

Abstract
DNA damage can cause (and result from) oxidative stress and mitochondrial impairment, both of which are implicated in the pathogenesis of Parkinson's disease (PD). We therefore examined the role of mitochondrial DNA (mtDNA) damage in human postmortem brain tissue and in in vivo and in vitro models of PD, using a newly adapted histochemical assay for abasic sites and a quantitative polymerase chain reaction (QPCR)-based assay. We identified the molecular identity of mtDNA damage to be apurinic/apyrimidinic (abasic) sites in substantia nigra dopamine neurons, but not in cortical neurons from postmortem PD specimens. To model the systemic mitochondrial impairment of PD, rats were exposed to the pesticide rotenone. After rotenone treatment that does not cause neurodegeneration, abasic sites were visualized in nigral neurons, but not in cortex. Using a QPCR-based assay, a single rotenone dose induced mtDNA damage in midbrain neurons, but not in cortical neurons; similar results were obtained in vitro in cultured neurons. Importantly, these results indicate that mtDNA damage is detectable prior to any signs of degeneration – and is produced selectively in midbrain neurons under conditions of mitochondrial impairment. The selective vulnerability of midbrain neurons to mtDNA damage was not due to differential effects of rotenone on complex I since rotenone suppressed respiration equally in midbrain and cortical neurons. However, in response to complex I inhibition, midbrain neurons produced more mitochondrial H2O2 than cortical neurons. We report selective mtDNA damage as a molecular marker of vulnerable nigral neurons in PD and suggest that this may result from intrinsic differences in how these neurons respond to complex I defects. Further, the persistence of abasic sites suggests an ineffective base excision repair response in PD.
Introduction
Parkinson's disease (PD) is the most common neurodegenerative movement disorder. A central pathological hallmark of PD is the loss of dopamine neurons in the substantia nigra pars compacta. These dopaminergic neurons are required for proper motor function, and their loss is associated with tremor, rigidity, bradykinesia and postural instability. The initial underlying mechanisms that trigger neurodegeneration in PD are complex and not completely understood. To date, treatments are only symptomatic; they do not alter the inexorable progression of the disease. Even with expert treatment, PD patients typically deteriorate over time and endure considerable motor and cognitive disability in the years after diagnosis.
High levels of reactive oxygen species (ROS) are an intrinsic property of the vulnerable subpopulation of ventral midbrain (VMB) dopaminergic neurons (1). Despite strong evidence that oxidative damage to proteins and lipids is a contributing factor in PD pathogenesis (2-5), very little is known about DNA damage in PD (6). DNA damage is defined as a modification that either alters its coding properties or interferes with normal cell metabolism (7,8), and many different forms of DNA damage can be generated by both normal cellular functions and exogenous stressors (9). DNA damage is distinct from DNA mutations, which are changes in the base sequence of the DNA. Higher levels of mitochondrial mutations have been found in dopaminergic neurons in the substantia nigra of PD patients relative to healthy controls and this has raised speculation for a causal relationship between mutations and neurodegeneration (10-12). However, methodological questions remain about the analysis of variations in mitochondrial DNA sequence and its functional significance (13). The specific role of mitochondrial DNA (mtDNA) damage in PD is unclear (14-16).
PD is widely accepted as a multifactorial disease, with both genetic and environmental contributions. The majority of cases are ‘idiopathic’, with ∼10% of PD cases having a genetic cause. Of these, mutations in LRRK2 are the most common. We recently reported increased mtDNA damage in induced pluripotent stem cell (iPSC)-derived neural cells from patients carrying PD-associated LRRK2 mutations and zinc finger nuclease-mediated gene editing of the LRRK2 G2019S mutation reversed the mtDNA damage phenotype (17). Whether mtDNA damage is found in idiopathic forms of PD is unknown.
One of the primary classes of environmental agents associated with PD is pesticides (18). One such pesticide that inhibits complex I of the mitochondria is rotenone, which is a prototypical example of how an exogenous toxin can mimic clinical and pathological features of PD in an animal model (19). Recently, in a rigorous, case-control study, rotenone was revealed as a bonafide risk factor for PD (20). We found with rotenone treatment, both in vitro and in vivo, that mitochondrial dysfunction was associated with robust mtDNA damage selectively in the midbrain – but not in cortex – and that this occurred well before there was any frank degeneration of the nigrostriatal system. In addition, we identified the molecular identity of the mtDNA damage to be apurinic/apyrimidinic sites (in which a segment of DNA has lost either a purine nor a pyrimidine base), which persist specifically in dopaminergic neurons in rotenone models and in the human PD brain.
Materials and Methods
PD patient samples
Paraffin-embedded midbrain and cortex sections from 6 PD and 5 control subjects, 50-80 yrs of age, were obtained from the University of Pittsburgh Brain Bank for detection of abasic sites by the histochemical approach. All banked specimens have undergone standardized premortem neurological and postmortem neuropathological assessments, including tau and α-synuclein immunohistochemistry. One PD case was excluded due to the presence of an acute midbrain infarct. Braak AD and PD staging and McKeith scores were performed according to (21,22). Midbrain and cortex sections from 5 PD and 5 control subjects, matched for age and postmortem intervals, were used for analysis (Supplemental Table I).
Histochemical assay for detection of abasic sites in situ in tissue sections
For detection of abasic sites in human midbrain and cortex samples, sections were deparaffinized and then endogenous biotin was blocked using the Avidin/Biotin blocking kit according to manufacturer's instructions (Vector Laboratories). Heat mediated antigen retrieval was performed for 20 min at 100°C (10 mM sodium citrate, 0.05% tween-20, pH 6.0). Slides were allowed to cool for five min at room temperature (RT) and then washed in H2O for 3 min. Sections were incubated with 10 mM aldehyde reactive probe (ARP) (Invitrogen) for 1 h at 37°C, followed by streptavidin-conjugated Alexa 488 (Invitrogen) for 1 h at 37°C. ARP reacts with a ring-opened sugar moiety resulting from the loss of a base in the DNA (abasic sites). Sections were blocked for 1 h in 10% normal donkey serum containing 0.3% Triton X-100. Midbrain sections were incubated in primary antibodies for TH and TOM20 overnight without shaking at 4°C (1:1,000; mouse anti-tyrosine hydroxylase; Millipore MAB318, 1:500; rabbit anti-TOM20; Santa Cruz sc-11415). Cortex sections were incubated with primary antibodies for MAP2 and TOM20 (1:1000; mouse anti-MAP2; Millipore MAB378, 1:500; rabbit anti-TOM20; Santa Cruz sc-11415). Primary antibodies were re-applied for 2 h at RT without agitation. Sections were then washed three times in PBS for 10 min, incubated for 1 h in donkey anti-mouse Cy3 secondary antibody (1:500; Jackson ImmunoResearch) and donkey anti-rabbit 647 secondary antibody (1:500, Jackson ImmunoResearch). Sections were then washed three times in PBS for 10 min and mounted onto slides using gelvatol mounting media. To quantify midbrain and cortex sections for neurons that were ARP positive, in sections from PD and control subjects either the total number of TH or MAP2 positive neurons were scored for ARP signal. The number of neurons that had detectable ARP signal was divided by the total number of neurons counted for each case to determine the percentage that were overall ARP positive. Staining and quantification were performed blinded and then results were categorized by disease status after unblinding.
For detection of abasic sites in the rat substantia nigra, sections were washed six times in 1X PBS for 10 min, incubated in either 50 mM Tris-HCL, DNase I (Invitrogen), or 5 μg/uL RNAse A (Fischer Scientific) for 1 h at 37°C. Sections were washed three times in 1X PBS for 5 min, and then endogenous biotin blocked using the Avidin/Biotin blocking kit according to manufacturer's instructions (Vector Laboratories). Sections were then incubated with 10 mM ARP (Invitrogen) for 1 h at 37°C, followed by streptavidin-conjugated Alexa 488 (Invitrogen) for 1 h at 37°C. Sections were blocked for 1 h in 10% normal donkey serum containing 0.3% Triton X-100, then incubated in primary antibody for TH and TOM20 for 48 h at 4°C (1:2,000; sheep anti-tyrosine hydroxylase; Millipore AB1542, 1:1000; rabbit anti-TOM20; Santa Cruz sc-11415). Sections were washed three times in PBS for 10 min, incubated for 2 h in donkey anti-sheep Cy3 secondary antibody (1:500; Jackson ImmunoResearch) and donkey anti-rabbit 647 secondary antibody (1:500, Jackson ImmunoResearch). Sections were then washed three times in PBS for 10 min and mounted onto slides using gelvatol mounting media. Pretreatment of tissue sections with DNase I abolished the signal, but RNase A had no effect, confirming the ARP signal derives from DNA (Supplemental Fig. 2).
ELISA quantitative measurements of abasic sites in total DNA
DNA was isolated as described below (see DNA isolation and quantification section) from ventral midbrain (n = 6 for vehicle and n = 7 for rotenone injected). Quantitative measurements of abasic sites were performed using the colorimetric assay according to manufacturer's instructions (Dojindo Molecular Technologies). ARP assays from DNA from each animal were performed in technical triplicate and the mean calculated. Data is expressed as the number of abasic sites per 106 nucleotides, which was calculated based on the linear calibration curve generated for each experiment using the ARP-DNA standard solutions.
In vivo rotenone treatment and tissue isolation
All experiments utilizing animals were approved by the Institutional Animal Care and Use Committee of the University of Pittsburgh. Male Lewis rats (7-9 months old, Hilltop Lab Animals, Inc., Scottsdale, PA, USA) were injected intraperitoneally with vehicle or 3.0 mg/kg/day rotenone (Sigma-Aldrich) either a single time or for five daily injections and then euthanized as described (23). Three separate cohorts of animals (for a total of 9 vehicle- and 9 rotenone-treated), were analyzed for each in vivo experiment. Brains were dissected to obtain the ventral midbrain (VMB) and frontal cortex for DNA isolation. Tissues were resuspended in Isolation Buffer I (Sucrose 80 mM, EDTA 1 mM, HEPES-K-Salt 10 mM, pH 7.4) and then incubated on ice for 10 min. Equal volume of Isolation Buffer II (Sucrose 300 mM, EDTA 1 mM, HEPES-K-Salt 10 mM, pH 7.4) was added and tissue was homogenized using a glass pestle. The homogenate was centrifuged at 10,000 × g for 20 min at 4 °C. The pellet was either stored at -20 °C or immediately processed further as described below.
Rat primary neuronal enriched cultures
Primary neuronal cultures were prepared from rat as described (24). Briefly, the primary ventral midbrain (VMB) and cortical tissues from embryonic day 17 Sprague-Dawley rats were dissected in L-15 medium: Leibovitz's 1X medium (Invitrogen), and penicillin-streptomycin (200 units / 200 μg / mL respectively, Cellgro). The tissues were then placed in 1X trypsin– ethylenediaminetetraacetic acid (EDTA) at 37°C for 20 minutes. The cells were mechanically dissociated with a Pasteur pipette in complete medium that includes neurobasal medium (Invitrogen), B-27 supplement 1X (Invitrogen), Glutamax 1X (Invitrogen), penicillin-streptomycin (50 units / 50 μg/mL respectively, Invitrogen), and Albumax 0.5 mg / mL (Invitrogen). Cell viability was evaluated using trypan blue dye exclusion and a hemocytometer. Cells were seeded to 6-well (1.0 × 106/well) culture plate pre-coated with poly-D-lysine (PDL, 0.1 mg/mL, Sigma). Maintenance of the cultures took place at 37°C with 5% CO2/95% air. One half of the medium was replenished twice a week. At 7 days after plating, cells were treated with vehicle (DMSO) or rotenone (10 nM) for 24 h.
DNA isolation and quantification
DNA isolation was performed using a high molecular weight genomic DNA purification kit according to the manufacturer's protocol (QIAGEN Genomic tip). DNA was quantified using the Picogreen dsDNA quantification assay as suggested by the manufacturer (Molecular Probes). The fluorescence from the Picogreen was measured with a 485 nm excitation filter and a 530 nm emission filter using a microplate reader (SpectraMax Gemini EM). Lambda DNA was used to construct a standard curve in order to determine the concentration of unknown samples. Quality of the DNA prior to QPCR analysis was verified by running the DNA on a 0.6% ethidium bromide-stained agarose gel. Only DNA of intact high molecular weight, which showed negligible evidence of degradation, was used in the DNA damage assays. DNA samples were aliquoted and stored at -20°C. Samples were thawed only once prior to downstream assays.
Quantifying mtDNA damage using the quantitative polymerase chain reaction (QPCR)
To measure levels of mtDNA damage, we adapted a quantitative polymerase chain reaction (QPCR)-based assay to various neuronal systems (25). This method is based on the principle that various forms of DNA damage have the propensity to slow down or block DNA polymerase progression (26). Thus, if equal amounts of mtDNA from experimental and control specimens are amplified under identical conditions, the mtDNA sample with the least mtDNA damage will produce the greatest amount of PCR product (26). The PCR product used here amplifies almost the entire mitochondrial genome, excluding the D-loop to avoid bias from this mutation hotspot (27). In addition to mtDNA damage, the QPCR-based assay measures certain mtDNA mutations, such as the common 4.9kb and other large deletions (based on a size change of the product), as well as smaller deletions or rearrangements that compromise primer complementarity. In contrast, typical sequencing strategies only detect mutations and not mtDNA damage.
To confirm that QPCR assays were performed in the linear range, optimal number of cycles for each template was conducted to show that 50% reduction in the amount of template resulted in about 50% reduction in amplification. QPCR products that demonstrated 40-60% in the amplification of the target sequence when using 50% of the original template were considered acceptable. The PCR amplification profile for a 13.4 kb rat mitochondrial fragment was as follows: hot start for 10 min at 75°C when the DNA polymerase was added, an initial denaturation step for 1 min at 94°C, followed by 18 cycles of denaturation for 15 s at 94°C, and then annealing/extension at 66°C for 14 min. To complete the profile a final extension for 10 min at 72°C was performed. Primers 10633 and 13559 were used for the amplification of the rat mitochondrial fragment and the primer nucleotide sequences were described previously (25). The final concentration of magnesium in the PCR was 1.1 mM. All QPCR-based experiments were performed in technical triplicate for each biological replicate.
To ensure quality and specificity, all QPCR products were resolved on a 0.6% agarose gel and UV light used to visualize ethidium bromide-stained gels. Relative fluorescence of QPCR products was quantified using Picogreen. Assuming a random distribution, the Poisson equation was used to calculate the number of DNA lesions. Based on this equation, the amplification is directly proportional to the fraction of undamaged DNA templates. As such, the average lesion frequency per strand is calculated as –lnAD/AO, where AD is the amplification of the damaged or experimental template, while AO is the amplification of the undamaged or control template. Therefore, the results are shown as the number of lesions per strand normalized to 10kb. DNA damage can then be expressed mathematically as the number of DNA lesions per kilobase (28). By normalizing this way, direct comparisons of different tissues or different chemical exposures are facilitated.
QPCR of a small mtDNA fragment
To ensure that the amplification of the large mtDNA fragment was not due to possible changes in mtDNA steady state levels, we amplified a rat (235-bp) small mtDNA fragment, since the amplification of this small fragment should be independent of damage. The profile for this PCR amplification was as follows: hot start for 10 min at 75°C when the DNA polymerase was added, an initial denaturation step for 1 min at 94°C, followed by 18 cycles of denaturation for 1 min at 94°C and then annealing at 60°C for 45 seconds and extension at 72°C for 45 seconds. To complete the profile, a final extension for 10 min at 72°C was performed. The primer nucleotide sequences were 14678 and 14885 (26). The PCR product was resolved on a 1.5 % agarose gel and UV light used to visualize ethidium bromide-stained gels. Relative fluorescence of PCR products were quantified using Picogreen.
Measurement of cellular oxygen consumption rate (OCR)
Cellular oxygen consumption rate (OCR) was measured using an extracellular flux analyzer (Seahorse Bioscience XF24) as described (29) with minor modifications. Intact primary VMB and cortical neurons were grown in XF24 plates seeded at 80,000 cells/well for 1 week in growth medium. Neuronal cultures were then treated with either vehicle (DMSO) or rotenone (10 nM) for 24 hours. Plates were then incubated in Calibrant medium (Seahorse Bioscience) for at least 30 min. Media containing final concentrations of 10 μM oligomycin, 300 nM carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP) and 1 μM rotenone were pre-loaded into the drug delivery system. Once the basal OCR was measured, the compounds were added sequentially and the effects on OCR measured every 8 minutes.
Mitochondrial specific hydrogen peroxide production
Primary neurons were plated at 1.0 × 106 on 35 mm glass bottom culture PDL coated dishes (MatTek). On DIV 7, neurons were transfected with CellLight mitochondria GFP (Invitrogen) for 16-18 h. On DIV 8, neurons were incubated with 5 μM MitoPY1 (30-32) for 40 min in 1X HBSS medium (136.9 mM NaCl, 5 mM KCl, 20 nM HEPES, 5.5mM glucose, 0.59 mM KH2PO4, 0.56 mM Na2HPO4, 0.9 mM MgSO4 and 1.4 mM CaCl2) at 37 °C. Either vehicle (DMSO) or 10 nM rotenone was added at time zero; the signal was allowed to stabilize for ∼100,000 ms, after which, the data were normalized to this baseline. Regions of interest (ROI) with only CellLight positive mitochondria were analyzed. Fluorescence intensities (excitation 555nm and emission 625nm) and images were captured with a 60X lens using an Olympus Fluoview FV1000 inverted confocal microscope.
Statistical analysis
The statistical software package GraphPad Prism was used for statistical computation. Data were analyzed by Student's t-tests and p < 0.05 was deemed significant. For all graphs, the error bars represent mean ± standard error of the mean (SEM). For ELISA quantitative abasic site measurements the 95% confidence interval was used to assess statistical significance.
Results
MtDNA abasic sites accumulate selectively in nigral dopamine neurons in PD
Currently there are no tools with which to examine specific kinds of mtDNA damage in an intact cell-specific manner (33). Therefore, we developed a new histochemical approach to detect apurinic/apyrimidinic (abasic or AP) sites in situ in tissue sections. Abasic sites are a type of DNA damage, often arising from oxidative stress, in which there is loss of a purine or a pyrimidine base. Given that polymorphisms in DNA repair genes, including a protein involved in the repair of abasic sites (22), were recently reported to be risk factors for developing PD, we investigated the role of abasic sites in human PD brain postmortem tissue.
Coded midbrain and cortex brain sections from control and PD cases were obtained for abasic site detection, and only after analysis were samples unblinded (Supplemental Table 1). Aldehyde reactive probe (ARP) reacts with a ‘ring-opened’ sugar moiety that results from the loss of a base in DNA (i.e., abasic sites). A robust ARP fluorescence signal was detected in nigral tyrosine hydroxylase (TH)-positive dopamine neurons in PD cases (Fig. 1a), which co-localized with TOM20 staining (Fig. 1a) and not DAPI (not shown), indicating that the accumulated abasic sites were in mtDNA, not nuclear DNA. As noted below, control experiments in rat tissue showed that the ARP signal was abolished by pretreatment of tissue with DNase I. In PD cases, the majority of remaining nigral dopamine neurons (55.1%) demonstrated ARP fluorescence; in contrast, less than 8% of TH neurons were positive for ARP in controls (Fig. 1b). To investigate a brain region affected by PD – but not subject to apparent degeneration – postmortem cortical samples from the same PD and control subjects were assayed for abasic sites. ARP staining was rarely detectable in cortical neurons from either control subjects or PD patients (Supplemental Fig. 1). Therefore, significant abasic site accumulation was observed only in nigral dopaminergic neurons from PD patients.
Figure 1.
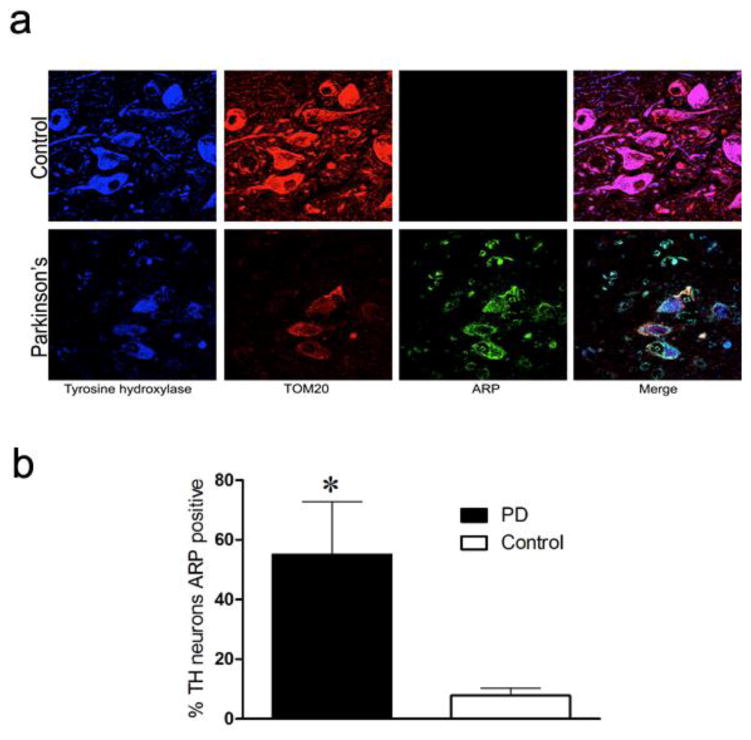
Mitochondrial abasic sites in substantia nigra dopamine neurons in human PD. (a) Representative fluorescence images of the SN from a healthy control and PD patient. Tissue was incubated with antibodies against TH (blue), a mitochondrial marker TOM20 (red), and probed with aldehyde-reactive probe (ARP, green). (b) Quantification of the proportion of TH neurons that were ARP positive. In contrast to controls, the majority of TH neurons from PD patients accumulated abasic sites (black bar, * p < 0.05, Student's t-test). Data are presented as mean ± SEM from five control and five PD midbrain sections.
Rotenone induces mitochondrial abasic sites selectively in nigral dopaminergic neurons
To model the systemic mitochondrial impairment in PD, we exposed rats to the pesticide rotenone, which has been linked epidemiologically to PD (20). Following rotenone treatment in rats (five daily injections), ARP fluorescence was detected in TH neurons (Fig. 2a, Supplemental Fig. 2). The majority of TH neurons demonstrated ARP fluorescence, which largely co-localized with TOM20 staining, indicating accumulated abasic sites were in mitochondria (Fig. 2a). In addition, ARP fluorescence did not co-localize with DAPI (not shown). The ARP signal was abolished by pre-treatment of tissue with DNase I, but not RNase A (Supplemental Fig. 3), further confirming that this non-nuclear signal is derived from mtDNA. ARP fluorescence was generally undetectable in vehicle-treated animals (Fig. 2a) and was likewise absent in cortical neurons from rotenone-treated or control animals (Supplemental Fig. 4). An increase in ventral midbrain total abasic sites following rotenone administration was further confirmed using a conventional quantitative ELISA-based method (Fig. 2b).
Figure 2.

Mitochondrial abasic sites in substantia nigra dopamine neurons of rotenone treated rats. (a) Representative fluorescence images of the SN from rats either treated with vehicle or rotenone (five daily injections). Tissue was incubated with antibodies against TH (blue), a mitochondrial marker TOM20 (red), and probed with aldehyde-reactive probe (ARP, green). Few neurons showed ARP fluorescence in vehicle treated animals. Bright ARP fluorescence that co-localized with TOM20 was observed in rats treated with rotenone. (b) Quantitative ELISA measurements using DNA from VMB extracts (black bar, * p < 0.05, Student's t-test) demonstrated increased levels of total abasic sites in rats injected with rotenone compared to vehicle. (c) Abasic sites form quickly with rotenone exposure in vitro. Rotenone treatment (10 nM) for two hours increased the number of abasic sites in rat primary VMB neurons relative to vehicle by ELISA measurement (black bar, * p < 0.05, Student's t-test). Data are presented as mean ± SEM.
To examine whether abasic sites are induced with rotenone exposure in vitro, rat primary neurons from the ventral midbrain (VMB) were treated with vehicle or 10 nM rotenone, which does not cause cell death (Fig. 4b). Abasic sites were increased within two hours of rotenone exposure relative to vehicle-treated VMB neurons (Fig. 2c). Together, these results indicate that mtDNA abasic sites are detectable prior to any signs of degeneration – and are produced selectively in nigral dopaminergic neurons under conditions of mitochondrial impairment.
Figure 4.
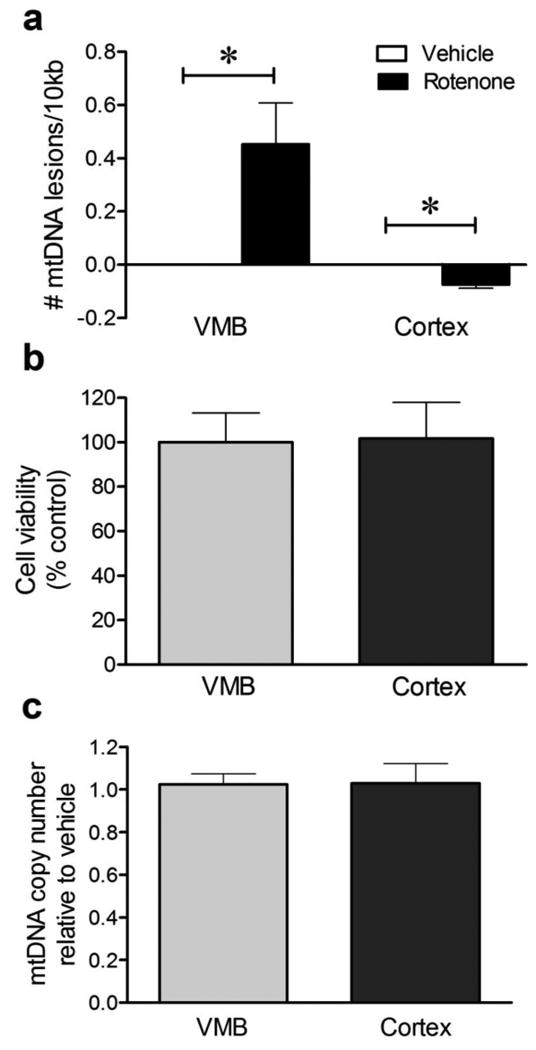
Sublethal rotenone induced mtDNA damage in rat primary VMB but not cortical neurons in vitro. (a) 24 hours of exposure to 10 nM rotenone increased mtDNA lesions in rat primary VMB neurons (*p < 0.05, Student's t-test). In contrast, rotenone reduced mtDNA lesions in rat primary cortical neurons (*p < 0.01, Student's t-test). (b) In these cultures, rotenone exposure did not change VMB or cortical neuron viability relative to vehicle. (c) Rotenone exposure did not alter mtDNA copy number in rat primary VMB or cortical neurons relative to vehicle. Data are presented as mean ± SEM. The QPCR-based assay was performed in technical triplicate for each biological replicate (n = 3 DMSO, n = 3 rotenone treated cultures).
Regionally selective mtDNA damage prior to nigrostriatal degeneration
Consistent with the ARP results described above, we recently observed increased mtDNA damage associated with LRRK2 mutations in human iPSC-derived neural cells (34). Oxidative damage, which is thought to be important in PD pathogenesis, results in a spectrum of DNA lesions, such as strand breaks, abasic sites and base damage. Although our new ARP assay detects an increase in a specific form of mtDNA damage – abasic sites – in human PD and with experimental mitochondrial impairment, it is important to evaluate mtDNA damage more broadly. For this purpose, we used a quantitative polymerase chain reaction (QPCR)-based assay, which was applied after rotenone-induced mitochondrial impairment in vivo and in vitro. By primer design, this assay involves the amplification of a fragment specific for the mitochondrial (13.4 kb) genome. The large mtDNA amplicon constitutes ∼ 77% of the genome, excluding the D-loop to avoid bias from this mutation hotspot (27). Many of the DNA lesions associated with oxidative stress interfere with the ability of the polymerase to replicate template DNA. Therefore, this assay simultaneously assesses a wide variety of DNA damage and DNA repair intermediates that have the propensity to slow down or block DNA polymerase progression (26). Thus, if equal amounts of DNA from experimental and control specimens are amplified under identical conditions, the DNA sample with the least DNA damage will produce the greatest amount of PCR product (26). Thus, the final amplified PCR product is inversely proportional to the number of DNA lesions.
Despite rapid and uniform distribution of rotenone throughout the body, chronic exposure to rotenone is selectively toxic to the nigrostriatal dopamine system in experimental animals and it causes a pattern of cell-type specific degeneration similar to PD (35). However, a single rotenone injection causes neither behavioral symptoms nor detectable neuropathology (23). Rats were injected with a single dose of rotenone (3 mg/kg) or vehicle, and 24 hours later the ventral midbrain (VMB) was dissected, DNA was purified and the QPCR-based assay performed. We found that rotenone induced mtDNA damage (lesions) in rat VMB even after a single injection (Fig. 3a; 0.31 ± 0.06 lesions/10kb; p < 0.001). To determine if rotenone-induced mtDNA damage is brain region specific, we assessed mtDNA damage in the cerebral cortex. In contrast to VMB, cortex showed fewer mtDNA lesions after rotenone compared to vehicle (Fig. 3a; -0.10 ± 0.06 lesions/10kb, p < 0.05). To control for a potential loss of mtDNA in response to cellular stress that could influence our interpretation of mtDNA damage, we amplified a short mtDNA fragment (235bp) that due to its small size is less likely to contain a lesion, and this was used to assay for mitochondrial copy DNA number. Mitochondrial DNA copy number was comparable in rotenone and vehicle injected rats in both the VMB and cortex (Fig. 3b). DNA damage in the rat VMB was specific to the mitochondria, since fewer nuclear DNA lesions were detectable after a single rotenone injection relative to vehicle (Supplemental Fig. 5).
Figure 3.
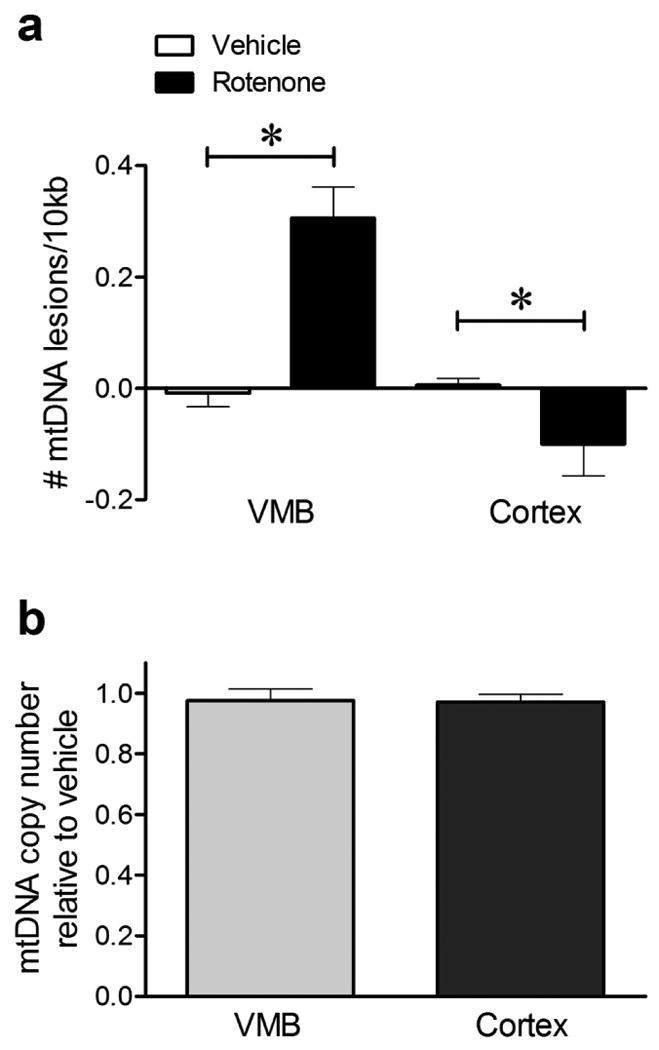
A single subclinical dose of rotenone induced mtDNA damage in rat VMB but not cortex in vivo. (a) 24 hours following a single injection of rotenone, mtDNA lesions were induced in the VMB (*p < 0.001, Student's t-test). In contrast, rotenone reduced mtDNA lesions in the cortex (*p < 0.05, Student's t-test). (b) Sublethal rotenone exposure did not alter mtDNA copy number in VMB or cortex relative to vehicle with a single rotenone injection. Data are presented as mean ± SEM. The QPCR-based assay was performed in technical triplicate for each biological replicate (n = 8 vehicle, n = 9 rotenone for VMB; n = 6 vehicle, n = 6 rotenone for cortex).
To examine whether mtDNA damage occurs prior to cell type specific degeneration in vitro, rat primary neurons from the VMB or cortex were treated with rotenone (10 nM) or vehicle. After 24 hours of rotenone treatment, DNA was purified and the QPCR-based assay performed. Similar to our in vivo data, mtDNA damage was increased in VMB neurons after exposure to rotenone relative to vehicle-treated neurons (Fig. 4a; 0.45 ± 0.15 lesions/10kb; p < 0.05). Also similar to the in vivo results, primary cortical neurons exhibited fewer mtDNA lesions after rotenone exposure than after vehicle treatment (Fig. 4a; -0.07 ± 0.02 lesions /10kb; p < 0.01). Neither VMB nor cortical neuronal viability was affected by this low concentration of rotenone (Fig. 4b). Furthermore, the differences in mtDNA damage in VMB vs. cortical neurons in response to rotenone were not attributable to changes in steady state mtDNA levels (Fig. 4c). Together, results from the QPCR-based assay indicate that rotenone-induced mtDNA damage is brain region-selective and occurs well before nigrostriatal neurodegeneration.
Rotenone increases mitochondrial H2O2 in ventral midbrain but not cortical neurons despite similar inhibition of oxygen consumption rate
Since mtDNA abasic sites were detected selectively in nigral dopamine neurons (but not cortical neurons) in human PD – and similar results were seen in rats exposed to rotenone – it raises the question of whether the differences in mtDNA damage between nigral and cortical neurons might simply reflect differences in the degree of mitochondrial impairment between these cell types. To examine this issue, VMB and cortical neurons were both exposed to the same concentration of rotenone, and mitochondrial respiration was assessed in the intact neurons by measuring oxygen consumption rates (OCRs) (36). Treatment with 10 nM rotenone for 24 hours (which does not affect neuronal viability or mtDNA copy number; Fig. 4b,c) reduced the baseline OCR in VMB and cortical neuronal cultures to a similar extent (Fig. 5a). Furthermore, detailed analysis mitochondrial respiration after sequential treatments with oligomycin, FCCP and 1 μM rotenone showed that VMB and cortical neurons behaved identically (Fig. 5b, c). Thus, although dopamine neurons selectively accumulate mtDNA damage in PD and after exposure to rotenone, this does not necessarily indicate that mitochondrial respiration is more impaired in the dopamine neurons. Instead, other factors must be involved.
Figure 5.

Sublethal rotenone impaired mitochondrial respiration equivalently in rat primary VMB and cortical neurons in vitro. (a) Treatment with 10 nM rotenone for 24 hours decreased the basal oxygen consumption rate (OCR) of rat primary VMB and cortical neurons (black and white bars, *p < 0.01, Student's t test). (b) The OCRs of VMB (black circles) and cortical neurons (white circles) pre-exposed to vehicle for 24 hours and then to oligomycin, FCCP and rotenone were similar to each other. (c) The OCRs of VMB and cortical neurons pre-exposed to 10 nM rotenone for 24 hours and then to oligomycin, FCCP and rotenone were also similar to each other. Data are presented as mean ± SEM.
Oxidative stress has been implicated in PD, and rotenone is known to increase reactive oxygen species. We therefore measured real time levels of mitochondrial hydrogen peroxide (H2O2) in VMB and cortical neurons in response to a sublethal concentration of rotenone (10 nM) using the MitoPY1 probe (30-32). Increased fluorescence of MitoPY1 indicates proportionally higher levels of mitochondrial H2O2. Using confocal microscopy, low levels of MitoPY1 H2O2-associated fluorescence co-localized with mitochondria in primary VMB neurons that were exposed to vehicle (Fig. 6a, b). After acute exposure to 10 nM rotenone, VMB neurons had a rapid and pronounced increase in the MitoPY1 mitochondrial (H2O2) fluorescence. Within 5 minutes of rotenone exposure, MitoPY1 fluorescence in VMB mitochondria was greater than vehicle – and it increased progressively for the duration of measurement (Fig. 6b). In contrast, in cortical neurons treated with either vehicle or 10 nM rotenone, MitoPY1 fluorescence was generally undetectable (Fig. 6a). Thus, despite equivalent inhibition of respiration (Fig. 5), VMB neurons respond to complex I inhibition with a rapid increase in H2O2 levels and cortical neurons do not. These results suggest that for a given level of complex I inhibition (with rotenone or in PD), dopamine neurons experience more oxidative stress – and this is associated with early, selective accumulation of mtDNA damage.
Figure 6.
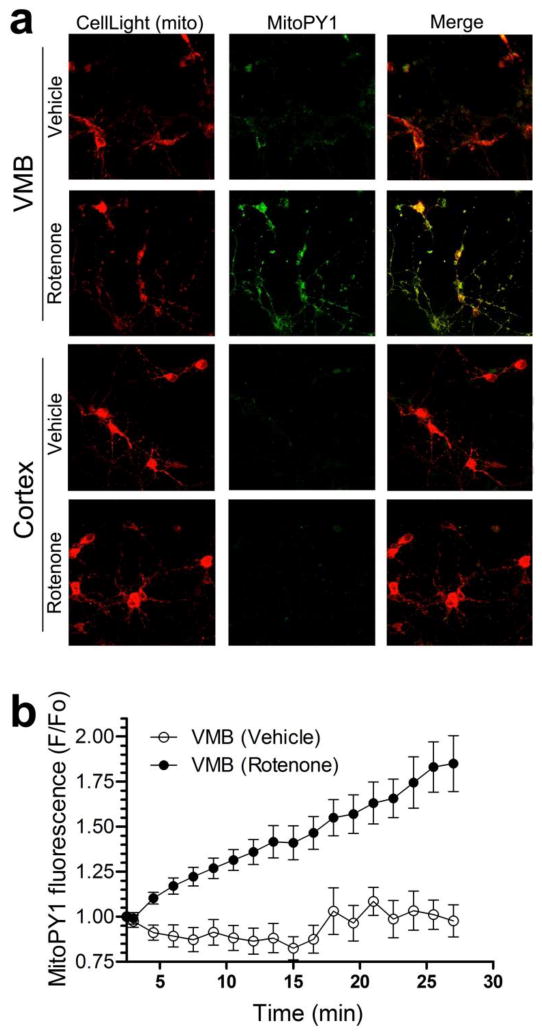
Increased levels of mitochondrial H2O2 in rat primary VMB but not cortical neurons after rotenone treatment in vitro. (a) Representative fluorescence images of mitochondria (red) and the mitochondrial H2O2 indicator, MitoPY1 (green), in live primary VMB and cortical neurons. Administration of 10 nM rotenone to VMB neurons generated bright MitoPY1 fluorescence that co-localized with mitochondria. Few cortical neurons showed bright MitoPY1 fluorescence after rotenone administration. Similarly, few VMB or cortical neurons contained bright MitoPY1 fluorescence after vehicle administration. (b) Kinetic studies of mitochondrial H2O2 accumulation in VMB neurons showed increased MitoPY1 fluorescence within 5 minutes of rotenone administration (black circles) relative to vehicle (white circles). MitoPY1 fluorescence continued to increase for at least 27 minutes after 10 nM rotenone exposure. Data are presented as mean ± SEM.
Discussion
There is an urgent need to define early events in PD pathogenesis for both diagnostic and therapeutic purposes. While both genetic and environmental factors contribute to an individual's risk for developing PD, mitochondrial dysfunction has been proposed as a central mechanism shared by sporadic and familial forms of PD. Nevertheless, the mechanisms by which mitochondrial dysfunction result in PD are unclear. Here we showed that mtDNA damage (i) occurs quickly (within 2 hr) after modest complex I impairment; (ii) accumulates selectively in nigral dopamine neurons; and (iii) is enduring (i.e., not repaired effectively), as it is found in the majority of nigral dopamine neurons in postmortem specimens from PD cases. Mitochondrial DNA damage may cause compromised bioenergetic function, genetic and protein instability, increased ROS and it may trigger cell death. Accumulation of DNA damage is particularly deleterious to post-mitotic, differentiated tissues, including neurons, which generally cannot rely on self-renewal through cell proliferation (37). However, to the best of our knowledge, mtDNA damage has not been specifically examined in idiopathic PD.
Although there have been previous reports of general DNA damage in late-stage PD, mtDNA has not been examined specifically or rigorously, and cellular specificity has not been reported (16,38,39). Applying a newly developed histochemical assay for abasic sites in DNA in postmortem specimens from PD cases and the rotenone model, we have shown that dopaminergic neurons – but not cortical neurons – accumulate mtDNA damage. This is the first report of mtDNA abasic sites in human PD. Abasic sites, whether generated through spontaneous base loss or caused by ROS, are potentially cytotoxic or genotoxic as they can block the polymerase during replication and transcription. While abasic site-induced toxicity in the nuclear genome poses a threat, this type of lesion is particularly detrimental for mitochondria because the mitochondrial DNA polymerase, polymerase-gamma, stalls in the presence of abasic sites causing replication blockage (40). The inability to effectively repair abasic sites can compromise mitochondrial function and contribute to neurodegeneration (41,42). While mtDNA damage is an integrated function of ongoing damage, repair mechanisms, mtDNA replication and other cytoprotective mechanisms, including mitophagy (43), our finding of persistent abasic sites in PD suggests ongoing base excision repair defects (9). These results complement reports that components of the base excision repair pathway are up-regulated in the substantia nigra of PD patients (44-46).
Occupational exposure to pesticides is a well-characterized environmental risk factor for PD (47). Such epidemiological studies suggest the importance of multiple low concentration occupational exposures in causing cumulative neural damage. In this regard, occupational exposure to rotenone is an independent risk factor for PD (20). Experimentally, chronic administration of rotenone causes selective nigrostriatal neurodegeneration despite systemic inhibition of complex I (35). To model in rats the systemic mitochondrial impairment and environmental component associated with PD (48), rats were exposed to the pesticide rotenone. We found that mitochondrial dysfunction was associated with robust mtDNA damage selectively in substantia nigra – but not in cortex – and that this occurred well before there was any frank degeneration of the nigrostriatal system. While we had previously shown in the MPTP model of PD that mtDNA damage is associated with extant neurodegeneration (49), the current study demonstrates that mtDNA damage in the nigrostriatal system occurs before the onset of neurodegeneration. Further support for the idea that increased DNA damage contributes to neurodegeneration comes from murine knockouts of the DNA repair enzymes MTH1, an oxidized purine nucleoside triphosphatase, and OGG1 (8-oxoguanine DNA glycosylase), which showed age-related degeneration of the nigrostriatal system and/or increased sensitivity to dopaminergic toxins (50,51). Lastly, genetic murine models that lead to mtDNA depletion either by a mitochondrially targeted endonuclease or by disruption in transcription factor A mitochondrial (TFAM), recapitulate many of the key features of PD (52).
The basis for the relatively selective loss of dopamine neurons in PD is uncertain, but high levels of ROS are an intrinsic property of the vulnerable subpopulation of dopaminergic neurons in the substantia nigra (1), and this may render dopaminergic neurons particularly prone to DNA damage. Just as in PD, we found that exposure to rotenone selectively produces mtDNA damage in nigral dopamine neurons, but not cortical neurons. To rule out the possibility that rotenone simply inhibits complex I more effectively in VMB neurons than in cortical neurons in our model system, we measured mitochondrial respiration in both types of neurons before and after application of rotenone. This experiment showed that rotenone impairs mitochondrial respiration equally effectively in both cell types. Since, like rotenone, at least some forms of PD (e.g., PINK1 and Parkin mutations) are associated with systemic mitochondrial defects, this result raises the possibility that VMB neurons and cortical neurons respond differently to mitochondrial impairment. For example, in response to a complex I defect, mitochondria in VMB neurons might produce more ROS – or buffer them less effectively. To test this possibility, we used a probe for mitochondrial H2O2, MitoPY1, and found a robust rotenone-induced H2O2 signal in VMB neurons but not cortical neurons. Whether this reflects differences in production or degradation of ROS remains to be determined, but these results are consistent with other recent work showing that VMB neurons handle ROS differently than cortical neurons (53). In fact, rotenone-exposed cortical neurons compared with vehicle showed less mtDNA damage both in vitro and in vivo. Similarly, it has been shown that after a brief exposure to hydrogen peroxide, mtDNA actually amplifies better than control (54). In addition, a previous study in mice showed that the cortex has lower basal levels of DNA damage and higher levels of DNA repair relative to other brain regions (55). In this context, reduced mtDNA lesions in cortical neurons suggests (i) an enhanced mtDNA repair response, (ii) a less oxidizing cellular environment, and/or (iii) a more efficient ROS scavenging system (56).
In conclusion, the present study provides the first demonstration that abasic sites are readily detected in nigral dopaminergic, but not cortical neurons, in PD and a model thereof. Abasic sites accumulate very quickly after even modest complex I inhibition and apparently persist to the final stages of the disease. As such, mitochondrial abasic sites may represent a new ‘molecular signature’ of vulnerable nigral neurons in PD. Although relatively little is currently known about DNA repair in neurons, future work will elucidate the critical mtDNA repair pathways involved in the accumulation of mtDNA damage in PD and may point to new therapeutic strategies.
Supplementary Material
Highlights.
mtDNA abasic sites selectively accumulate in dopaminergic neurons in PD
mtDNA damage is detected prior to nigrostriatal degeneration in PD models
mtDNA damage is likely a critical early event in neuronal dysfunction in sporadic PD
The persistence of abasic sites suggests ineffective base excision repair in PD
Acknowledgments
Funding: This work was supported by grants from the National Institutes of Health: T32MH18273 (L.H.S.), 1F32ES019009-01 (L.H.S.), 1R01ES020718 (J.T.G.), 2R01AG026389 (C.T.C.), 1R01NS065789 (C.T.C.), and R01GM97465 (C.J.C.). The University of Pittsburgh Brain Bank is supported in part by P50AG005133, and the Parkinson's Disease Neuropathology Core (C.T.C.) is supported by P01NS059806 (J.T.G.). C.J.C. is an Investigator with the Howard Hughes Medical Institute. This work was also supported by the American Parkinson Disease Association (J.T.G) and the JPB Foundation (J.T.G.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Guzman JN, Sanchez-Padilla J, Wokosin D, Kondapalli J, Ilijic E, Schumacker PT, Surmeier DJ. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature. 2010;468:696–700. doi: 10.1038/nature09536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mariani E, Polidori MC, Cherubini A, Mecocci P. Oxidative stress in brain aging, neurodegenerative and vascular diseases: an overview. Journal of chromatography. B, Analytical technologies in the biomedical and life sciences. 2005;827:65–75. doi: 10.1016/j.jchromb.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 3.Sherer TB, Greenamyre JT. Oxidative damage in Parkinson's disease. Antioxid Redox Signal. 2005;7:627–629. doi: 10.1089/ars.2005.7.627. [DOI] [PubMed] [Google Scholar]
- 4.Cardoso SM, Moreira PI, Agostinho P, Pereira C, Oliveira CR. Neurodegenerative pathways in Parkinson's disease: therapeutic strategies. Curr Drug Targets CNS Neurol Disord. 2005;4:405–419. doi: 10.2174/1568007054546072. [DOI] [PubMed] [Google Scholar]
- 5.Jenner P. Oxidative mechanisms in nigral cell death in Parkinson's disease. Mov Disord. 1998;13(Suppl 1):24–34. [PubMed] [Google Scholar]
- 6.Sanders LH, Timothy Greenamyre J. Oxidative damage to macromolecules in human Parkinson disease and the rotenone model. Free Radic Biol Med. 2013 doi: 10.1016/j.freeradbiomed.2013.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362:709–715. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- 8.Rao KS. Genomic damage and its repair in young and aging brain. Mol Neurobiol. 1993;7:23–48. doi: 10.1007/BF02780607. [DOI] [PubMed] [Google Scholar]
- 9.Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger . DNA Repair and Mutagenesis. ASM Press; 2006. [Google Scholar]
- 10.Elstner M, Muller SK, Leidolt L, Laub C, Krieg L, Schlaudraff F, Liss B, Morris C, Turnbull DM, Masliah E, et al. Neuromelanin, neurotransmitter status and brainstem location determine the differential vulnerability of catecholaminergic neurons to mitochondrial DNA deletions. Mol Brain. 2011;4:43. doi: 10.1186/1756-6606-4-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kraytsberg Y, Kudryavtseva E, McKee AC, Geula C, Kowall NW, Khrapko K. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat Genet. 2006;38:518–520. doi: 10.1038/ng1778. [DOI] [PubMed] [Google Scholar]
- 12.Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, Perry RH, Jaros E, Hersheson JS, Betts J, Klopstock T, et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nature genetics. 2006;38:515–517. doi: 10.1038/ng1769. [DOI] [PubMed] [Google Scholar]
- 13.Vermulst M, Wanagat J, Loeb LA. On mitochondria, mutations, and methodology. Cell Metab. 2009;10:437. doi: 10.1016/j.cmet.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 14.Zhang J, Perry G, Smith MA, Robertson D, Olson SJ, Graham DG, Montine TJ. Parkinson's disease is associated with oxidative damage to cytoplasmic DNA and RNA in substantia nigra neurons. The American journal of pathology. 1999;154:1423–1429. doi: 10.1016/S0002-9440(10)65396-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alam ZI, Jenner A, Daniel SE, Lees AJ, Cairns N, Marsden CD, Jenner P, Halliwell B. Oxidative DNA damage in the parkinsonian brain: an apparent selective increase in 8-hydroxyguanine levels in substantia nigra. J Neurochem. 1997;69:1196–1203. doi: 10.1046/j.1471-4159.1997.69031196.x. [DOI] [PubMed] [Google Scholar]
- 16.Hegde ML, Gupta VB, Anitha M, Harikrishna T, Shankar SK, Muthane U, Subba Rao K, Jagannatha Rao KS. Studies on genomic DNA topology and stability in brain regions of Parkinson's disease. Archives of biochemistry and biophysics. 2006;449:143–156. doi: 10.1016/j.abb.2006.02.018. [DOI] [PubMed] [Google Scholar]
- 17.Sanders LH, Laganiere J, Cooper O, Mak SK, Vu BJ, Huang YA, Paschon DE, Vangipuram M, Sundararajan R, Urnov FD, et al. LRRK2 mutations cause mitochondrial DNA damage in iPSC-derived neural cells from Parkinson's disease patients: Reversal by gene correction. Neurobiology of disease. 2013 doi: 10.1016/j.nbd.2013.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cannon JR, Greenamyre JT. Gene-environment interactions in Parkinson's disease: Specific evidence in humans and mammalian models. Neurobiology of disease. 2012 doi: 10.1016/j.nbd.2012.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martinez TN, Greenamyre JT. Toxin models of mitochondrial dysfunction in Parkinson's disease. Antioxid Redox Signal. 2012;16:920–934. doi: 10.1089/ars.2011.4033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tanner CM, Kamel F, Ross GW, Hoppin JA, Goldman SM, Korell M, Marras C, Bhudhikanok GS, Kasten M, Chade AR, et al. Rotenone, paraquat, and Parkinson's disease. Environ Health Perspect. 2011;119:866–872. doi: 10.1289/ehp.1002839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chu CT, Caruso JL, Cummings TJ, Ervin J, Rosenberg C, Hulette CM. Ubiquitin immunochemistry as a diagnostic aid for community pathologists evaluating patients who have dementia. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2000;13:420–426. doi: 10.1038/modpathol.3880072. [DOI] [PubMed] [Google Scholar]
- 22.Alafuzoff I, Ince PG, Arzberger T, Al-Sarraj S, Bell J, Bodi I, Bogdanovic N, Bugiani O, Ferrer I, Gelpi E, et al. Staging/typing of Lewy body related alpha-synuclein pathology: a study of the BrainNet Europe Consortium. Acta neuropathologica. 2009;117:635–652. doi: 10.1007/s00401-009-0523-2. [DOI] [PubMed] [Google Scholar]
- 23.Cannon JR, Tapias V, Na HM, Honick AS, Drolet RE, Greenamyre JT. A highly reproducible rotenone model of Parkinson's disease. Neurobiol Dis. 2009;34:279–290. doi: 10.1016/j.nbd.2009.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ghosh A, Greenberg ME. Distinct roles for bFGF and NT-3 in the regulation of cortical neurogenesis. Neuron. 1995;15:89–103. doi: 10.1016/0896-6273(95)90067-5. [DOI] [PubMed] [Google Scholar]
- 25.Ayala-Torres S, Chen Y, Svoboda T, Rosenblatt J, Van Houten B. Analysis of gene-specific DNA damage and repair using quantitative polymerase chain reaction. Methods. 2000;22:135–147. doi: 10.1006/meth.2000.1054. [DOI] [PubMed] [Google Scholar]
- 26.Santos JH, Meyer JN, Mandavilli BS, Van Houten B. Quantitative PCR-based measurement of nuclear and mitochondrial DNA damage and repair in mammalian cells. Methods in molecular biology. 2006;314:183–199. doi: 10.1385/1-59259-973-7:183. [DOI] [PubMed] [Google Scholar]
- 27.Sanchez-Cespedes M, Parrella P, Nomoto S, Cohen D, Xiao Y, Esteller M, Jeronimo C, Jordan RC, Nicol T, Koch WM, et al. Identification of a mononucleotide repeat as a major target for mitochondrial DNA alterations in human tumors. Cancer Res. 2001;61:7015–7019. [PubMed] [Google Scholar]
- 28.Furda AM, Bess AS, Meyer JN, Van Houten B. Analysis of DNA damage and repair in nuclear and mitochondrial DNA of animal cells using quantitative PCR. Methods in molecular biology. 2012;920:111–132. doi: 10.1007/978-1-61779-998-3_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Diers AR, Higdon AN, Ricart KC, Johnson MS, Agarwal A, Kalyanaraman B, Landar A, Darley-Usmar VM. Mitochondrial targeting of the electrophilic lipid 15-deoxy-Delta12,14-prostaglandin J2 increases apoptotic efficacy via redox cell signalling mechanisms. Biochem J. 426:31–41. doi: 10.1042/BJ20091293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dickinson BC, Chang CJ. A targetable fluorescent probe for imaging hydrogen peroxide in the mitochondria of living cells. Journal of the American Chemical Society. 2008;130:9638–9639. doi: 10.1021/ja802355u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dickinson BC, Lin VS, Chang CJ. Preparation and use of MitoPY1 for imaging hydrogen peroxide in mitochondria of live cells. Nature protocols. 2013;8:1249–1259. doi: 10.1038/nprot.2013.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dickinson BC, Chang CJ. Chemistry and biology of reactive oxygen species in signaling or stress responses. Nature chemical biology. 2011;7:504–511. doi: 10.1038/nchembio.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maynard S, de Souza-Pinto NC, Scheibye-Knudsen M, Bohr VA. Mitochondrial base excision repair assays. Methods. 2010;51:416–425. doi: 10.1016/j.ymeth.2010.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sanders LH, Laganiere J, Cooper O, Mak SK, Vu BJ, Huang YA, Paschon DE, Vangipuram M, Sundararajan R, Urnov FD, et al. LRRK2 mutations cause mitochondrial DNA damage in iPSC-derived neural cells from Parkinson's disease patients: Reversal by gene correction. Neurobiology of disease. 2013;62C:381–386. doi: 10.1016/j.nbd.2013.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nature neuroscience. 2000;3:1301–1306. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- 36.Qian W, Van Houten B. Alterations in bioenergetics due to changes in mitochondrial DNA copy number. Methods. 2010;51:452–457. doi: 10.1016/j.ymeth.2010.03.006. [DOI] [PubMed] [Google Scholar]
- 37.Van Houten B, Woshner V, Santos JH. Role of mitochondrial DNA in toxic responses to oxidative stress. DNA repair. 2006;5:145–152. doi: 10.1016/j.dnarep.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 38.Zhang J, Perry G, Smith MA, Robertson D, Olson SJ, Graham DG, Montine TJ. Parkinson's disease is associated with oxidative damage to cytoplasmic DNA and RNA in substantia nigra neurons. The American journal of pathology. 1999;154:1423–1429. doi: 10.1016/S0002-9440(10)65396-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Alam ZI, Jenner A, Daniel SE, Lees AJ, Cairns N, Marsden CD, Jenner P, Halliwell B. Oxidative DNA damage in the parkinsonian brain: an apparent selective increase in 8-hydroxyguanine levels in substantia nigra. J Neurochem. 1997;69:1196–1203. doi: 10.1046/j.1471-4159.1997.69031196.x. [DOI] [PubMed] [Google Scholar]
- 40.Pinz KG, Shibutani S, Bogenhagen DF. Action of mitochondrial DNA polymerase gamma at sites of base loss or oxidative damage. J Biol Chem. 1995;270:9202–9206. doi: 10.1074/jbc.270.16.9202. [DOI] [PubMed] [Google Scholar]
- 41.Boiteux S, Guillet M. Abasic sites in DNA: repair and biological consequences in Saccharomyces cerevisiae. DNA Repair (Amst) 2004;3:1–12. doi: 10.1016/j.dnarep.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 42.Lauritzen KH, Dalhus B, Storm JF, Bjoras M, Klungland A. Modeling the impact of mitochondrial DNA damage in forebrain neurons and beyond. Mech Ageing Dev. 132:424–428. doi: 10.1016/j.mad.2011.02.006. [DOI] [PubMed] [Google Scholar]
- 43.Zhu J, Wang KZ, Chu CT. After the banquet: Mitochondrial biogenesis, mitophagy and cell survival. Autophagy. 2013;9 doi: 10.4161/auto.24135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shimura-Miura H, Hattori N, Kang D, Miyako K, Nakabeppu Y, Mizuno Y. Increased 8-oxo-dGTPase in the mitochondria of substantia nigral neurons in Parkinson's disease. Annals of neurology. 1999;46:920–924. [PubMed] [Google Scholar]
- 45.Arai T, Fukae J, Hatano T, Kubo S, Ohtsubo T, Nakabeppu Y, Mori H, Mizuno Y, Hattori N. Up-regulation of hMUTYH, a DNA repair enzyme, in the mitochondria of substantia nigra in Parkinson's disease. Acta neuropathologica. 2006;112:139–145. doi: 10.1007/s00401-006-0081-9. [DOI] [PubMed] [Google Scholar]
- 46.Fukae J, Takanashi M, Kubo S, Nishioka K, Nakabeppu Y, Mori H, Mizuno Y, Hattori N. Expression of 8-oxoguanine DNA glycosylase (OGG1) in Parkinson's disease and related neurodegenerative disorders. Acta neuropathologica. 2005;109:256–262. doi: 10.1007/s00401-004-0937-9. [DOI] [PubMed] [Google Scholar]
- 47.Cannon JR, Greenamyre JT. The role of environmental exposures in neurodegeneration and neurodegenerative diseases. Toxicological sciences : an official journal of the Society of Toxicology. 2011;124:225–250. doi: 10.1093/toxsci/kfr239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Henchcliffe C, Beal MF. Mitochondrial biology and oxidative stress in Parkinson disease pathogenesis. Nat Clin Pract Neurol. 2008;4:600–609. doi: 10.1038/ncpneuro0924. [DOI] [PubMed] [Google Scholar]
- 49.Mandavilli BS, Ali SF, Van Houten B. DNA damage in brain mitochondria caused by aging and MPTP treatment. Brain research. 2000;885:45–52. doi: 10.1016/s0006-8993(00)02926-7. [DOI] [PubMed] [Google Scholar]
- 50.Cardozo-Pelaez F, Sanchez-Contreras M, Nevin AB. Ogg1 null mice exhibit age-associated loss of the nigrostriatal pathway and increased sensitivity to MPTP. Neurochem Int. 61:721–730. doi: 10.1016/j.neuint.2012.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yamaguchi H, Kajitani K, Dan Y, Furuichi M, Ohno M, Sakumi K, Kang D, Nakabeppu Y. MTH1, an oxidized purine nucleoside triphosphatase, protects the dopamine neurons from oxidative damage in nucleic acids caused by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Cell Death Differ. 2006;13:551–563. doi: 10.1038/sj.cdd.4401788. [DOI] [PubMed] [Google Scholar]
- 52.Pickrell AM, Pinto M, Hida A, Moraes CT. Striatal dysfunctions associated with mitochondrial DNA damage in dopaminergic neurons in a mouse model of Parkinson's disease. J Neurosci. 31:17649–17658. doi: 10.1523/JNEUROSCI.4871-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Horowitz M, Milanese C, Di Maio R, Hu X, Montero LM, Sanders LH, Tapias V, Sepe S, van Capellen WA, Burton EA, et al. Single-cell redox imaging demonstrates a distinctive response of dopaminergic neurons to oxidative insults. Antioxid Redox Signal. 2011 doi: 10.1089/ars.2010.3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Santos JH, Hunakova L, Chen Y, Bortner C, Van Houten B. Cell sorting experiments link persistent mitochondrial DNA damage with loss of mitochondrial membrane potential and apoptotic cell death. The Journal of biological chemistry. 2003;278:1728–1734. doi: 10.1074/jbc.M208752200. [DOI] [PubMed] [Google Scholar]
- 55.Cardozo-Pelaez F, Brooks PJ, Stedeford T, Song S, Sanchez-Ramos J. DNA damage, repair, and antioxidant systems in brain regions: a correlative study. Free radical biology & medicine. 2000;28:779–785. doi: 10.1016/s0891-5849(00)00172-6. [DOI] [PubMed] [Google Scholar]
- 56.Chen KH, Yakes FM, Srivastava DK, Singhal RK, Sobol RW, Horton JK, Van Houten B, Wilson SH. Up-regulation of base excision repair correlates with enhanced protection against a DNA damaging agent in mouse cell lines. Nucleic Acids Res. 1998;26:2001–2007. doi: 10.1093/nar/26.8.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.