at its core—during hematopoiesis.
Tissue-Resident Phagocytes Mediate Protection by Commensal Microbes
Innate immune cells are the first responders to infection, mediating early pathogen control and coordinating downstream immune reactions(Kastenmuller et al., 2012; Shi and Pamer, 2011). We sought to test the impact of commensal microbes on myeloid cell differentiation by employing infection models where innate immunity is vital for an effective immune response. SPF and GF mice were infected intravenously (i.v.) with the model pathogen, Listeria monocytogenes. SPF mice challenged systemically with L. monocytogenes effectively control infection, as previously described (Figure 3A) (Serbina et al., 2012; Shi et al., 2011). However, GF mice rapidly succumb at the same inoculum (Figure 3A). Heightened susceptibility to infection among GF mice was associated with a significant increase in splenic and liver bacterial burden 24 and 72 hours post-infection (hpi), demonstrating a defect in early resistance to Listeria infection (Figures 3B, C and Figure S3A). Susceptibility to infection is not restricted to L. monocytogenes, as GF mice also displayed increased disease burden following systemic challenge with Staphylococcus aureus (Figure S3B). Interestingly, SPF mice treated orally with broad-spectrum antibiotics were also impaired in controlling Listeria, indicating protection by commensal microbes is an active process and is subject to loss following depletion of gut microbiota (Figure 3D). Collectively, these data reveal that commensal microbes are critical for rapid and potent systemic immune responses to acute bacterial infection.
Figure 3. The Microbiota Promotes Early Resistance to Systemic Infection by L. monocytogenes via Tissue-Resident Cells.
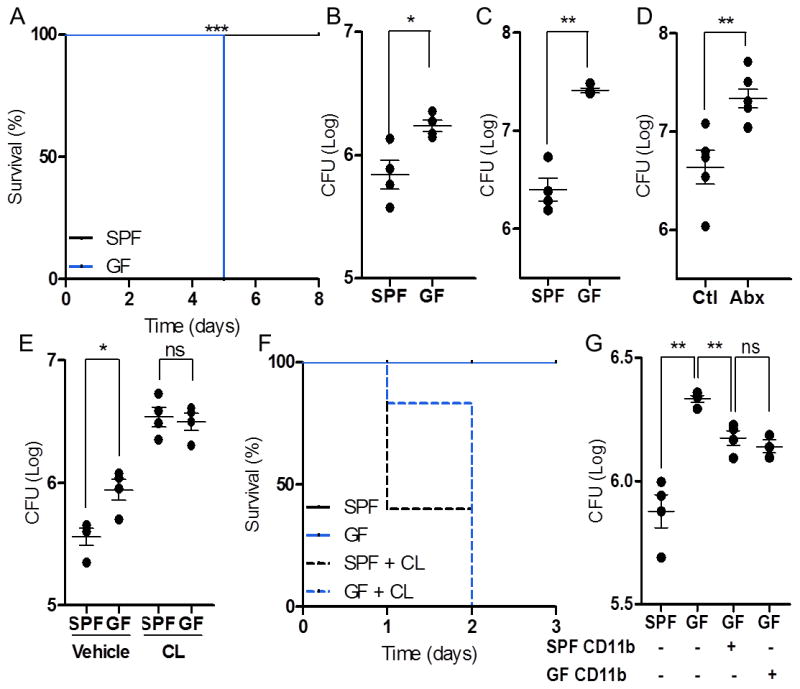
(A-C) SPF and GF mice infected with L. monocytogenes and assessed for survival (A) and splenic bacterial burden at 24 (B) and 72 (C) hours post- infection (hpi). SPF mice treated with antibiotics (Abx) and untreated controls (Ctl) were infected with L. monocytogenes and splenic bacterial burden was measured 72 hpi (D). SPF and GF mice depleted of tissue-resident cells prior to infection with L. monocytogenes and assessed for splenic bacterial burden 24 hpi (E) and survival (F). Splenic bacterial burden, 24 hpi, following transfer of splenic CD11b+ cells from SPF or GF donors (G). For all panels, data are representative of at least 2-3 independent trials with n≥ 4/ group. Each symbol represents data from a single animal. Error bars represent SEM. *p<0.05, **p<0.01, *** p<0.05 log-rank test used for survival curves in (A). CL: clodronate-loaded liposomes. See also Figure S3.
To confirm that defects in myelopoiesis contribute to increased disease burden in GF mice, phagocytic cells were depleted with clodronate-loaded liposomes (CL) prior to infection with L. monocytogenes(van Rooijen et al., 1996). CL pre-treatment increased susceptibility to Listeria infection (Figure 3E), confirming the importance of resident cells in pathogen resistance(Aichele et al., 2003; Kastenmuller et al., 2012). Importantly, depletion of resident phagocytes rendered both SPF and GF mice equally susceptible to infection, resulting in similar splenic disease burden 24 hpi (Figure 3E), and rapid death within 48 hpi (Figure 3F). While functional defects inmyeloid cellsmay potentially contribute to increased disease in GF mice, we did not detect differences during in vitroListeria killing by macrophages from SPF or GF mice (Figure S3C). Furthermore, CD11b+ myeloid cells isolated from either SPF or GF donors were equally sufficient in providing protection when transferred into GF mice prior to infection (Figure 3G),suggesting reduced cell proportions are likely the primary defect in GF mice. These studies confirm the importance of microbiota-driven myelopoiesis in promoting host resistance during systemic infection.
Effective responses to L. monocytogenes requires coordination between innate and adaptive immune cells, resulting in pathogen clearance and protective immunity (Pamer, 2004). Thus, we investigated whether additional immune cells beyond tissue-resident phagocytes may mediate commensal-derived protection to Listeria infection. We show that adaptive immunity is not required for protection by the microbiota during acute infection (Figure S3D), nor are GF mice deficient in developing long-term protective immunity against subsequent infection (Figure S3E). Furthermore, the selective expansion of myeloid cells during acute infection, called emergency hematopoiesis, which is necessary for mediating delayed resistance to L. monocytogenes (following 48 hpi), was maintained in GF mice (Figure S3F). Finally, while there are fewer inflammatory neutrophils and monocytes recruited to the spleen following infection (Figure S3G), a possible consequence of increased apoptosis (Figure S3H), these cells were not required for commensal-mediated protection against L. monocytogenes (Figure S3I, J). Together, these findings demonstrate that hematopoietic defects in tissue-resident myeloid cells prior to infection of GF mice (i.e., during cell development) is the primary cause of impaired control of Listeria.
CommensalBacterialSignals Mediate Maintenance of Myelopoiesis
The molecular mechanism(s) by which commensal microbes promote steady-state expansion of bone marrow- and yolk sac-derived myeloid cells remains unknown. Microbial associated molecular patterns (MAMPs) and microbial metabolites, such as short chain fatty acids (SCFAs) have been shown to modulate various aspects of the host immune response (Chu and Mazmanian, 2013; Clarke et al., 2010; Smith et al., 2013). Furthermore, MyD88 (an adaptor for recognition of many MAMPs) was recently shown to promote GMP expansion and differentiation (Fiedler et al., 2013). Accordingly, we sought to address whether commensal-derived factors are involved in the maintenance of myeloid cells under naïve conditions. Re-colonization of GF mice with a complex microbiota andoral treatment with MAMPs, but not SCFAs, was sufficient to promote recovery of GMP-derived myeloid cells (neutrophils and monocytes) within the bone marrow (Figure 4A, B). Importantly, only re-colonization of GF mice with an SPF microbiota was sufficient to restore splenic populations of F4/80hi macrophagesand F4/80lo splenocytes (i.e., neutrophils, monocytes and macrophages) (Figure 4C and Figures S4A-D). Therefore, while MAMP treatmentis necessaryfor the maintenance of bone marrow-derived myeloid cells, colonization with a live and complex microbiota is required to promote complete myelopoiesis (including yolk sac-derived macrophages). Finally, only re-colonization of GF animals, and not oral MAMP treatment, was sufficient to restore the defect in GF mice to systemic challenge with L. monocytogenes (Figure 4D and data not shown).Collectively, these studiesreveal that the microbiota provides complex molecular signals that actively promote the hematopoietic differentiation of myeloid cells, resulting in peripheral phagocyte populationsthat function as sentinels for the early detection and control of systemic bacterial infection.
Figure 4. Re-colonization of GF Mice Restores Immune Integrity Against Systemic Listeriosis.
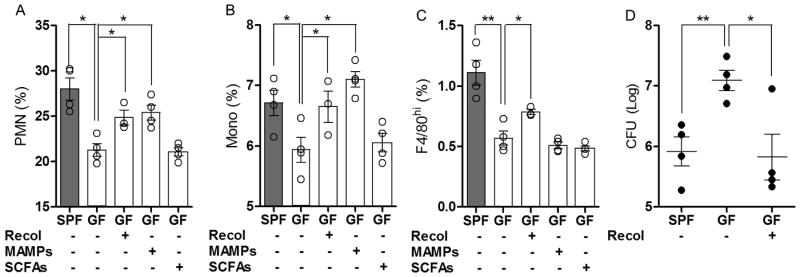
(A) Neutrophil (GR1hi CD115-) and (B) monocyte (GR1hi CD115+) bone marrow profiles from SPF, GF, re-colonized GF mice and MAMP or SCFA-treated GF mice. (C) F4/80hi splenic macrophage profile among SPF, GF, re-colonized GF mice and GF mice treated with MAMPs or SCFAs. (D) Splenic bacterial burden 72 hpi among SPF, GF and re-colonized GF mice infected with L. monocytogenes. For all panels, data are representative of at least 2 independent trials with n≥4/ group. Each symbol represents data from a single animal. Error bars represent standard error of mean (SEM). *p<0.05, **p<0.01. Recol: re-colonized; MAMPs: molecular associated molecular patterns; SCFAs: short chain fatty acids. See also Figure S4.
Discussion
Advances in understanding host-microbial symbiosis have revealed that, remarkably, the gut microbiota controls the phenotype, migration and activity of multiple innate and adaptive cells(Belkaid and Naik, 2013; Chu and Mazmanian, 2013). Disruption or alteration of commensal communities impacts host susceptibility to various disorders, particularly at sites of microbial colonization such as the intestines, respiratory mucosa and skin epithelium(Kamada et al., 2013). In addition to modulating functional immune outcomes, the microbiota is necessary for maintaining systemic populations of neutrophils in the circulation and CD4+ T cells in the spleen(Bugl et al., 2013; Mazmanian et al., 2005), suggesting a possible contribution by gut microbiota to the development of the immune system. Herein, we revealthat gut bacteria regulatehematopoiesiswithin primary immune sites, providing a unifying explanation for previous observations of the widespread effectsby the microbiota on the immune system. Specifically, our study uncovers that the microbiota promotes steady-state myeloid cell development by driving the expansion of yolk sac-derived macrophages, as well as enhancing the numbers and differentiation potential of GMPs in the bone marrow. We propose a model whereby a primary defect in hematopoiesis in GF or antibiotic-treated mice compromisesmultiple tissue-resident innate immune cell populations prior to infection, leading to blunted early responses upon subsequent pathogen encounter (see diagram in Figure S4E). While our studies focus on innate immunity due to its role in rapid control of early Listeria infection, impaired microbiota-mediated hematopoiesis may alsoextend to the adaptive immune system, providing an explanation for observations that peripheral T, B and iNKT cell populations are altered in GF mice (Ivanov et al., 2008; Macpherson and Uhr, 2004; Mazmanian et al., 2005; Olszak et al., 2012).
How commensal microbes (presumably in the gut) are able to control immune responses in distant sites such as the bone marrow remains incompletely understood. It has recently been shown that mice deficient in MyD88 signaling display reductions in systemic myeloid cell populations and GMP numbers(Fiedler et al., 2013; Yanez et al., 2013), similar to our findings in GF mice. Further, as microbial ligands have been detected in systemic sites, including the bone marrow (Clarke et al., 2010), commensal-derived MAMPs that originate in the gut may mediate steady-state myelopoiesis in primary immune sites. Accordingly, we show that oral treatment with MAMPs is sufficient to rescue GMP-mediated expansion of neutrophils and monocytes. However, MAMP treatment aloneis inadequate to expand splenic F4/80hiand F4/80lo cells, indicating additional commensal-derived signals are necessary to influence site-specific HSC and yolk sac-derived myeloid cells. Interestingly, re-colonization of adult GF mice with SPF microbiota is insufficient to restore splenic F4/80hi macrophages to the levels found in SPF mice. This may suggest that complete rescue requires either colonization from birth or colonization with specific microbes that were not transferred into GF mice. In addition to MAMPs translocating from the gut into the circulation, other explanations for how the microbiota affects hematopoiesis may include a role for myeloid cell growth factors. In support of this notion, preliminary data suggest that GF mice are reduced in M-CSF transcript levels in the gut (data not shown), though further work is need to uncoverthe complex molecular mechanism(s) by which commensal bacteria signal from the gut to distant primary immune organs.
Finally, we speculate that these findings may be relevant to human infections. The spread of antibiotic-resistance among pathogens, paired with a dwindling supply of effective antibiotics,has necessitated alternative strategies to combatinfections (Khosravi and Mazmanian, 2013). Evidence that depletion of the microbiota leads to transient immune suppression suggests factors that disrupt commensal microbes, including clinical antibiotic use may, paradoxically, be a risk factor for susceptibility to opportunistic pathogens. The concepts proposed herein, if validated in humans, may herald future medical approaches that combine antibiotics with immunomodulatorymicrobial molecules as revolutionary combination treatments to address the reemerging crisis of infectious diseases.
Experimental Procedures
Animal Studies
Specific pathogen-free (SPF) C57BL/6 mice were purchased from Taconic Farms. Germ-free (GF) C57BL/6 and C57BL/6 Rag-/- mice were bred and raised in sterile gnotobiotic flexible film isolators at the California Institute of Technology. Mice at 8-12 weeks of age were infected via retro-orbital injection with 3×104 colony forming units (CFU) of Listeriamonocytogenes 10403S. Splenic and liver bacterial CFU were assessed 24-72 hpi by microbiological plating. For microbiota depletion studies, SPF mice were treated with 1 mg/ml of ampicillin (Auromedics), neomycin sulfate (Fisher), streptomycin (Sigma) and 0.5 mg/ml of vancomycin (Sagent) in the drinking water for 4-5 weeks. Mice were taken off antibiotics 4 days prior to infection. Antibiotic-treated and untreated SPF mice were infected with 3×104 CFU of L. monocytogenes and splenic bacterial burden was assessed 72 hpi. GF mice were recolonized by gavage with cecal contents of SPF mice. Alternatively, GF mice were treated with MAMPs through the addition of heat killed Escherichia coli strain Nissle(Lodinova-Zadnikova and Sonnenborn, 1997)or autoclaved cecal contents from SPF mice in water (∼1×109 CFU/ml indrinking water). For treatment with short chain fatty acids,sodium proprionate (Sigma), sodium butyrate (Sigma), and sodium acetate (Sigma) was added to drinking water at previously described concentrations (25mM, 40mM and 67.5mM, respectively)(Smith et al., 2013). Mice were re-colonized or treated with microbial ligands or metabolites for 4 weeks prior to cellular analysis and infectious studies. Animals were cared for under established protocols and IACUC guidelines from the California Institute of Technology.
Cellular Analysis
Spleens were either mechanically disrupted viapassage through 100 μm mesh filters (BD Biosciences) or digested in 0.5 mg/ml of Collagenase D (Roche) and 0.5 mg/ml of DNase I (Worthington). Bone marrow was collected by flushing femurs with PBS containing 0.5% BSA and 5mM EDTA. Single cell suspensions were removed of red blood cells (RBC lysis buffer, Sigma). Mature myeloid cells were evaluated by staining with antibodies to GR1 (RB6-8C5), Ly6C (HK 1.4), CD11b (M1/70), CD115 (AFS98) and F4/80 (BM8). Mouse hematopoietic stem and progenitor cells (HSPCs) were isolated from bone marrow by a combination of MACS magnetic bead purification (Miltenyi) and fluorescence activated cell sorting (FACS). Lineage marker-negative cells (Lin-) were first separated using a MACS lineage cell depletion kit (containing antibodies against CD5 (53-7.3), CD45R (B220; RA3-6B2), CD11b, Gr-1, 7-4 (15BS) and Ter-119 (Ter-119)) and an autoMACS Separator (Miltentyi). Lin- cells were then further stained with c-Kit (CD117; 3C1), Sca-1 (D7), CD16/CD32 (93), CD34 (RAM34). Populations of LKS+ cells (Lin- c-Kit+ Sca-1+; HSCs and MPPs), Lin- c-Kit+ Sca-1- (LKS-) CD34+ FcγRlo cells (CMPs) and LKS- CD34+ FcγRhi cells (GMPs) were analyzed by flow cytometry. LKS- CD34+ FcγRhi cells were FACS sorted using an Aria cell sorter (BD Biosciences). Steady-state cell proliferation was measured by intraperitoneal (i.p.) injection of 500 μg EdU (Life Technologies) and EdU incorporation among splenic myeloid cells was measured 24 hours later via Click-it EdU assay kit (Life Technologies). Antibodies were purchased from eBioscience, BD Bioscience, Miltenyi or Biolegend. Data were collected on a FACSCalibur or LSR Fortessa (BD Bioscience) and analyzed with FlowJo software (TreeStar).
Cell Depletion and Adoptive Transfer
Resident phagocytes were depleted by intravenous(i.v.) treatment with 100 μl of clodronate-loaded liposomes (CL; FormuMax) 48 hours prior to infection. CD11b+ splenocytes were isolated from naïve SPF and GF mice using CD11b microbeads (Miltenyi). 2×106 CD11b+ cells (>90% purity) were transferred into GF recipients, 24 hours prior to infection with L. monocytogenes. CFU burden was assessed 24 hpi.
CFU Assays
To evaluate hematopoietic potential, 1×103 Lin- or 1×103 LKS- CD34+ FcγRhi cells or 2×105 splenocytes were plated in triplicate in MethoCult GF M3434 (StemCell Technologies) methylcellulose-based medium and incubated for 7 days in 37°C with 5% CO2, after which the colonies were counted on the basis of their morphological characteristics in accordance with the manufacturer's instructions. On the same day, cells were harvested, counted and stained for c-Kit and CD11b expression for progenitor quantification by flow cytometry. For re-plating assays, 5×104 cells from the first culture were plated in triplicate in a secondary culture of fresh MethoCult GF M3434, and colonies were counted after 7 days of incubation.
Supplementary Material
Highlights.
Germ-free mice are deficient in spleen and bone marrow resident myeloid cell populations
Gut microbes impact both yolk sac- and stem cell-derived myeloid cell development
Microbiota promotes early resistance to systemic Listeria monocytogenes infection
Re-colonizationrestores immune integrity against systemic Listeriosis in germ-free mice
Acknowledgments
We thank H. Chu, Y. K. Lee, G. Sharon, A. Nanz (Caltech), and N. Hassanzadeh-Kiabi (Cedars-Sinai) for technical assistance, T. Thron for animal care, and S. McBride, D. Majumdar, S. Damle, P. Mehrabian (Caltech) and members of the Mazmanian laboratory for critical reading of the manuscript. We thank U. Sonnenborn (Ardeypharm GmbH) for the generous gift of Escherichia coli Nissle 1917. A.K. was supported by a pre-doctoral training grant from the National Institute of Health (GM007616). This research was supported by a Burroughs Wellcome Fund in the Pathogenesis of Infectious Disease award to S.K.M.
Footnotes
The authors declare no conflicts of interest related to this work.
Author notes: A.K., M.M., H.S.G. and S.K.M. designed the research. A.K., A.Y., J.G.P. and A.C. carried out the experiments. A.K., M.M. and H.S.G. and S.K.M. wrote the manuscript.
Supplemental Information: Supplemental Information includes four figures and can be found with this article online
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aichele P, Zinke J, Grode L, Schwendener RA, Kaufmann SH, Seiler P. Macrophages of the splenic marginal zone are essential for trapping of blood-borne particulate antigen but dispensable for induction of specific T cell responses. J Immunol. 2003;171:1148–1155. doi: 10.4049/jimmunol.171.3.1148. [DOI] [PubMed] [Google Scholar]
- Akashi K, Traver D, Miyamoto T, Weissman IL. A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature. 2000;404:193–197. doi: 10.1038/35004599. [DOI] [PubMed] [Google Scholar]
- Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, Fukuda S, Saito T, Narushima S, Hase K, et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature. 2013;500:232–236. doi: 10.1038/nature12331. [DOI] [PubMed] [Google Scholar]
- Belkaid Y, Naik S. Compartmentalized and systemic control of tissue immunity by commensals. Nat Immunol. 2013;14:646–653. doi: 10.1038/ni.2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugl S, Wirths S, Radsak MP, Schild H, Stein P, André MC, Müller MR, Malenke E, Wiesner T, Märklin M, et al. Steady-state neutrophil homeostasis is dependent on TLR4/TRIF signaling. Blood. 2013;121:723–733. doi: 10.1182/blood-2012-05-429589. [DOI] [PubMed] [Google Scholar]
- Chu H, Mazmanian SK. Innate immune recognition of the microbiota promotes host-microbial symbiosis. Nat Immunol. 2013;14:668–675. doi: 10.1038/ni.2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke TB, Davis KM, Lysenko ES, Zhou AY, Yu Y, Weiser JN. Recognition of peptidoglycan from the microbiota by Nod1 enhances systemic innate immunity. Nat Med. 2010;16:228–231. doi: 10.1038/nm.2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiedler K, Kokai E, Bresch S, Brunner C. MyD88 is involved in myeloid as well as lymphoid hematopoiesis independent of the presence of a pathogen. American journal of blood research. 2013;3:124–140. [PMC free article] [PubMed] [Google Scholar]
- Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. Development of Monocytes, Macrophages, and Dendritic Cells. Science. 2010;327:656–661. doi: 10.1126/science.1178331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgopoulos K. Haematopoietic cell-fate decisions, chromatin regulation and ikaros. Nat Rev Immunol. 2002;2:162–174. doi: 10.1038/nri747. [DOI] [PubMed] [Google Scholar]
- Hill DA, Siracusa MC, Abt MC, Kim BS, Kobuley D, Kubo M, Kambayashi T, LaRosa DF, Renner ED, Orange JS, et al. Commensal bacteria-derived signals regulate basophil hematopoiesis and allergic inflammation. Nat Med. 2012;18:538–546. doi: 10.1038/nm.2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov II, Frutos RdL, Manel N, Yoshinaga K, Rifkin DB, Sartor RB, Finlay BB, Littman DR. Specific Microbiota Direct the Differentiation of IL-17-Producing T-Helper Cells in the Mucosa of the Small Intestine. Cell Host & Microbe. 2008;4:337–349. doi: 10.1016/j.chom.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins SJ, Ruckerl D, Cook PC, Jones LH, Finkelman FD, van Rooijen N, MacDonald AS, Allen JE. Local macrophage proliferation, rather than recruitment from the blood, is a signature of TH2 inflammation. Science. 2011;332:1284–1288. doi: 10.1126/science.1204351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamada N, Kim YG, Sham HP, Vallance BA, Puente JL, Martens EC, Nunez G. Regulated virulence controls the ability of a pathogen to compete with the gut microbiota. Science. 2012;336:1325–1329. doi: 10.1126/science.1222195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamada N, Seo SU, Chen GY, Nunez G. Role of the gut microbiota in immunity and inflammatory disease. Nat Rev Immunol. 2013;13:321–335. doi: 10.1038/nri3430. [DOI] [PubMed] [Google Scholar]
- Kastenmuller W, Torabi-Parizi P, Subramanian N, Lammermann T, Germain RN. A spatially-organized multicellular innate immune response in lymph nodes limits systemic pathogen spread. Cell. 2012;150:1235–1248. doi: 10.1016/j.cell.2012.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khosravi A, Mazmanian SK. Disruption of the gut microbiome as a risk factor for microbial infections. Current Opinion in Microbiology. 2013;16:221–227. doi: 10.1016/j.mib.2013.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YK, Menezes JS, Umesaki Y, Mazmanian SK. Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2011;108(Suppl 1):4615–4622. doi: 10.1073/pnas.1000082107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodinova-Zadnikova R, Sonnenborn U. Effect of preventive administration of a nonpathogenic Escherichia coli strain on the colonization of the intestine with microbial pathogens in newborn infants. Biology of the neonate. 1997;71:224–232. doi: 10.1159/000244421. [DOI] [PubMed] [Google Scholar]
- Macpherson AJ, Uhr T. Induction of Protective IgA by Intestinal Dendritic Cells Carrying Commensal Bacteria. Science. 2004;303:1662–1665. doi: 10.1126/science.1091334. [DOI] [PubMed] [Google Scholar]
- Markle JG, Mortin-Toth S, Wong AS, Geng L, Hayday A, Danska JS. gammadelta T cells are essential effectors of type 1 diabetes in the nonobese diabetic mouse model. J Immunol. 2013;190:5392–5401. doi: 10.4049/jimmunol.1203502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massberg S, Schaerli P, Knezevic-Maramica I, Kollnberger M, Tubo N, Moseman EA, Huff IV, Junt T, Wagers AJ, Mazo IB, et al. Immunosurveillance by hematopoietic progenitor cells trafficking through blood, lymph, and peripheral tissues. Cell. 2007;131:994–1008. doi: 10.1016/j.cell.2007.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazmanian SK, Liu CH, Tzianabos AO, Kasper DL. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell. 2005;122:107–118. doi: 10.1016/j.cell.2005.05.007. [DOI] [PubMed] [Google Scholar]
- Mazmanian SK, Round JL, Kasper DL. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature. 2008;453:620–625. doi: 10.1038/nature07008. [DOI] [PubMed] [Google Scholar]
- Naik S, Bouladoux N, Wilhelm C, Molloy MJ, Salcedo R, Kastenmuller W, Deming C, Quinones M, Koo L, Conlan S, et al. Compartmentalized control of skin immunity by resident commensals. Science. 2012;337:1115–1119. doi: 10.1126/science.1225152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olszak T, An D, Zeissig S, Vera MP, Richter J, Franke A, Glickman JN, Siebert R, Baron RM, Kasper DL, et al. Microbial Exposure During Early Life Has Persistent Effects on Natural Killer T Cell Function. Science. 2012;336:489–493. doi: 10.1126/science.1219328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pamer EG. Immune responses to Listeria monocytogenes. Nat Rev Immunol. 2004;4:812–823. doi: 10.1038/nri1461. [DOI] [PubMed] [Google Scholar]
- Robbins CS, Chudnovskiy A, Rauch PJ, Figueiredo JL, Iwamoto Y, Gorbatov R, Etzrodt M, Weber GF, Ueno T, van Rooijen N, et al. Extramedullary Hematopoiesis Generates Ly-6Chigh Monocytes That Infiltrate Atherosclerotic Lesions. Circulation. 2012;125:364–374. doi: 10.1161/CIRCULATIONAHA.111.061986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues NP, Boyd AS, Fugazza C, May GE, Guo Y, Tipping AJ, Scadden DT, Vyas P, Enver T. GATA-2 regulates granulocyte-macrophage progenitor cell function. Blood. 2008;112:4862–4873. doi: 10.1182/blood-2008-01-136564. [DOI] [PubMed] [Google Scholar]
- Round JL, Mazmanian SK. The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol. 2009;9:313–323. doi: 10.1038/nri2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Round JL, Mazmanian SK. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc Natl Acad Sci U S A. 2010;107:12204–12209. doi: 10.1073/pnas.0909122107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz C, Gomez Perdiguero E, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, Prinz M, Wu B, Jacobsen SE, Pollard JW, et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science. 2012;336:86–90. doi: 10.1126/science.1219179. [DOI] [PubMed] [Google Scholar]
- Serbina NV, Shi C, Pamer EG. Monocyte-mediated immune defense against murine Listeria monocytogenes infection. Adv Immunol. 2012;113:119–134. doi: 10.1016/B978-0-12-394590-7.00003-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi C, Hohl TM, Leiner I, Equinda MJ, Fan X, Pamer EG. Ly6G+ neutrophils are dispensable for defense against systemic Listeria monocytogenes infection. J Immunol. 2011;187:5293–5298. doi: 10.4049/jimmunol.1101721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol. 2011;11:762–774. doi: 10.1038/nri3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieweke MH, Allen JE. Beyond Stem Cells: Self-Renewal of Differentiated Macrophages. Science. 2013;342 doi: 10.1126/science.1242974. [DOI] [PubMed] [Google Scholar]
- Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly YM, Glickman JN, Garrett WS. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science. 2013;341:569–573. doi: 10.1126/science.1241165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, Figueiredo JL, Kohler RH, Chudnovskiy A, Waterman P, et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009;325:612–616. doi: 10.1126/science.1175202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rooijen N, Sanders A, van den Berg TK. Apoptosis of macrophages induced by liposome-mediated intracellular delivery of clodronate and propamidine. J Immunol Methods. 1996;193:93–99. doi: 10.1016/0022-1759(96)00056-7. [DOI] [PubMed] [Google Scholar]
- Weissman IL. Developmental switches in the immune system. Cell. 1994;76:207–218. doi: 10.1016/0092-8674(94)90329-8. [DOI] [PubMed] [Google Scholar]
- Wu HJ, Ivanov II, Darce J, Hattori K, Shima T, Umesaki Y, Littman DR, Benoist C, Mathis D. Gut-Residing Segmented Filamentous Bacteria Drive Autoimmune Arthritis via T Helper 17 Cells. Immunity. 2010;32:815–827. doi: 10.1016/j.immuni.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanez A, Goodridge HS, Gozalbo D, Gil ML. TLRs control hematopoiesis during infection. European journal of immunology. 2013;43:2526–2533. doi: 10.1002/eji.201343833. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.