

Abstract
Protein ubiquitination and deubiquitination are central to the control of a large number of cellular pathways and signaling networks in eukaryotes. Although the essential roles of ubiquitination have been established in the eukaryotic DNA damage response, the deubiquitination process remains poorly defined. Chemical probes that perturb the activity of deubiquitinases (DUBs) are needed to characterize the cellular function of deubiquitination. Here we report ML323 (2), a highly potent inhibitor of the USP1-UAF1 deubiquitinase complex with excellent selectivity against human DUBs, deSUMOylase, deneddylase and unrelated proteases. Using ML323, we interrogated deubiquitination in the cellular response to UV- and cisplatin-induced DNA damage and revealed new insights into the requirement of deubiquitination in the DNA translesion synthesis and Fanconi anemia pathways. Moreover, ML323 potentiates cisplatin cytotoxicity in non-small cell lung cancer and osteosarcoma cells. Our findings point to USP1-UAF1 as a key regulator of the DNA damage response and a target for overcoming resistance to the platinum-based anticancer drugs.
Ubiquitin is a critical signaling protein that controls a vast number of cellular processes in eukaryotes, including proteasomal degradation, the DNA damage response, cell cycle regulation and chromatin remodeling1. Ubiquitin modification is reversible, and ubiquitin removal is mediated by a large group of proteins known as deubiquitinases or DUBs. DUBs counteract ubiquitin ligase activity, edit ubiquitin chains and rescue ubiquitin from proteins that have been targeted for proteasomal degradation2,3. Genetic deficiencies in DUBs have been associated with a number of human diseases, particularly cancer4.
Our knowledge of the nondegradative functions of ubiquitin has grown considerably in recent years, particularly in the arena of the DNA damage response. The Fanconi anemia (FA) and DNA translesion synthesis (TLS) pathways are among the first DNA damage tolerance and repair pathways that have been found to be regulated by reversible ubiquitination. Genetic deficiency in the FA pathway impairs the cellular repair of DNA interstrand crosslinks (ICLs) and predisposes patients to cancer. Fifteen FA genes have been identified, and eight of them (FANCA, FANCB, FANCC, FANCE, FANCF, FANCG, FANCL and FANCM) encode proteins forming a FA core complex that monoubiquitinates FA complementation group D2 (FANCD2) and FANCI5,6. As a result of monoubiquitination, FANCD2 and FANCI are directed to the nuclear DNA damage foci, where as a complex they colocalize with BRCA1 and RAD51 and interact with the downstream FA proteins (FANCD1, FANCN, FANCJ, FANCO and FANCP)6. Although the functions of these cellular events are not fully understood, the monoubiquitinated FANCD2–FANCI complex may act as a ‘landing pad’ on chromatin for multiple DNA nucleases such as FA-associated nuclease 1 (FAN1) and FANCP (also called SLX4) that function in ICL repair7. In addition to FANCD2–FANCI monoubiquitination, timely deubiquitination of modified FANCD2– FANCI was recently suggested to be required for the completion of ICL repair6.
TLS is another DNA damage tolerance pathway that requires reversible ubiquitination for its regulation. TLS allows DNA replication past DNA lesions that block high-fidelity DNA polymerases and is under tight control because of the intrinsic low fidelity of TLS polymerases. Ubiquitination of proliferating cell nuclear antigen (PCNA) has a central role in regulating TLS. PCNA undergoes monoubiquitination by RAD6 and RAD18 in response to replication fork stalling8,9. Monoubiquitinated PCNA allows for the recruitment of the TLS polymerases for lesion-bypass DNA synthesis10,11. Although the role of PCNA monoubiquitination in TLS has been established, the function of PCNA deubiquitination is largely unexplored. We and others have proposed that deubiquitination of PCNA may control the timely removal of TLS polymerases after lesion-bypass synthesis and restore the normal DNA replication polymerases (Pols)12-15. However, direct evidence for this notion remains elusive.
Human ubiquitin-specific protease 1 (USP1), which is associated with UAF1, has been identified as the DUB responsible for deubiquitinating PCNA, FANCD2 and FANCI in the DNA damage response6,13. USP1 is also required for the FANCD2 foci formation in both mouse and human cells16,17. A high level of genomic instability has been linked to deficiency in human ATAD5 (the human ortholog of yeast Elg1), which mediates PCNA deubiquitination by USP1-UAF1 (refs. 15,18). Together, these observations suggest that the DUB activity of USP1-UAF1 is important for the normal cellular response to DNA damage.
An in-depth understanding of the cellular roles of deubiquitination in the FA and TLS pathways requires an effective means of modulating the cellular activity of USP1. A small-molecule inhibitor that antagonizes the enzymatic activity of USP1 without perturbing its cellular protein level is highly desirable given that many human USPs are engaged in large physical interaction networks19. Besides being used as a chemical probe to decipher the deubiquitination process in the DNA damage response, a small-molecule inhibitor of USP1 also has the potential to be developed into new anticancer therapeutics, as supported by the observations that disruption of USP1 in chicken DT40 cells and knockout of the mouse Usp1 gene increase the sensitivity toward DNA crosslinking agents16,20.
We recently identified pimozide and GW7647, two known compounds, as inhibitors of human USP1-UAF1 with low micromolar affinity21. To our knowledge, these compounds are the first reported inhibitors against USP1-UAF1, and they demonstrate the feasibility of inhibiting the USP1-UAF1 complex with small molecules. However, both compounds are known to bind proteins unrelated to DUBs22,23, which has limited their utility as USP1-UAF1 chemical probes. Here we report the development and characterization of ML323, a nanomolar inhibitor of USP1-UAF1 with remarkable selectivity and no substantial cytotoxicity to the human cells tested. This study provides ML323 as a best-in-class chemical probe to investigate the role of USP1-UAF1 in the cellular response to DNA damage.
RESULTS
ML323 is a potent and reversible USP1-UAF1 inhibitor
Through quantitative high-throughput screening (qHTS) (Supplementary Results, Supplementary Table 1) and subsequent medicinal chemistry optimization of the hit compound 1, N-(thiophen-2-ylmethyl)-2-(2-(trifluoromethyl)phenyl)quinazolin-4-amine, we developed ML323 (2) as a small-molecule inhibitor of USP1-UAF1(Fig. 1a). Our results show that ML323 is a potent USP1-UAF1 inhibitor with half-maximal inhibitory concentration (IC50) values of 76 nM in a ubiquitin-rhodamine (Ub-Rho) assay and 174 nM and 820 nM in orthogonal gel-based assays using K63-linked diubiquitin (di-Ub) and monoubiquitinated PCNA (Ub-PCNA) as substrates, respectively (Fig. 1b,c and Supplementary Fig. 1a,b). Replacement of the isopropyl group on ML323 with the more polar oxetane group led to NCGC-959 (3) and a complete loss of activity (Supplementary Fig. 1c–f). NCGC-959 was thus used as a negative control compound in our studies.
Figure 1. ML323 is a potent USP1-UAF1 inhibitor through a mixed inhibition mechanism.

(a) Medicinal chemistry optimization and structure-activity relationship exploration of HTS hit 1 led to ML323. (b) Dose-dependent inhibition of USP1-UAF1 by ML323 in a Ub-Rho assay, a di-Ub assay and a Ub-PCNA assay. The results shown represent the mean ± s.d. of three independent experiments. (c) SDS-PAGE analysis of the cleavage of K63-linked diubiquitin by USP1-UAF1 in the presence of different concentrations of ML323. (d) ML323 inhibits USP1-UAF1 through a mixed inhibition mechanism. Shown are Lineweaver-Burk plots of USP1-UAF1–catalyzed cleavage of diubiquitin in the presence of 0 (diamond), 100 (square), 300 (triangle) or 500 (circle) nM of ML323. The data represent the mean ± s.d. The full SDS-PAGE gel for this figure is shown in Supplementary Figure 13.
The reversibility of the inhibition of USP1-UAF1 by ML323 was assessed by a rapid dilution assay, which measured the recovery of enzymatic activity after a 100-fold dilution of USP1-UAF1 after treatment with ML323 (at ten times the IC50) for 15, 30 and 60 min. The results indicate that ML323 is a reversible inhibitor (Supplementary Fig. 1g). Further analyses of the USP1-UAF1 inhibition kinetics by ML323 supported a mixed inhibition mechanism (Fig. 1d). These data suggest that ML323 probably exerts its inhibitory effect through an allosteric mechanism. The measured inhibition constants of ML323 for the free enzyme (Ki) and the enzyme-substrate complex (K’i) were 68 nM and 183 nM (Supplementary Fig. 1h,i), respectively, which is in accordance with the IC50 determination. We ruled out the possibility that ML323 acts as a disruptor of the USP1-UAF1 complex by native gel analysis of USP1-UAF1 incubated with varied concentrations of ML323 up to 100 times its IC50 (Supplementary Fig. 1j).
To further define the mode of action, we conducted a hydrogen-deuterium exchange (HDX) analysis of USP1-UAF1 after ML323 binding. HDX has been used increasingly to investigate the local and global effects of small-molecule inhibitors on the exposure and dynamics of polypeptide segments in proteins24-26. We determined the ML323-induced change in deuterium incorporation (ΔHDX) of the USP1-UAF1 complex in the presence and absence of ML323. The peptides identified by mass spectrometry covered 76% and 55% of the USP1 and UAF1 sequences, respectively. We observed two strongly protected and four modestly protected peptides in USP1 (Supplementary Fig. 2). Mapping the peptides to a modeled USP1 structure revealed that they are located outside the USP1 catalytic site27. The peptides comprising residues 412–448, 451–481, 524–563 and 553–566 are in the finger and palm domains that form the ubiquitin-binding site. The peptide comprising residues 137–151 is in the thumb domain, and the peptide comprising residues 367–383 is located in an inserted domain that interacts with UAF1 (ref. 27). ML323 binding to USP1-UAF1 also induced alterations in deuterium incorporation in the UAF1 subunit. Two UAF1 peptides with modest to strong protection after ML323 binding (residues 298-301 and 395–430) map to the two blades in the modeled eight-bladed β-propeller structure, whereas the peptides comprising residues 471–496 and 522–547 are located in the C-terminal region of UAF1 that is not included in the model. The HDX results provide further support for ML323 as an allosteric inhibitor of USP1-UAF1.
ML323 is a selective inhibitor of USP1-UAF1
The selectivity of ML323 was first assessed by determining the inhibitory effect of ML323 against 18 DUBs and the related deSUMOylase and deneddylase, 70 unrelated proteases and 451 kinases in in vitro assays (Supplementary Tables 2 and 3 and Supplementary Figs. 3 and 4a,b). ML323 showed little to no inhibition against the USPs tested, including USP2, USP5, USP7, USP8, USP10, USP11, USP14 and USP21. Notably, ML323 did not inhibit the USP46-UAF1 complex that contains the same UAF1 subunit as the USP1-UAF1 complex and demonstrated little inhibition against USP1 alone (IC50 > 200 μM) in a Ub-Rho assay in which USP1-UAF1 and USP1 demonstrated similar Km values. Furthermore, ML323 displayed little to no inhibition against DUBs in the ubiquitin C-terminal hydrolase (UCH), ovarian tumor protease (OTU) and Machado-Joseph domain (MJD) families and did not inhibit a deSUMOylase, SENP1, and a deneddylase, NEDP1 (Supplementary Tables 2 and 3). Lastly, no substantial inhibition was detected for ML323 against other proteases and kinases (Supplementary Table 3 and Supplementary Fig. 3).
To assess the on-target effect of ML323 in cells, we carried out activity-based profiling against cellular DUBs using hemagglutinin (HA)-tagged ubiquitin vinyl methyl ester (HA-Ub-VME) as a probe. We first demonstrated that ML323 inhibited the labeling of USP1-UAF1 by HA-Ub-VME in a biochemical assay (Supplementary Fig. 4c). We then treated HEK293T cells with 100 μM cisplatin in the absence or presence of ML323 at increasing concentrations from 0.5 to 50 μM. The cell lysates of the treated cells were separated on a denaturing SDS-PAGE gel, and the labeled DUBs were detected by HA-specific antibody (Fig. 2a). The profile of the labeled DUBs was in accordance with those reported in previous DUB profiling studies using HA-Ub-VME as a probe28,29. A progressive decrease in the intensity of a single band at ~100 kDa was observed with increasing concentrations of ML323, and no appreciable diminishment of other probe-reactive bands was detected in the same samples treated with ML323. Using a USP1-specific antibody, we confirmed that the labeled band close to 100 kDa was USP1 (Fig. 2b). With increasing concentrations of ML323, we observed a clear decrease in the intensity of the labeled USP1 band, a result that agrees with inhibition of the USP1 enzymatic activity by ML323 in a cellular context. To further demonstrate the selectivity of ML323, we detected a number of cellular DUBs, including USP8, USP7, CYLD, USP5, USP14, UCH-L3 and UCH-L1, using antibodies specific for each DUB. ML323 did not inhibit the labeling of the DUBs tested by HA-Ub-VME (Fig. 2b).
Figure 2. Selective inhibition of USP1 by ML323 revealed by DUB profiling.

(a,b) Immunoblot analysis using HA-specific antibody (a) or antibodies to USP1, USP8, USP7, CYLD, USP5, USP14, UCH-L3 and UCH-L1 (b) of HEK293T cells that were treated with ML323 (0.5, 5 or 50 μM) or DMSO in the presence of 100 μM cisplatin, lysed, labeled with HA-Ub-VME and separated by SDS-PAGE. α-tubulin served as a loading control. The full immunoblot gels for this figure are shown in Supplementary Figure 13.
ML323 inhibits the deubiquitination of PCNA and FANCD2
To further confirm that ML323 inhibited USP1-UAF1 activity in cells, we monitored the level of monoubiquitinated PCNA and FANCD2 in H596 cells (Fig. 3a and Supplementary Fig. 5a). Cells were treated with ML323 at increasing concentrations in the absence or presence of cisplatin. The increase in Ub-PCNA level could be seen starting at an ML323 concentration of 5 μM (Supplementary Fig. 5b,c). A time profile analysis showed that ML323 alone or in combination with cisplatin exerted an effect on PCNA ubiquitination in 3–6 h after treatment (Supplementary Fig. 5d,e). When cells were treated with cisplatin alone for 6 h, a small increase in the levels of Ub-PCNA and Ub-FANCD2 was detected, whereas a combination of cisplatin and ML323 resulted in a threefold increase in these levels. Moreover, an increase in the monoubiquitination of PCNA and FANCD2 was also observed when the cells were treated with ML323 alone, albeit a lower increase than that with combination treatment (Fig. 3a). Notably, this effect could be fully reversed after removal of ML323 and cisplatin from the cell culture (Supplementary Fig. 5f–h). ML323 alone or in combination with cisplatin did not affect the level of Ub-H2A in either the absence or presence of cisplatin, which agrees with an on-target effect of ML323. Moreover, the protein level of USP1 was not affected in any treatment (Supplementary Fig. 5a). Similar observations were made in U2OS (Supplementary Fig. 6a,b) and HEK293T (Supplementary Fig. 6c,d) cells. Notably, the negative control compound NCGC-959 exerted no effect on the ubiquitination of PCNA and FANCD2 in H596 cells (Supplementary Fig. 6e,f).
Figure 3. ML323 inhibits the cellular activity of USP1-UAF1 and sensitizes cisplatin-resistant cells to cisplatin killing.
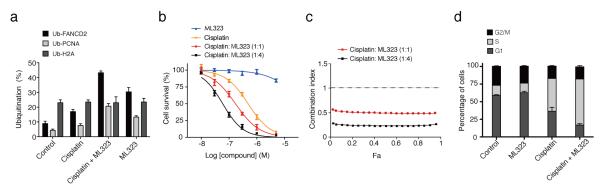
(a) The percentages of monoubiquitinated PCNA, FANCD2 and H2A in H596 cells that were treated with cisplatin (100 μM), ML323 (30 μM) or a combination of cisplatin (100 μM) and ML323 (30 μM). The data represent the mean ± s.d. of three independent experiments. (b) Cytotoxicity in H596 cells treated with cisplatin, ML323 or a combination of cisplatin and ML323 at a ratio of 1:1 or 1:4 determined by colony-forming assay. The data represent the mean ± s.d. of three independent experiments. (c) Synergistic interaction of cisplatin and ML323 at ratios of 1:1 and 1:4 in H596 cells in a colony-forming assay. The horizontal line in the figure represents a combination index of 1. (Fa, the fraction of cell affected). (d) The distribution of cell cycle phases of H596 cells that were synchronized in S phase and treated with DMSO, ML323 (20 μM), cisplatin (5 μM) or a combination of ML323 (20 μM) and cisplatin (5 μM). The data represent the mean ± s.d. of three independent experiments.
To further confirm the on-target effect of ML323 on USP1, we used short hairpin RNA (shRNA) to generate a USP1 knockdown H596 cell line. H596 cells transfected with the control shRNA behaved similarly to the parent H596 cells in response to the treatments (Supplementary Fig. 6g,h). In particular, combination treatment with cisplatin and ML323 led to an increased level of Ub-PCNA when compared to treatment with cisplatin alone. In marked contrast, in the USP1 shRNA knockdown H596 cells, little difference in Ub-PCNA levels was observed between treatment with cisplatin and treatment with the combination of ML323 and cisplatin (Supplementary Fig. 6g,h). Thus, removal of USP1 annulled the effect of ML323 on PCNA ubiquitination, which supports the on-target effect of ML323 in inhibiting USP1 in cells.
ML323 sensitizes resistant cells to cisplatin
Small molecules that disrupt DNA damage repair and tolerance pathways are promising in overcoming cancer cells’ resistance to cisplatin30. To investigate whether ML323 can potentiate cisplatin-resistant non-small cell lung cancer (NSCLC) H596 cells to cisplatin killing, we conducted a colony-forming assay. Cisplatin alone killed H596 cells with an effector concentration for half-maximum response (EC50) of 486 nM, whereas no substantial cell killing was observed for ML323 alone (EC50 > 10 μM) (Fig. 3b). For the combination of cisplatin and ML323 at ratios of 1:1 and 1:4, the EC50 values for H596 cell killing decreased to 171 and 59 nM, respectively. The 1:4 combination of cisplatin to ML323 represents an eightfold increase in the efficacy of cancer cell killing, which is comparable to the EC50 (74 nM) for the cisplatin-sensitive NSCLC H460 cell line (Supplementary Fig. 7a). The interaction between cisplatin and ML323 was further analyzed using CalcuSyn31. Strong synergistic effects were observed for the combination of cisplatin and ML323 in H596 cells (Fig. 3c). Similar potentiation and synergistic effects of ML323 were also observed in U2OS cells (Supplementary Fig. 7b,c). In comparison, NCGC-959 showed no potentiation to cisplatin cell killing (Supplementary Fig. 7d). To rule out off-target effects of ML323 in cell cytotoxicity, we used the USP1 shRNA knockout H596 cell strain (Supplementary Fig. 7e). In the same colony-forming assay, we observed no further sensitization to cisplatin after treatment with ML323 (Supplementary Fig. 7f). In contrast, the H596 cells transfected with the control shRNA showed similar potentiation by ML323 in cisplatin killing as compared to the parent H596 cells (Supplementary Fig. 7g).
Next we analyzed how inhibition of USP1-UAF1 by ML323 affects cell cycle distribution in NSCLC H596 cells by propidium iodide incorporation (Fig. 3d and Supplementary Fig. 8). Treatment of H596 cells with ML323 alone did not result in cell cycle delay or arrest, whereas treating H596 cells with cisplatin alone led to an increased population of cells in S phase (45%), which is consistent with the effect of cisplatin DNA adducts in blocking DNA replication32. Remarkably, treatment of H596 cells with a combination of ML323 and cisplatin further increased the percentage of S-phase cells to 65%. These observations suggest that deubiquitination by USP1-UAF1 is required for S-phase cells to overcome the damage induced by cisplatin and continue the replication of genomic DNA.
Inhibition of USP1-UAF1 compromises TLS
We next investigated deubiquitination of PCNA in TLS using ML323. Polη is the specialized DNA polymerase responsible for the translesion synthesis past UV-induced DNA lesion, cyclobutane pyrimidine dimer (CPD)33. We determined the UV sensitivity of the Polη-deficient xeroderma pigmentosum variant XP115LO (XPV) and Polη-complemented XP115LO cells (XPV + Polη) in the absence or presence of ML323 treatment. In the absence of ML323, a dose lethal to 50% of the cells tested (LD50) of 3.1 J m−2 was observed for XPV cells (Fig. 4a). Complementing the XPV cells with ectopically expressed Polη increased the LD50 to 8.1 J m−2 (Fig. 4b). This observation supports the idea that Polη has an essential role in TLS-mediated cellular tolerance of UV-induced DNA damage. We then determined the LD50 of UV sensitivity for XPV and XPV + Polη cells in the presence of 20 μM ML323. For XPV cells, a LD50 of 3.2 J m−2 was obtained, a value that is virtually unchanged from that determined for XPV cells in the absence of ML323 (Fig. 4a). This observation suggests that ML323 had no off-target effects that would lead to further sensitization of human cells to UV-induced DNA damage. In contrast, the LD50 of the XPV+ Polη cells was decreased to 4.7 J m−2 with the combination of UV and ML323, which is a 42% decrease from the LD50 of 8.1 J m−2 for UV treatment alone (Fig. 4b). We observed similar effects using small interfering RNA (siRNA)-mediated knockdown of USP1 in XPV and XPV+ Polη cells when irradiated with UV (Supplementary Fig. 9a–d). Collectively, these observations support the notion that inhibition of cellular USP1-UAF1 activity by ML323 compromises Polη-mediated TLS of UV-induced DNA damage.
Figure 4. The effect of inhibiting USP1-UAF1 by ML323 in the TLS and FA pathways.

(a,b) Colony-forming assay of XPV cells (a) and XPV + Polη cells (b) irradiated with UV alone (black) or a combination of UV and 20 μM ML323 (red). The data represent the mean ± s.d. of three independent experiments. (c,d) Cytotoxicity assay of PD20 cells (c) and PD20 + D2 cells (d) treated with cisplatin alone (black), ML323 (blue) or a combination of cisplatin with ML323 at a ratio of 1:4 (red). The data represent the mean ± s.d. of three independent experiments. (e) Colony-forming assay of PD20 cells irradiated with UV alone (black) or a combination of UV and 20 μM ML323 (red). The data represent the mean ± s.d. of three independent experiments. (f) Comparison of FANCD2 foci formation in U2OS cells treated with 30 μM ML323, 5 μM cisplatin or a combination of 30 μM ML323 and 5 μM cisplatin. Scale bars, 10 μm. DAPI, 4′,6-diamidino-2-phenylindole.
To determine whether treatment with ML323 alters TLS efficiency in human cells, we exploited a cellular DNA replication assay to quantify the TLS efficiency of a defined DNA lesion in human cells (Supplementary Fig. 10a,b)34. We found that inhibition of USP1-UAF1 by ML323 led to a 31% decrease in cellular replication efficiency past a defined CPD lesion (Supplementary Fig. 10c,d). This result from the cellular TLS assay supports the notion that USP1-UAF1 is required for TLS synthesis past a CPD lesion induced by UV irradiation.
USP1-UAF1 is important for the FA pathway
FANCD2, another substrate of USP1, functions in the FA pathway in response to DNA damage. The exact role of deubiquitination of FANCD2 in the FA-mediated DNA damage response is not fully understood, although USP1-UAF1-catalyzed deubiquitination of FANCD2 has been shown to be required for DNA ICL repair6,20. We have demonstrated that ML323 sensitized human cells to cisplatin. However, it is not clear whether ML323 exerted its effect by disrupting the FA pathway, the TLS pathway or both. To investigate this, we tested the cisplatin sensitivity of FANCD2-deficient PD20 cells and FANCD2-complemented PD20 cells (PD20 + D2) in the presence of ML323 and found that the PD20 cells were sensitive to cisplatin. A small but significant increase in sensitivity was observed in PD20 cells treated with a combination of cisplatin and ML323 (1:4), as evidenced by a shift of the dose-response curve to the lower cisplatin concentration regime for the combination of ML323 and cisplatin (P < 0.05) (Fig. 4c). In PD20 + D2 cells, we observed a higher EC50 (12.3 μM) when the cells were treated with cisplatin alone(Fig. 4d) , a result that is in agreement with the role of FANCD2 and the FA pathway in tolerance of cisplatin-induced DNA damage. Notably, treating PD20 + D2 cells with a combination of cisplatin and ML323 (1:4) shifted the EC50 from 12.3 to 4.2 μM (Fig. 4d). A similar effect of USP1 knockdown by siRNA was observed in PD20 and PD20 + D2 cells in response to cisplatin treatment (Supplementary Fig. 11a–d). These observations support the notion that deubiquitination of FANCD2 by USP1-UAF1 is essential for cellular tolerance to cisplatin. In the absence of FANCD2, USP1-UAF1 may still contribute to the tolerance of cisplatin-induced intrastrand crosslink, likely through deubiquitinating PCNA in TLS. Notably, there was no further change in sensitization to cisplatin in FANCC- and FANCG-deficient cells (FANCC and FANCG are subunits of the FA core complex that ubiquitinates FANCD2) when treated with a combination of ML323 and cisplatin (Supplementary Fig. 11e,f). This observation suggests that the FA core complex may also contribute to the tolerance of cisplatin-induced DNA damage through an indirect mechanism by facilitating TLS. Indeed, multiple lines of evidence have suggested that the FA core complex is required for the expression of Polη and the formation of Rev1 nuclear foci after UV irradiation35,36.
To further elucidate the role of USP1-UAF1 in the FA pathway, we assessed the UV sensitivity of PD20 and PD20 + D2 cells in the presence of ML323. In the absence of ML323 treatment, the LD50 of PD20 cells was 5.1 J m−2 (Fig. 4e), which is comparable to that of PD20 + D2 cells (5.7 J m−2) (Supplementary Fig. 11g). This observation indicates that FANCD2 is not required for the cellular tolerance of UV-induced DNA damage. We then investigated whether ML323 could further sensitize PD20 and PD20 + D2 cells to UV-induced DNA damage. ML323 led to a similar decrease in LD50 in both PD20 and PD20 + D2 cells (to 2.8 and 3.4 J m−2, respectively) (Fig. 4e and Supplementary Fig. 11g). The facts that ectopic expression of FANCD2 did not alter UV sensitivity in human cells and that the inhibition of USP1-UAF1 by ML323 exhibited similar UV sensitization in both PD20 and PD20 + D2 cells suggests that FANCD2 was not directly involved in tolerance to UV-induced damage.
We also examined FANCD2 subnuclear foci formation in U2OS cells treated with ML323, cisplatin or a combination thereof (Fig. 4f and Supplementary Fig. 11h). Treatment of U2OS cells with cisplatin alone increased FANCD2 subnuclear foci formation (59.4% as compared to 13.1% in cells treated with vehicle). A combination of cisplatin and ML323 led to a decrease in FANCD2 subnuclear foci to 34.2%, demonstrating that the deubiquitinating activity of USP1 is required for FANCD2 foci formation, which is in agreement with earlier reports that used siRNA knockdown of USP1 in human cells17 and USP1 knockout in mouse embryonic fibroblast cells16. However, when we treated U2OS cells with ML323 alone, no substantial change in the percentage of FANCD2 foci was observed, which is different from a previous study that showed a decrease in FANCD2 foci when USP1 was knocked down by siRNA17. The exact cause of this discrepancy is not clear; however, given that ML323 only inhibits the DUB activity of USP1 without changing the USP1 protein level in treated cells, such a difference may suggest a structural role of USP1 or USP1-UAF1 in FANCD2 foci formation.
ML323 impairs homologous recombination and sister chromatid exchange
Previous studies have linked the FA pathway to DNA repair through homologous recombination (HR)37,38. To investigate whether USP1’s DUB activity is required for HR, we used DR-U2OS cells, which carry a single and stable copy of the HR reporter DR-GFP39. DR-U2OS cells were treated with either DMSO or ML323, and HR was induced by transfection with an I-SceI expression plasmid (pCBASce). In the absence of ML323, I-SceI expression led to an HR frequency of 2.56%, which is comparable to the value in a previous report39. In contrast, inhibition of USP1 by ML323 decreased the I-SceI–induced HR frequency to 0.81% (Fig. 5a and Supplementary Fig. 12a). As negative controls, treatment with transfection reagent or transfection with the control vector (pCAGGS) gave background HR frequencies (0.04%) in the absence or presence of ML323 (Fig. 5a and Supplementary Fig. 12a).
Figure 5. Inhibition of USP1-UAF1 by ML323 decreases HR and SCE in U2OS cells.

(a) HR efficiency as reported by flow cytometry of GFP-positive cells. DR-U2OS cells were treated with DMSO or ML323 (30 μM) for 6 h and then transfected with either a control vector (pCAGGS) or an I-SceI expression plasmid (pCBASce) to induce double-strand breaks. Control cells were treated with the transfection reagent. The bar graph represents the mean ± s.d. of three independent tests. (b) Comparison of SCE frequencies in U2OS cells treated with DMSO, ML323 (30 μM), cisplatin (15 μM) or combined ML323 (30 μM) and cisplatin (15 μM). The dot graph shows the SCE events per cell under indicated treatment. Horizontal lines indicate the mean ± s.d. of SCE events per chromosome. NS (nonsignificant), P > 0.05; ***P < 0.001 by Student’s t-test (n > 40 per group).
Sister chromatid exchange (SCE) has been used to assess impairment in DNA repair through the HR, TLS and FA pathway38,40,41. To further decipher the mode of action of ML323 in modulating DNA repair, we performed a SCE assay using U2OS cells. Treating U2OS cells with ML323 alone had no effect on SCE frequencies (0.13 and 0.12 SCE events per chromosome for ML323 treatment and control, respectively) (Fig. 5b and Supplementary Fig. 12b), a result that is in line with an absence of cytotoxicity of the compound. However, treatment of U2OS cells with a combination of cisplatin and ML323 led to 0.84 SCE events per chromosome, which is significantly lower than that of cells treated with cisplatin alone (0.99 SCE events per chromosome; P < 0.001).
DISCUSSION
In this study we report the discovery and characterization of a new USP1-UAF1 inhibitor, ML323, with nanomolar potency and high selectivity. Inhibition mechanism and HDX studies suggest that ML323 binds away from the USP1 active site, which probably forms the basis for the selectivity against USP1-UAF1 over other USPs. This notion is further supported by the observation that ML323 is a poor inhibitor against USP1 alone. Notably, ML323 did not disrupt the association of the USP1 and UAF1 subunits in the complex.
In addition to the in vitro enzymatic assays, we also assessed the selectivity of ML323 against USP1 in the cellular environment. Using HA-tagged Ub-VME, which has been used to profile DUB activity in cell lysates28,29, we showed that ML323 efficiently inhibited the labeling of USP1 by the Ub-VME probe and exhibited selectivity toward other DUBs in human cells. We further demonstrated the on-target effect of ML323 in inhibiting the deubiquitination of monoubiquitinated PCNA and FANCD2, which are known substrates of USP1-UAF1, in NSCLC H596, U2OS and HEK293T cells. Using a USP1 shRNA knockdown H596 cell line, we showed that removal of the target protein USP1 in cells nullified the effect of ML323, thus ruling out an off-target effect of ML323 in the cells.
Using the USP1-UAF1–specific inhibitor ML323, we investigated the role of deubiquitination of PCNA in TLS past UV-induced DNA damage. We showed that treatment of Polη-complemented XPV cells with ML323 sensitized the cells to UV to a level similar to that in XPV cells. In marked contrast, no difference in UV sensitivity was observed between FANCD2-deficient and FANCD2-complemented PD20 cells with or without ML323 treatment. These observations support an important role of USP1-UAF1– catalyzed PCNA deubiquitination in successful TLS. At a molecular level, failed deubiquitination of PCNA may lead to impaired DNA replication due to an inefficient reverse polymerase switching after lesion-bypass synthesis, as proposed in a ‘polymerase switching’ model (Fig. 6). To obtain further evidence of this notion, we employed a cellular DNA replication assay that allowed us to quantify the lesion-bypass synthesis efficiency across a CPD lesion. We found that the treatment with ML323 led to a 31% decrease in CPD-lesion bypass efficiency in cells, agreeing with the model described above.
Figure 6.

ML323 inhibits the deubiquitination by USP1-UAF1 in the TLS and FA pathways in response to DNA damage. D2, FANCD2; I, FANCI; J, FANCJ; N, FANCN; O, FANCO; D1, FANCD1.
Cisplatin, one of the most widely used anticancer drugs, has been the first-line treatment of lung, ovarian, testicular and bladder cancers42. However, the therapeutic efficacy of cisplatin is limited by resistance developed during the treatment. DNA damage response pathways, particularly TLS and ICL repair, probably contribute to this resistance43,44. Disruption of these pathways may be a new therapeutic strategy for overcoming cisplatin resistance. In this study we demonstrated that ML323 effectively sensitized cisplatin-resistant NSCLC H596 cells and U2OS osteosarcoma cells to cisplatin. A strong synergistic effect between cisplatin and ML323 was evident in inhibiting the proliferation of H596 and U2OS cells. Inhibiting USP1 was also recently suggested as a useful strategy for treating squamous cell cancers45.
A notable feature of ML323 is that it simultaneously targets two major DNA damage response pathways (TLS and FA) by inhibiting a common deubiquitinase. Given that cisplatin can generate both intrastrand crosslinks and ICLs that are tolerated and repaired by the TLS and FA pathways, respectively, inhibiting USP1-UAF1 may be an effective way to overcome cancer cells’ resistance to cisplatin. We found similar potentiation to cisplatin treatment by ML323 for both NSCLC and osteosarcoma cells, suggesting that combining a USP1-UAF1 inhibitor with cisplatin can be a general strategy in blocking the growth of different types of cancers.
Notably, ML323 as a chemical probe provides a new means for investigating the cellular DNA damage response and its regulation by USP1-UAF1–catalyzed deubiquitination. Moreover, given that USP1-UAF1 has been recently found to deubiquitinate inhibitor of DNA binding (ID) proteins that are abundant in embryonic and adult stem cells46, ML323 may be adapted to investigate the cellular processes in stem cell differentiation. As a tool compound, ML323 can also be used to identify potential new USP1-UAF1 target proteins through a proteomic approach using ubiquitin remnant immunoaffinity profiling47. All of these applications of ML323 will propel understanding of the function and regulation of a key DUB complex, USP1-UAF1, and the related cellular pathways.
Online Methods
Expression and purification of recombinant enzymes and Ub-PCNA
USP1, USP1-UAF1, USP46-UAF1 and USP11 were generated as previously described21,48. Ub-PCNA was prepared as previously reported49. A USP21 (residues 209–563) expression plasmid was obtained from the laboratory of C. Arrowsmith through Addgene and transformed into Escherichia coli BL21 (DE3) competent cells. Transformed cells were grown at 37 °C in LB medium supplemented with 50 μg ml−1 kanamycin until the optical density at 600 nm (OD600) reached 0.6. Induction was performed overnight with 0.1 mM IPTG at 15 °C. Cell pellets were resuspended in a lysis buffer containing 50 mM NaH2PO4 (pH 8.0), 500 mM NaCl, 5% glycerol, 10 mM 2-mercaptoethanol and 10 mM imidazole, sonicated on ice and centrifuged at 4 °C. The supernatant was bound to nickel–nitrilotriacetic acid (Ni-NTA) resin (Invitrogen) for 1 h at 4 °C. Recombinant USP21 was eluted with lysis buffer containing 100 mM imidazole and subsequently dialyzed into 50 mM NaH2PO4 (pH 7.0), 300 mM NaCl, 5% glycerol and 1 mM DTT and loaded on a HiLoad 16/60 Superdex 200 (GE Life Sciences) gel filtration column. Eluted protein was diluted to reduce NaCl concentration to 50 mM and further purified using a HiTrap SP FF column (GE Life Sciences).
Reagents and antibody sources
K63-linked diubiquitin, SUMO1-AMC, UCH-L1, UCH-L3, SENP1, full-length USP7 (HAUSP), USP5 (Isopeptidase T), USP8 and the catalytic domain of USP2 (residues 259– 605) were purchased from Boston Biochem, and papain was obtained from Sigma-Aldrich. Z-FR-AMC was from Invitrogen. Caspase-1, caspase-3 and caspase-6 were from Abcam. HA-Ub-VME, Ac-WEHD-AFC, Ac-DEVD-AFC and Ac-VEID-AFC were purchased from Enzo Life Science. Anti–5-bromodeoxyuridine (BrdU)-conjugated (sc-56259) (dilution of 1: 150), anti-mouse fluorescein isothiocyanate (FITC)-conjugated (sc-2010) (dilution of 1: 100), anti- human PCNA (sc-56) (dilution of 1: 500) and anti-FANCD2 (sc-20022) (dilution of 1: 750) antibodies were from Santa Cruz. H2A-specific (ab18255) (dilution of 1: 1000) and α-tubulin–specific (ab7291) (dilution of 1: 5000) antibodies were from Abcam. Antibodies to USP7 (A310-006A) (dilution of 1: 1000), USP5 (A310-542A) (dilution of 1: 2000) and USP14 (A310-293A) (dilution of 1: 2000) were from Bethyl Laboratories. Antibodies to CYLD (8462S) (dilution of 1: 1000), UCH-L1 (3524S) (dilution of 1: 1000) and UCH-L3 (8141S) (dilution of 1: 1000) were from Cell Signaling. USP8-specific (U2385-200UL) (dilution of 1: 500), HA-tag–specific (H9658) (dilution of 1: 10000), horseradish peroxidase (HRP)-conjugated anti-mouse (A9044) (dilution of 1: 80000) and HRP-conjugated anti-rabbit (A0545) (dilution of 1: 80000) antibodies were from Sigma-Aldrich. Anti-mouse Alexa Fluor 488–conjugated (A11001) (dilution of 1: 400) antibody was from Invitrogen. The USP1 antibody used in Supplementary Figures 5f-h and 7e was from Proteintech (14346-1-AP) (dilution of 1: 200); the USP1 antibody used in the other figures was from Abcam (ab108104) (dilution of 1: 200). USP1 siRNAs were from Santa Cruz. USP1 shRNA and scramble shRNA were from Applied Biological Materials (ABM) Inc.
High-throughput screening
For HTS, USP1-UAF1 activity was monitored using ubiquitin–rhodamine 110 as a substrate, where hydrolysis of the amide bond between the C-terminal glycine of ubiquitin and rhodamine results in an increase in fluorescence21. The assay was miniaturized to a 4 μl volume in a 1,536-well format and was used to screen approximately 402,701 compounds in quantitative HTS mode, with each compound tested over a range of four to five concentrations50. The assay showed robust performance with an average Z’ factor of 0.8 throughout the screen.
Cell cultures
The human NSCLC cell lines H596 and H460 were purchased from American Type Culture Collection (ATCC) and cultured in RPMI 1640 medium containing 10% FBS, 100 units ml−1 penicillin and 0.1 mg ml−1 streptomycin. The HEK293T cell line and human osteosarcoma U2OS cells were cultured in DMEM supplemented with 10% FBS, 100 units ml−1 penicillin and 0.1 mg ml−1 streptomycin. FANCC-deficient cells (PD331), FANCG-deficient cells (PD352), FANCD2-deficient cells (PD20) and FANCD2-reconstituted PD20 cells (PD20 + D2) were from Fanconi Anemia Research Fund, INC. and were cultured in DMEM supplemented with 10% FBS, 100 units ml−1 penicillin and 0.1 mg ml−1 streptomycin. XPV human fibroblasts GM02359-hTERT (XP115LO) and Polη-complemented GM02359-hTERT (XPV + Polη) were gifts from M. Cordeiro-Stone (University of North Carolina at Chapel Hill) and were cultured in DMEM supplemented with 10% FBS, two times the concentration of MEM nonessential amino acids (Invitrogen) and 200 μg ml−1 G418 (Invitrogen). DR-GFP U2OS cells were gifts from M. Jasin (Memorial Sloan-Kettering Cancer Center) and were cultured in DMEM supplemented with 10% FBS, 100 units ml−1 penicillin and 0.1 mg ml−1 streptomycin. All the cell cultures were maintained at 37 °C and 5% CO2 in a humidified incubator.
K63-linked diubiquitin and Ub-PCNA gel-based assay
To determine the IC50 of ML323 in inhibiting USP1-UAF1, the inhibitor was added at seven different concentrations to the assay containing 150 nM USP1-UAF1 and 3 μM K63-linked diubiquitin or 300 nM USP1-UAF1 and 3 μM Ub-PCNA in a buffer containing 50 mM HEPES (pH 7.8), 0.1 mg ml−1 BSA, 0.5 mM EDTA and 1 mM or 5 mM DTT for 1–2 h at 37 °C. The reaction was quenched by the addition of Laemmli sample buffer. In the in vitro ML323 selectivity test, six different concentrations (0.08–114 μM) of ML323 and 3 μM K63-linked diubiquitin were incubated individually with 7.5 nM USP7, 30 nM USP2, 15 nM USP5, 255 nM USP8, 100 nM USP11, 600 nM USP21 and 600 nM USP46-UAF1. The results were analyzed as previously described21.
In vitro profiling of DUBs and proteases
For DUB profiling, ML323 was tested at a single-dose of 10 μM in duplicate. The DUB activities were monitored using Ub–7-amido-4-methylcoumarin (AMC) as a substrate. The increase in fluorescent signal from free AMC was monitored over time, although only the initial linear portion of slope (signal min−1) was used for analysis. The activity of enzyme with no compound was treated as 100%. For protease profiling, ML323 was tested using threefold serial dilutions starting at 20 μM against 70 proteases. Proteases were pre-incubated with the compound for 5–15 min before the addition of the appropriate enzyme substrates. The enzyme activities were measured by reading the fluorescent signal from fluorescently labeled peptides.
Reversibility assay
The inhibitor ML323 at a concentration of ten times the IC50 was pre-incubated with 200 nM USP1-UAF1 at room temperature for 15, 30 or 60 min. This solution was then diluted 100-fold in assay buffer (50 mM HEPES (pH 7.8), 0.1 mg ml−1 BSA, 0.5 mM EDTA and 1 mM DTT) and incubated at room temperature for an additional 5 min. Ub-AMC was then added to the solution at a final concentration of 200 nM to measure enzymatic activity. USP1-UAF1 was incubated with DMSO, and the activity measured for USP1-UAF1 was treated as 100%. USP1-UAF1 treated with rottlerin served as a control.
Inhibition mechanism determination
The standard reaction contained 150 nM USP1-UAF1, 7–80 μM K63-linked diubiquitin and variable concentrations of inhibitor in a buffer containing 50 mM HEPES, pH 7.8, 0.1 mg ml−1 BSA, 0.5 mM EDTA and 1 mM DTT. The reaction was allowed to proceed for 10–90 min at 37 °C and was quenched by the addition of Laemmli sample buffer at a given time point. The reaction product was separated on a 20% denaturing SDS-PAGE gel and stained with Coomassie Blue. The intensity of the individual diubiquitin and monoubiquitin bands were quantified using Quantity One 4.3.1 (Bio-Rad). The percentage of the conversion was determined and used to calculate the reaction rate. The Lineweaver-Burk plot was obtained by plotting 1/v against 1/[di-Ub] at four different inhibitor concentrations. For mixed inhibition, Ki and K’i were determined by fitting the data to the equations below:
Native gel electrophoresis
The USP1-UAF1 complex, UAF1 and USP1 were in a buffer containing 50 mM Tris (pH 8.0), 100 mM NaCl, 10% glycerol and 2 mM DTT. The USP1-UAF1 complex (2 μM) was incubated with ML323 at concentrations of 0.68, 3.4 and 17 μM at room temperature for 30 min. The samples were run on an 8% native gel at a temperature of 4 °C at 125 V for 2 h, and the gel was stained by Coomassie Blue.
HDX assay
The USP1-UAF1 complex was pre-incubated with 20 μM ML323 on ice for 30 min. Hydrogen-deuterium exchanges on proteins were initiated by diluting each sample fivefold in deuterated buffer containing 50 mM Tris (pD 8.0), 100 mM NaCl and 1 mM DTT (NaOD and DCl were used to adjust the pD) at 0 °C. Exchange was allowed to proceed for 10 min, after which the samples were quenched with deuterated denaturing buffer (0.5 M ammonium phosphate (pD 2.5), 100% (w/v) guanidinium hydrochloride and 2.5 mM Tris(2-carboxyethyl)phosphine (TCEP)) and subjected to a digestion with equimolar pepsin in solution (pH 2.1).
For the HDX LC/MS system and peptide identification, a Shimadzu HPLC with two LC-10AD pumps was used to generate a fast gradient that was optimized for best sequence coverage. Solvent A contained 5% acetonitrile and 0.1% formic acid in H2O, and solvent B consisted of 95% acetonitrile and 0.1% formic acid in H2O. All components of the setup, including tubing, injector and column, were submerged in an ice bath at all times in order to reduce back exchange. For analysis of proteolytic peptides, 50 μl of chilled digest was injected onto a 1.0 mm inner diameter × 50 mm C18 column (Phenomenex, Torrance, CA). After desalting for 5 min with 5% solvent B, the peptides were eluted with a gradient consisting of 5–10% solvent B in 0.01 min, 10–40% solvent B in 10 min, 40–50% solvent B in 1 min and 50–95% solvent B in 1 min at a flow rate of 50 μl min−1. The effluent was infused into a 12-T Varian IonSpec Fourier transform ion cyclotron resonance (FT-ICR) MS (Varian Inc.). For peptide identification, 30-s fractions were collected into a 96-well plate by coupling the HPLC with TriVersa NanoMate (Advion). Each fraction was spiked with a quadrupole Fourier transform (QFT) mass spectrometry standard (to allow internal mass calibration), and the mass spectra were collected by coupling chip-based infusion of the TriVersa NanoMate with the FT-ICR MS. The peptides were identified by searching MS-NonSpecific Protein Prospector (a University of California San Francisco web tool) against the combined sequences of USP1, UAF1 and pepsin (to eliminate pepsin self-digest peptides). The mass error threshold was set at 3 p.p.m., which is the instrument mass accuracy after internal calibration. Isobaric peptides, which are those whose masses are identical but whose sequences are different, were subjected to MS/MS analysis, and fragment ions were compared to those predicted by the Protein Prospector for each possible sequence. The two searches were combined to create a final list of USP1 and UAF1 peptides that were products of the peptic digest.
Changes in deuterium incorporation (ΔHDX) of each peptide were determined from the difference between the centroid masses of the peptides in the presence and absence of ML323 (control). Peptides that exhibited substantial changes in deuterium incorporation were mapped onto the modeled USP1 and UAF1 structures.
Activity-based DUB profiling
Purified USP1-UAF1 complex was incubated with 0.017, 0.17 or 10 μM of inhibitor at room temperature for 30 min. HA-Ub-VME (5 μM) was then added and incubated at 37 °C for an additional 4 h. Samples were separated by SDS-PAGE, transferred to nitrocellulose membrane and blotted with antibody against USP1.
For cellular DUB profiling, HEK293T cells were seeded in 10-cm plates and incubated at 37 °C overnight. Cells were treated with 100 μM cisplatin plus 0.5, 5 or 50 μM of ML323 for 6 h. Control cells were treated with an equal volume of saline and/or DMSO. After treatment, cells were washed with PBS, harvested and incubated on ice for 1 h. Samples were lysed on ice with lysis buffer containing 50 mM Tris (pH 7.4), 150 mM NaCl, 5 mM MgCl2, 0.5 mM EDTA, 5 mM DTT, 2 mM ATP, 0.5% NP40 and 10% glycerol. Cell lysates (100 μg) were then incubated with 0.5, 5 or 50 μM of ML323 or DMSO at room temperature for 1 h, which was followed by treatment with 2 μM HA-Ub-VME in a labeling buffer containing 50 mM Tris (pH 7.6), 5 mM MgCl2, 0.5 mM EDTA, 2 mM DTT, 2 mM ATP and 250 mM sucrose. The labeling reaction was incubated at 37 °C for an additional 2 h. Samples were then heated to 70 °C for 10 min and separated by SDS-PAGE. Gels were transferred onto a nitrocellulose membrane and blotted with antibodies against HA, USP1, USP8, USP7, CYLD, USP5, USP14, UCH-L1, UCH-L3 and α-tubulin. HRP-conjugated anti-mouse or HRP-conjugated anti-rabbit was used as the secondary antibody. Signals were detected using enhanced chemiluminescence Western blotting substrate (Thermo Fisher).
siRNA and shRNA knockdown of USP1
Cells were transfected with the USP1 siRNA, the USP1 shRNA or scramble shRNA using FuGENE 6 (Promega). For USP1 siRNA-transfected cells, treatments were applied 48 h after transfection. For stable USP1 shRNA knockdown, cells transfected with USP1 shRNA or scramble shRNA in FuGENE 6 were subjected to selection with 2 μg ml−1 puromycin in cell culture. A single colony was picked and grown to expand the cell culture. The USP1 knockdown efficiency was confirmed by western blotting. The cells grown from the single colony were used for the later experiments.
Cellular activity assay of USP1-UAF1
Cells were plated in 10-cm dishes and synchronized in S phase using the double-thymidine block method as described previously21. Cells were then treated with 100 μM cisplatin, 30 μM ML323 or 100 μM cisplatin plus 30 μM ML323 for 6 h at 37 °C. Cells treated with an equal volume of DMSO were used as a control. Treated cells were harvested, lysed, separated by SDS-PAGE and transferred to a nitrocellulose membrane. The membranes were blotted with PCNA antibody, FANCD2 antibody, H2A antibody and α-tubulin antibody. HRP-conjugated anti-mouse and anti-rabbit were the secondary antibodies. Images were quantified in FluorChem Q software (Imgen Technologies).
For the dose-dependent analysis of USP1-UAF1 inhibition in cells by ML323, synchronized H596 cells were plated in 10-cm dishes and treated with ML323 at the indicated concentrations in the absence or presence of 100 μM cisplatin for 6 h. For the time profile analysis of cellular PCNA ubiquitination after ML323 treatment, NSCLC H596 cells were plated in 10-cm dishes and synchronized in S phase with double-thymidine block. Cells were then treated with 100 μM cisplatin, 30 μM ML323 or 100 μM cisplatin plus 30 μM ML323 at the indicated times, and an equal volume of DMSO was used as a control. For the reversible ubiquitination assay, cells were treated with 100 μM cisplatin, 30 μM ML323 or 100 μM cisplatin plus 30 μM ML323 for 6 h. The compounds were removed by wash with PBS. Cells were incubated with fresh medium at the indicated times. Harvested cells were lysed, separated by SDS-PAGE and transferred to a nitrocellulose membrane. The membranes were blotted with anti-PCNA or anti-USP1 antibody. HRP-conjugated anti-mouse or anti-rabbit was used as the secondary antibody.
Cytotoxicity and colony-forming assays
Cell viability was measured by a cell counting kit (CCK) assay using CCK-8 solution (Dojindo Molecular Technologies, Inc.) as previously described21. For the colony-forming assay, cells were seeded at a density of 300–500 cells per well in six-well plates and grown overnight. Cells were then treated with ML323 alone, cisplatin alone or a combination of cisplatin and ML323 (1:1 or 1:4) at the indicated concentrations. Cells treated with an equal volume of DMSO and saline were used as a control. After 48 h of treatment, fresh growth medium was added, and cells were incubated for an additional 5–10 d to allow for colony formation. For UV combination treatment, the cells were treated with ML323 at the indicated concentrations or an equal volume of DMSO. After 48 h, the medium was removed, and cells were irradiated at 254 nm at the indicated dosage. Fresh growth medium was added, and the cells were incubated for an additional 5–10 d to allow for colony formation. The cells without UV irradiation but treated with ML323 or an equal volume of DMSO were used as controls and designated as 100%. After the formation of the colonies, cells were fixed with methanol and stained with 0.5% crystal violet (Sigma-Aldrich). Colonies consisting of >50 cells were scored. The number of colonies was determined from triplicate plates. The dose-response curves were generated using GraphPad Prism and analyzed by using CalcuSyn (Biosoft) to calculate the combination index, which was determined for the fraction of cells affected after the addition of fixed ratios of cisplatin and the USP1–UAF1 inhibitor.
Cell cycle analysis
H596 cells were synchronized in S phase with thymidine and treated with either DMSO or 20 μM ML323 for 3 h, which was followed by treatment with 5 μM cisplatin or an equal volume of saline for an additional 3 h. Cells were released from cisplatin by adding fresh medium containing 20 μM ML323 or DMSO. After 24 h of incubation, cells were pulsed with 10 μM BrdU for 1 h. The BrdU was removed, and cells were placed in the fresh medium containing ML323 or DMSO for 18 h. Cells were harvested, fixed in 70% ethanol and blocked in PBS containing 0.1% BSA and 0.05% Tween 20. Cells were then stained with mouse anti-BrdU antibody (Santa Cruz) and then with rabbit anti-mouse FITC-conjugated secondary antibody (Santa Cruz). Cells were then counterstained with propidium iodide (Invitrogen) in PBS containing RNase A (Sigma-Aldrich). Cells were acquired on the Accuri C6 flow cytometer system. The data were analyzed by FlowJo software (Treestar, Ashland, OR).
SCE assay
U2OS cells (1–5 × 105) were seeded 24 h before treatment. In the treatment with ML323 alone, cells were incubated in medium containing 30 μM of ML323 for 7 h and then switched to fresh medium containing 10 μM of BrdU. In the ML323 and cisplatin combination treatment group, cells were first pretreated with 30 μM ML323 for 6 h, and then 15 μM cisplatin was added to the culture, which was further incubated for 1 h before BrdU supplementation. In the treatment with cisplatin alone, cells were treated with 15 μM cisplatin for 1 h before BrdU addition. After 42 h of BrdU incubation, 0.1 μg ml−1 colcemid was added, and cells were incubated for an additional 6 h. Cells were harvested and treated with 75 mM KCl for 15 min and subsequently fixed with ice-cold methanol and acetic acid (3:1) for 30 min. Cells were dropped onto glass slides, air dried and incubated at 55 °C for 1 h. Dried slides were incubated with 10 μg ml−1 Hoechst 33258 for 20 min, which was followed by rinsing with MacIlvaine solution (164 mM Na2HPO4 (pH 7.0) and 16 mM citric acid). Slides were irradiated with UV light at 365 nm for 10 min and incubated in 2× SSC (0.3 M NaCl, 0.03 M Sodium Citrate) solution at 62 °C for 1 h before being stained with 0.3% Leishman’s stain solution and mounted for subsequent microscopy analysis. At least 40 metaphase chromosome spreads were analyzed for each treatment group.
Immunofluorescence microscopy
U2OS cells were seeded in eight-well chamber coverslips at a density of 1 × 104 cells per well 24 h before treatment. In the ML323-alone treatment group, cells were incubated in a medium containing 30 μM of ML323 for 22 h. In the combination treatment with ML323 and cisplatin, cells were first pretreated with 30 μM ML323 for 6 h, and 5 μM cisplatin was added to the culture and incubated for an additional 16 h. In the treatment with cisplatin alone, cells were incubated in medium containing 5 μM of cisplatin for 16 h. Cells were fixed with 4% paraformaldehyde in PBS for 10 min, which was followed by permeabilization with 0.3% Triton X-100 and PBS for 10 min. Cells were then subjected to blocking with 10% goat serum in PBS, which was followed by incubation with monoclonal FANCD2 antibody (Santa Cruz) (dilution of 1: 400) in 1% BSA and PBS for 16 h. Cells were subsequently washed three times in PBS and incubated with Alexa Fluor 488– conjugated anti-mouse antibody (Invitrogen) for 1 h. Images were captured on a Zeiss confocal microscope, and FANCD2 foci were analyzed using ImageJ software (NIH, Bethesda, MD).
HR assay
DR-U2OS cells were seeded in 60-mm dishes at 50–60% confluence overnight and then treated with 30 μM ML323 or an equal volume of DMSO. After 6 h of incubation, the culture medium was refreshed, and the cells were incubated with the transfection reagent FuGENE 6 (Promega) alone, the control vector (pCAGGS) or the I-SceI expression plasmid (pCBASce) for 24 h. ML323 (30 μM) or DMSO was then added into the dishes again. After incubation with ML323 for an additional 48 h, cells were trypsinized and analyzed by the Accuri C6 flow cytometer (BD Biosciences, San Jose, CA). Data were analyzed by FlowJo software (Treestar, Ashland, OR).
Strand-specific PCR-based competitive replication and adduct bypass assay
Strand-specific PCR-based competitive replication and adduct bypass (SSPCR-CRAB) assay was carried out as previously described51. All unmodified oligodeoxyribonucleotides (ODNs), chemicals, [γ-32P]ATP and shrimp alkaline phosphatase were purchased from Integrated DNA Technologies, Sigma-Aldrich, Perkin Elmer and USB Corporation, respectively. All other enzymes, unless otherwise specified, were purchased from New England BioLabs. We first constructed a parent vector for CPD by modifying the sequence of the original pTGFP-Hha10 plasmid, which contains an SV40 origin and is able to replicate in the SV40-transformed mammalian cells52,53. To this end, a 50-mer ODN with the sequence 5′AATTCGCAGCGAGTCATCCATGGCTTGGTATGAAGGAGTCGATGCATCCG-3′ was annealed with its complementary strand and ligated to an NheI-EcoRI restriction fragment from the pTGFP-Hha10 plasmid. We next constructed the CPD-bearing double-stranded shuttle vector using a previously described method (Supplementary Fig. 10a)52,53. Briefly, we nicked the parent vector with Nt.BstNBI to produce a gapped vector by removing a 25-mer single-stranded ODN, which was followed by filling the gap with a 12-mer CPD-bearing ODN (5′-ATGGCXXGCTAT-3′, XX = CPD), a 13-mer unmodified ODN (5′GAAGGAGTCGATG-3′) and T4-DNA ligase. The ligation mixture was incubated with ethidium bromide, and the resulting supercoiled lesion-bearing plasmid was isolated by agarose gel electrophoresis. Using the same method, we prepared the lesion-free control and competitor vectors for CPD, with the 12-mer CPD-containing ODN being replaced with a 12-mer ODN (5′-ATGGCTTGCTAT-3′) and a 15-mer ODN (5′-ATGGCTTGCAGCTAT-3′), respectively.
The lesion-bearing or the corresponding non-lesion control plasmids were premixed with the competitor genome for in vivo replication assay, with the molar ratios of competitor vector to control or lesion-bearing genome being 1:1 and 1:10, respectively. The HEK293T cells were seeded in 24-well plates and cultured overnight. The cells were subsequently treated with or without 50 μM ML323 (or DMSO as a control) for 6 h, after which they were transfected with 300 ng plasmid using Lipofectamine 2000 (Invitrogen) following the manufacturer’s instructions. The cells were harvested at 24 h after transfection, and the progenies of the plasmid were isolated using the Qiagen Spin kit (Qiagen) with minor changes54. The residual unreplicated plasmid was further removed by DpnI digestion, which was followed by digesting the resulting linear DNA with exonuclease III as described elsewhere55-57.
The progeny genomes arising from cellular replication were amplified in a PCR reaction (GoTaq Green Master Mix, Promega) containing a pair of primers whose products cover the initial lesion site and span eight DpnI recognition sites. The primers were 5′CTAGCGGATGCATCGACTCCTTCACAG-3′ and 5′GGCTCCCTTTAGGGTTCCGATTTAGTG-3′, and the PCR amplification started at 95 °C for 2 min followed by 35 cycles at 95 °C for 30 s, 65 °C for 30 s and 72 °C for 1.5 min and a final 5-min extension at 72 °C. The PCR products were purified by the QIAquick PCR Purification kit (Qiagen) and stored at –20 °C until use. For PAGE analysis, a portion of the PCR fragments was treated with 5 U NcoI and 1 U shrimp alkaline phosphatase at 37 °C in 10 μl NEB buffer 3 (New England Biolabs) for 1 h, which was followed by heating at 80 °C for 20 min to deactivate the shrimp alkaline phosphatase. The mixture was then treated in a 15 μl NEB buffer 3 with 5 mM DTT, ATP (50 pmol cold premixed with 1.66 pmol [γ- 32P]ATP) and 5 U T4 polynucleotide kinase. The reaction was continued at 37 °C for 1 h, which was followed by heating at 65 °C for 20 min to deactivate the T4 polynucleotide kinase. 5 U SfaNI was then added to the reaction mixture, and the solution was incubated at 37 °C for 1 h, which was followed by quenching with 15 μl formamide gel loading buffer containing xylene cyanol FF and bromophenol blue dyes. The mixture was loaded onto a 30% native polyacrylamide gel (acrylamide:bis-acrylamide = 19: 1), and the products were quantified by phosphorimager analysis. The bypass efficiency was calculated using the following equation58,59.
Summary of statistical analyses
The statistical analyses were carried out using GraphPad Prism. The data are presented as the mean ± s.d. Statistical significance was calculated using Student’s t-test (unpaired, two tailed). NS, P > 0.05; *P < 0.05, **P < 0.01, ***P < 0.001. The IC50 and EC50 values were calculated using GraphPad Prism and a sigmoidal dose-response equation with a variable slope.
Supplementary Material
ACKNOWLEDGMENTS
We thank M. Cordeiro-Stone (University of North Carolina at Chapel Hill) for xeroderma pigmentosum variant (XPV) human fibroblasts GM02359-hTERT (XP115LO) and Polη-complemented GM02359-hTERT (XPV + Polη) and M. Jasin (Memorial Sloan-Kettering Cancer Center) for DR-GFP U2OS cells. We also thank C. Arrowsmith (University of Toronto and Ontario Cancer Institute) for the USP21 plasmid; A. Tencer for assistance with protein purification; S. Michael and R. Jones for automation support; P. Shinn and D. van Leer for assistance with compound management; and W. Leister, H. Baker, C. Leclair and E. Fernandez for analytical chemistry and compound purification support. This work was supported by US National Institutes of Health (NIH) grant R03DA030552 and in part by NIH grant R01GM097468 to Z.Z. C.A.O. was supported by NIH training grant T32GM008550. Work in the laboratory of Y.W. was supported by NIH grant R01DK082779. T.S.D., A.S.R., D.K.L., A.S., A.J. and D.J.M. were supported by the intramural research program of the National Center for Advancing Translational Sciences and the Molecular Libraries Initiative of the National Institutes of Health Roadmap for Medical Research (U54MH084681).
Footnotes
Author Contributions Q.L., T.S.D. and J.C. designed, performed and analyzed in vitro biochemical assays for DUBs and other proteases. T.S.D. and A.S. designed and performed the HTS assay. Q.L. and P.Z. designed, performed and analyzed cell culture experiments. A.S.R., D.K.L. and D.J.M. performed chemical synthesis and oversaw the chemistry efforts. M.A.V. performed protein purification and native gel analysis. C.Y. and B.Y. performed the cellular DNA replication assay under the guidance of Y.W. Q.Z. and H.X. performed and analyzed HDX experiments. C.A.O. generated substrates for the DUB assay. H.S. and A.J. analyzed HTS data and structure-activity relationship results. Z.Z. and D.J.M. designed the research plan and wrote the manuscript with input from all authors.
Competing financial interests The authors declare competing financial interests: details accompany the online version of the paper.
Supplementary information, chemical compound information and chemical probe information is available in the online version of the paper. Reprints and permissions information is available online at http://www.nature.com/reprints/index.html. Correspondence and requests for materials should be addressed to Z.Z. or D.J.M.
REFERENCE
- 1.Chen ZJ, Sun LJ. Nonproteolytic functions of ubiquitin in cell signaling. Mol Cell. 2009;33:275–286. doi: 10.1016/j.molcel.2009.01.014. [DOI] [PubMed] [Google Scholar]
- 2.Amerik AY, Hochstrasser M. Mechanism and function of deubiquitinating enzymes. Biochim Biophys Acta. 2004;1695:189–207. doi: 10.1016/j.bbamcr.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 3.Nijman SM, et al. A genomic and functional inventory of deubiquitinating enzymes. Cell. 2005;123:773–786. doi: 10.1016/j.cell.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 4.Fraile JM, Quesada V, Rodriguez D, Freije JM, Lopez-Otin C. Deubiquitinases in cancer: new functions and therapeutic options. Oncogene. 2012;31:2373–2388. doi: 10.1038/onc.2011.443. [DOI] [PubMed] [Google Scholar]
- 5.Wang W. Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins. Nat Rev Genet. 2007;8:735–748. doi: 10.1038/nrg2159. [DOI] [PubMed] [Google Scholar]
- 6.Kim H, D’Andrea AD. Regulation of DNA cross-link repair by the Fanconi anemia/BRCA pathway. Genes Dev. 2012;26:1393–1408. doi: 10.1101/gad.195248.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Crossan GP, Patel KJ. The Fanconi anaemia pathway orchestrates incisions at sites of crosslinked DNA. J Pathol. 2012;226:326–337. doi: 10.1002/path.3002. [DOI] [PubMed] [Google Scholar]
- 8.Hoege C, Pfander B, Moldovan GL, Pyrowolakis G, Jentsch S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature. 2002;419:135–141. doi: 10.1038/nature00991. [DOI] [PubMed] [Google Scholar]
- 9.Stelter P, Ulrich HD. Control of spontaneous and damage-induced mutagenesis by SUMO and ubiquitin conjugation. Nature. 2003;425:188–191. doi: 10.1038/nature01965. [DOI] [PubMed] [Google Scholar]
- 10.Chen J, Bozza W, Zhuang Z. Ubiquitination of PCNA and its essential role in eukaryotic translesion synthesis. Cell Biochem Biophys. 2011;60:47–60. doi: 10.1007/s12013-011-9187-3. [DOI] [PubMed] [Google Scholar]
- 11.Chang DJ, Cimprich KA. DNA damage tolerance: when it’s OK to make mistakes. Nat Chem Biol. 2009;5:82–90. doi: 10.1038/nchembio.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haracska L, Torres-Ramos CA, Johnson RE, Prakash S, Prakash L. Opposing effects of ubiquitin conjugation and SUMO modification of PCNA on replicational bypass of DNA lesions in Saccharomyces cerevisiae. Mol Cell Biol. 2004;24:4267–4274. doi: 10.1128/MCB.24.10.4267-4274.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang TT, et al. Regulation of monoubiquitinated PCNA by DUB autocleavage. Nat Cell Biol. 2006;8:339–347. doi: 10.1038/ncb1378. [DOI] [PubMed] [Google Scholar]
- 14.Zhuang Z, et al. Regulation of polymerase exchange between Poleta and Poldelta by monoubiquitination of PCNA and the movement of DNA polymerase holoenzyme. Proc Natl Acad Sci U S A. 2008;105:5361–5366. doi: 10.1073/pnas.0801310105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee KY, et al. Human ELG1 regulates the level of ubiquitinated proliferating cell nuclear antigen (PCNA) through Its interactions with PCNA and USP1. J Biol Chem. 2010;285:10362–10369. doi: 10.1074/jbc.M109.092544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim JM, et al. Inactivation of murine Usp1 results in genomic instability and a Fanconi anemia phenotype. Dev Cell. 2009;16:314–320. doi: 10.1016/j.devcel.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guervilly JH, Renaud E, Takata M, Rosselli F. USP1 deubiquitinase maintains phosphorylated CHK1 by limiting its DDB1-dependent degradation. Hum Mol Genet. 2011;20:2171–2181. doi: 10.1093/hmg/ddr103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bell DW, et al. Predisposition to cancer caused by genetic and functional defects of mammalian Atad5. PLoS Genet. 2011;7:e1002245. doi: 10.1371/journal.pgen.1002245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sowa ME, Bennett EJ, Gygi SP, Harper JW. Defining the human deubiquitinating enzyme interaction landscape. Cell. 2009;138:389–403. doi: 10.1016/j.cell.2009.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oestergaard VH, et al. Deubiquitination of FANCD2 is required for DNA crosslink repair. Mol Cell. 2007;28:798–809. doi: 10.1016/j.molcel.2007.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen J, et al. Selective and cell-active inhibitors of the USP1/ UAF1 deubiquitinase complex reverse cisplatin resistance in non-small cell lung cancer cells. Chem Biol. 2011;18:1390–1400. doi: 10.1016/j.chembiol.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Post RM, Jimerson DC, Bunney WE, Jr., Goodwin FK. Dopamine and mania: behavioral and biochemical effects of the dopamine receptor blocker pimozide. Psychopharmacology (Berl) 1980;67:297–305. doi: 10.1007/BF00431272. [DOI] [PubMed] [Google Scholar]
- 23.Brown PJ, et al. Identification of a subtype selective human PPARalpha agonist through parallel-array synthesis. Bioorg Med Chem Lett. 2001;11:1225–1227. doi: 10.1016/s0960-894x(01)00188-3. [DOI] [PubMed] [Google Scholar]
- 24.Prage EB, et al. Location of inhibitor binding sites in the human inducible prostaglandin E synthase, MPGES1. Biochemistry. 2011;50:7684–7693. doi: 10.1021/bi2010448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiao H, et al. Insights into the mechanism of microtubule stabilization by Taxol. Proc Natl Acad Sci U S A. 2006;103:10166–10173. doi: 10.1073/pnas.0603704103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Khrapunovich-Baine M, et al. Distinct pose of discodermolide in taxol binding pocket drives a complementary mode of microtubule stabilization. Biochemistry. 2009;48:11664–11677. doi: 10.1021/bi901351q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Villamil MA, et al. Serine phosphorylation is critical for the activation of ubiquitin-specific protease 1 and its interaction with WD40-repeat protein UAF1. Biochemistry. 2012;51:9112–9123. doi: 10.1021/bi300845s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Borodovsky A, et al. Chemistry-based functional proteomics reveals novel members of the deubiquitinating enzyme family. Chem Biol. 2002;9:1149–1159. doi: 10.1016/s1074-5521(02)00248-x. [DOI] [PubMed] [Google Scholar]
- 29.Altun M, et al. Activity-based chemical proteomics accelerates inhibitor development for deubiquitylating enzymes. Chem Biol. 2011;18:1401–1412. doi: 10.1016/j.chembiol.2011.08.018. [DOI] [PubMed] [Google Scholar]
- 30.Koberle B, Tomicic MT, Usanova S, Kaina B. Cisplatin resistance: preclinical findings and clinical implications. Biochim Biophys Acta. 2010;1806:172–182. doi: 10.1016/j.bbcan.2010.07.004. [DOI] [PubMed] [Google Scholar]
- 31.Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70:440–446. doi: 10.1158/0008-5472.CAN-09-1947. [DOI] [PubMed] [Google Scholar]
- 32.Albertella MR, Green CM, Lehmann AR, O’Connor MJ. A role for polymerase eta in the cellular tolerance to cisplatin-induced damage. Cancer Res. 2005;65:9799–9806. doi: 10.1158/0008-5472.CAN-05-1095. [DOI] [PubMed] [Google Scholar]
- 33.Johnson RE, Prakash S, Prakash L. Efficient bypass of a thymine-thymine dimer by yeast DNA polymerase, Poleta. Science. 1999;283:1001–1004. doi: 10.1126/science.283.5404.1001. [DOI] [PubMed] [Google Scholar]
- 34.You C, et al. A quantitative assay for assessing the effects of DNA lesions on transcription. Nat Chem Biol. 2012;8:817–822. doi: 10.1038/nchembio.1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim H, Yang K, Dejsuphong D, D’Andrea AD. Regulation of Rev1 by the Fanconi anemia core complex. Nat Struct Mol Biol. 2012;19:164–170. doi: 10.1038/nsmb.2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Renaud E, Rosselli F. FANC pathway promotes UV-induced stalled replication forks recovery by acting both upstream and downstream Poleta and Rev1. PLoS One. 2013;8:e53693. doi: 10.1371/journal.pone.0053693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li X, Heyer WD. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008;18:99–113. doi: 10.1038/cr.2008.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Niedzwiedz W, et al. The Fanconi anaemia gene FANCC promotes homologous recombination and error-prone DNA repair. Mol Cell. 2004;15:607–620. doi: 10.1016/j.molcel.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 39.Xia B, et al. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol Cell. 2006;22:719–729. doi: 10.1016/j.molcel.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 40.Sonoda E, et al. Multiple roles of Rev3, the catalytic subunit of polzeta in maintaining genome stability in vertebrates. Embo J. 2003;22:3188–3197. doi: 10.1093/emboj/cdg308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Takata M, Ishiai M, Kitao H. The Fanconi anemia pathway: insights from somatic cell genetics using DT40 cell line. Mutat Res. 2009;668:92–102. doi: 10.1016/j.mrfmmm.2008.12.012. [DOI] [PubMed] [Google Scholar]
- 42.Lebwohl D, Canetta R. Clinical development of platinum complexes in cancer therapy: an historical perspective and an update. Eur J Cancer. 1998;34:1522–1534. doi: 10.1016/s0959-8049(98)00224-x. [DOI] [PubMed] [Google Scholar]
- 43.Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat Rev Cancer. 2007;7:573–584. doi: 10.1038/nrc2167. [DOI] [PubMed] [Google Scholar]
- 44.Brabec V, Kasparkova J. Molecular aspects of resistance to antitumor platinum drugs. Drug Resist Updat. 2002;5:147–161. doi: 10.1016/s1368-7646(02)00047-x. [DOI] [PubMed] [Google Scholar]
- 45.Park E, et al. FANCD2 activates transcription of TAp63 and suppresses tumorigenesis. Mol Cell. 2013;50:908–918. doi: 10.1016/j.molcel.2013.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Williams SA, et al. USP1 deubiquitinates ID proteins to preserve a mesenchymal stem cell program in osteosarcoma. Cell. 2011;146:918–930. doi: 10.1016/j.cell.2011.07.040. [DOI] [PubMed] [Google Scholar]
- 47.Xu G, Paige JS, Jaffrey SR. Global analysis of lysine ubiquitination by ubiquitin remnant immunoaffinity profiling. Nat Biotechnol. 2010;28:868–873. doi: 10.1038/nbt.1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
Reference
- 48.Burkhart RA, et al. Mitoxantrone targets human ubiquitin-specific peptidase 11 (USP11) and is a potent inhibitor of pancreatic cancer cell survival. Mol Cancer Res. 2013;11:901–911. doi: 10.1158/1541-7786.MCR-12-0699. [DOI] [PubMed] [Google Scholar]
- 49.Hibbert RG, Sixma TK. Intrinsic flexibility of ubiquitin on proliferating cell nuclear antigen (PCNA) in translesion synthesis. J Biol Chem. 2012;287:39216–39223. doi: 10.1074/jbc.M112.389890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Inglese J, et al. Quantitative high-throughput screening: a titration-based approach that efficiently identifies biological activities in large chemical libraries. Proc Natl Acad Sci U S A. 2006;103:11473–11478. doi: 10.1073/pnas.0604348103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.You C, et al. Translesion synthesis of 8,5′-cyclopurine-2′-deoxynucleosides by DNA polymerases eta, iota, and zeta. J Biol Chem. 2013;288:28548–28556. doi: 10.1074/jbc.M113.480459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Baker DJ, et al. Nucleotide excision repair eliminates unique DNA-protein cross-links from mammalian cells. J Biol Chem. 2007;282:22592–22604. doi: 10.1074/jbc.M702856200. [DOI] [PubMed] [Google Scholar]
- 53.Yuan B, et al. The roles of DNA polymerases kappa and iota in the error-free bypass of N2-carboxyalkyl-2′-deoxyguanosine lesions in mammalian cells. J Biol Chem. 2011;286:17503–17511. doi: 10.1074/jbc.M111.232835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mitra D, et al. An ultraviolet-radiation-independent pathway to melanoma carcinogenesis in the red hair/fair skin background. Nature. 2012;491:449–453. doi: 10.1038/nature11624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Burns JA, Dreij K, Cartularo L, Scicchitano DA. O6-methylguanine induces altered proteins at the level of transcription in human cells. Nucleic Acids Res. 2010;38:8178–8187. doi: 10.1093/nar/gkq706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sanchez JA, Marek D, Wangh LJ. The efficiency and timing of plasmid DNA replication in Xenopus eggs: correlations to the extent of prior chromatin assembly. J Cell Sci. 1992;103:907–918. doi: 10.1242/jcs.103.4.907. [DOI] [PubMed] [Google Scholar]
- 57.Taylor ER, Morgan IM. A novel technique with enhanced detection and quantitation of HPV-16 E1- and E2-mediated DNA replication. Virology. 2003;315:103–109. doi: 10.1016/s0042-6822(03)00588-9. [DOI] [PubMed] [Google Scholar]
- 58.Delaney JC, Essigmann JM. Mutagenesis, genotoxicity, and repair of 1-methyladenine, 3-alkylcytosines, 1-methylguanine, and 3-methylthymine in alkB Escherichia coli. Proc Natl Acad Sci U S A. 2004;101:14051–14056. doi: 10.1073/pnas.0403489101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Delaney JC, Essigmann JM. Assays for determining lesion bypass efficiency and mutagenicity of site-specific DNA lesions in vivo. Methods Enzymol. 2006;408:1–15. doi: 10.1016/S0076-6879(06)08001-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.