

Abstract
Heavy metal contamination of surface waters at mining sites often involves complex interactions of multiple sources and varying biogeochemical conditions. We compared surface and subsurface metal loading from mine waste pile runoff and mine drainage discharge and characterized the influence of iron oxides on metal fate along a 0.9-km stretch of Tar Creek (Oklahoma, USA), which drains an abandoned Zn/Pb mining area. The importance of each source varied by metal: mine waste pile runoff contributed 70% of Cd, while mine drainage contributed 90% of Pb, and both sources contributed similarly to Zn loading. Subsurface inputs accounted for 40% of flow and 40-70% of metal loading along this stretch. Streambed iron oxide aggregate material contained highly elevated Zn (up to 27,000 μg g−1), Pb (up to 550 μg g−1) and Cd (up to 200 μg g−1) and was characterized as a heterogeneous mixture of iron oxides, fine-grain mine waste, and organic material. Sequential extractions confirmed preferential sequestration of Pb by iron oxides, as well as substantial concentrations of Zn and Cd in iron oxide fractions, with additional accumulation of Zn, Pb, and Cd during downstream transport. Comparisons with historical data show that while metal concentrations in mine drainage have decreased by more than an order of magnitude in recent decades, the chemical composition of mine waste pile runoff has remained relatively constant, indicating less attenuation and increased relative importance of pile runoff. These results highlight the importance of monitoring temporal changes at contaminated sites associated with evolving speciation and simultaneously addressing surface and subsurface contamination from both mine waste piles and mine drainage.
Keywords: heavy metals, iron oxides, metal loading, mine drainage, mine waste piles, sequential extractions
1. Introduction
Abandoned mining sites cause substantial impairment to water quality worldwide (Fields 2003; Younger et al. 2002). After mining operations cease, oxygen-rich groundwater floods abandoned mine workings and can promote oxidation of pyrite (FeS2) and other metal sulfide minerals, producing acidity and sulfate and releasing co-occurring trace metals. Over time, seepage from mines can emerge aboveground as metal-rich acid mine drainage. In addition, runoff from large piles of fine-grained tailings and other mining wastes can carry large metal loads as precipitation infiltrates the piles and promotes geochemical weathering of metal-containing primary minerals (Tonkin et al. 2002).
Mitigating impacts from abandoned mines on water quality requires an understanding of the major sources of metal loading and subsequent fate of trace metals. The chemical composition of drainage from underground mines and mine waste piles (MWPs) can vary considerably within a watershed, based on host rock composition and flow conditions (Balistrieri et al. 1999; España et al. 2005). At many sites, discrete surface inputs of metals from underground mines and mine waste piles are carried through adits, mine tunnels, and naturally-forming streams. In addition, subsurface inputs also can contribute substantially to metal loading in mining-impacted streams (Kimball et al. 2001; Lachmar et al. 2006). Metals can be removed from the water column by secondary mineral precipitation (e.g., as carbonates or hydroxides), coprecipitation, or sorption onto organic matter (OM) or surface-reactive iron (oxyhydr)oxides, (hereafter referred to as iron oxides), that form following oxidation of ferrous iron (Balistrieri et al. 2003; Dzombak and Morel 1990; España et al. 2005; Hochella et al. 2005).
Natural iron oxide minerals vary widely in composition, morphology, and physicochemical properties (Perret et al. 2000; Thompson et al. 2011). The ability of iron oxides to sequester heavy metals depends on their mineralogy, size, extent of crystallinity, purity, and aggregation state, as well as competitive interactions between metals and other ions at the mineral surface (Balistrieri et al. 2003; Cismasu et al. 2011; Gilbert et al. 2009; Masue-Slowey et al. 2011; Ostergren et al. 2000). Furthermore, iron phase transformations in natural environmental conditions are common (Hansel et al. 2003; Thompson et al. 2006) and may alter sequestration capacity over time (Henneberry et al. 2012), either by changing the type and availability of sorption sites (Masue-Slowey et al. 2011) or by element-dependent exsolution or more permanent incorporation of metals into the crystal structure (Ford et al. 1997). Therefore, iron oxides formed in situ are heterogeneous and their reactivity towards contaminants is determined by the interplay of system-dependent physical and chemical parameters (e.g., incorporation of impurities such as Al, Si, and OM). Characterizing their composition and behavior is therefore necessary to determine the overall role of iron oxide sequestration in metal transport at a watershed scale.
In this study we employ a novel suite of analytical tools and geochemical speciation modeling to characterize iron oxide aggregate materials that form and evolve in composition as two geochemically distinct sources of heavy metal loading (mine drainage and mine waste pile runoff) mix and move down stream. Our goal is to understand the interplay of these two metal sources both spatially and temporally in order to enable better predictions of metal transport, inventories, and fate. We also present data that point to decadal-scale evolution of metal loading pathways and consider implications for long-term remediation strategies.
1.1 Tar Creek Superfund Site
Tar Creek (Oklahoma, USA) drains a portion of the abandoned Tri-State Mining District, a major Zn and Pb producing area from the mid-1800s to the mid-1900s. Ore minerals are primarily associated with the Mississippi Boone formation, with host rocks consisting of fossiliferous limestone and thick beds of nodular chert (Luza 1986). Metals are concentrated in large piles of mine waste, locally called chat, a combination of tailings and other mining waste. This mine waste material consists of chert (microcrystalline quartz), calcite, dolomite, marcasite, pyrite, sphalerite (ZnS), galena (PbS), and hemimorphite (Zn silicate) (Carroll et al. 1998; Schaider et al. 2007). Cadmium is present in sphalerite at around 0.5% (O'Day et al. 1998). The site has dozens of major piles, some up to 60 m in height (Luza 1986), which act as persistent sources of metals into surface water and groundwater. Chat is actively being removed from the site and incorporated into asphalt and other uses as part of remediation at the Tar Creek Superfund Site, which was added to the National Priority List in 1983. Groundwater was actively pumped out of the mines until the cessation of mining activities in 1970, after which the mines filled with metal-rich water that still seeps into Tar Creek. Although mine drainage (MD) is now only slightly acidic due to the acid neutralizing capacity of carbonate-bearing minerals in the host rock, the creek is nonetheless heavily impacted by both artesian seepage discharges from underground mines and runoff from MWPs. While MD is often considered the major source of water contamination at this site, runoff from MWPs adjacent to the creek provides surface and subsurface inputs of water, and discharges of runoff from MWPs and mill ponds provide most of the baseflow to Tar Creek and its main tributary, Lytle Creek (Cope et al. 2008).
Characterizing the chemical composition of MD and MWP runoff at the Tar Creek site is necessary in order to understand the interactions of these two sources and their relative metal loading. Cope et al. (2008) showed substantial loading of Zn, Pb, and Cd along stretches of Tar Creek affected by MWP and MD inputs. However, this study did not characterize these sources individually or distinguish between surface and subsurface inputs. Mine drainage contains highly elevated Fe concentrations, resulting in abundant iron oxide coatings on the Tar Creek streambed and buoyant flocculant material in the water column. Periodic flood events flush streambed iron oxides downstream and into nearby floodplain soils that support residential and agricultural land uses.
Initial work at the Tar Creek site explored metal associations with iron oxide phases in sediments and controls on water column solubility (Carroll et al. 1998; O'Day et al. 1998). X-ray absorption spectroscopy (XAS) analyses of Pb, Cd, and Zn binding environments in streambed sediments (down to 5 cm) showed key differences among these metals (O'Day et al. 1998). Iron oxides appeared important for Zn binding and Pb was associated with both iron oxides and carbonate phases, while Cd was present in primary sphalerite or secondary carbonate phases. A companion paper (Carroll et al. 1998) used equilibrium modeling to explain trends in metal concentrations and to identify controls on metal speciation in the water column. Their results suggested that the parameters controlling metal speciation vary by metal and include degassing of CO2, solubility of carbonate phases, dissolution of galena and sphalerite catalyzed by dissolved iron, and sorption to iron oxides. These results highlight differing metal behavior, although they did not focus specifically on the particles (and particle surfaces) most closely in contact with the water column, including consideration of the role of iron oxides in transport. We build on their work by characterizing metal loading inputs and considering the roles of heavy metal loading from multiple sources and advective flow in determining changes in metal concentrations along the creek.
2. Materials and Methods
2.1 Field sampling
Water samples were collected from Tar Creek, Lytle Creek (a tributary), MWP runoff, and MD discharges during three sampling trips (January and May 2005, June 2006). Based on trace metal analyses of 2005 samples, a 0.9-km stretch of Tar Creek heavily impacted both by MD and MWP runoff was selected for additional sampling in 2006. This stretch had two distinct sections: the upstream portion, Stretch #1 (0.65 km), was primarily impacted by MWP runoff, and Stretch #2 (0.25 km) received substantial input of MD discharges and had extensive iron oxide formation in sediments and floc material in the water column. In June 2006, water samples were collected at 7 in-stream locations and from all surface inputs: 3 MD discharges, 4 MWP runoff, and Lytle Creek (Figure 1). At each sampling location, water samples were collected for analysis of total acid soluble metal concentrations and acid neutralizing capacity (ANC). Two aliquots of each sample were filtered through a 0.2 μm nylon filter for dissolved metal and anion analyses. Triplicate samples were collected at all 7 in-stream sites and a field blank was also collected. All samples were stored on ice in the field, and were frozen within 12 hours of collection. Temperature, pH, specific conductivity, and dissolved oxygen were determined in situ with a Hydrolab Quanta (Hach Company, Loveland, CO).
Figure 1.

Map of sampling locations. Shaded areas show outline of mine waste piles, based on an aerial photograph.
Because of the shallow depth and poorly-defined creek geometry, a conservative tracer was used to measure flow rate. Various conservative tracers have been used to measure flow in mining-impacted creeks, including Rhodamine WT, Cl−, Br−, and Li+. LiBr was selected because Li+ and Br− have been found to behave more conservatively than Rhodamine WT (Dierberg and DeBusk 2005; Lin et al. 2003) and because Tar Creek contained high background Cl− concentrations (9-47 mg/L, May 2005 data). Using Br− also permitted rapid assessment of steady state Br− concentrations downstream of the injection point using a Br− ion selective electrode. While some tracer studies use synoptic sampling at multiple locations along a stretch of stream after achieving steady-state (e.g., Kimball et al. 2001), individual LiBr injections (working downstream to upstream) were performed at multiple well-constrained and well-mixed locations within Tar Creek and in surface inputs close to their junction with the creek. This approach was more appropriate because large stagnant pools within Tar Creek would have required an impractically long time to achieve steady state and because some MD and MWP discharges entered into the creek in close proximity, with insufficient mixing zones to distinguish the flow rate of each input. Details are provided in the Supplementary Material.
Samples of iron oxide aggregate material were collected from the surface of Fe-rich sediments at two locations: immediately downstream of a major MD input (point E) and approximately 2 km downstream of all major MWP runoff and MD discharges (point H). Material was collected by pumping water near the sediment-water interface with a peristaltic pump through plastic tubing onto a GF/F filter (Whatman Inc., Piscataway, NJ). Pieces of mine waste and other large particles were avoided. Samples were immediately stored on ice, frozen within 12 hours, and remained frozen until they were processed in the laboratory. Additional samples used for mineralogical analyses were collected by a similar procedure, but onto 0.2 μm filters, in September 2009.
2.2 Analytical methods
2.2.1 Water samples
Water samples were stored frozen and thawed prior to analysis. Total acid soluble and dissolved metal samples were acidified with concentrated HNO 3(5% v:v) and refrigerated until analysis. Dissolved anion samples were re-filtered through a 0.2 μm nylon filter to remove precipitates that formed after thawing prior to analysis. Details of analytical methods are provided in Supplementary Material. In short, water samples were analyzed for: total acid soluble and dissolved metals (Zn, Cd, Pb, Cu, Ni, Mn: inductively-coupled plasma mass spectrometry; Ca: flame atomic absorption spectrophotometry; Fe: spectrophotometry), dissolved anions (SO42−, Cl−: ion chromatography (IC)); and ANC (pH titration). Li (graphite furnace atomic absorption spectrophotometry) and Br (IC) were measured in samples collected before and after LiBr tracer injections to estimate flow rate.
2.2.2 Iron oxide aggregate material samples
Solid phase samples of iron oxides collected from the Tar Creek streambed were freeze-dried and weighed before and after drying to calculate water content. Details of analytical methods are provided in Supplementary Material. In short, bulk elemental composition of five freeze-dried iron oxides collected in 2006 was analyzed using polarized energy-dispersive x-ray fluorescence (XRF). Sequential extractions of four of these samples were conducted in triplicate to quantify metal association with mineral phases according to an eight-step procedure hybridizing methods used in previous studies of mine waste from the Tar Creek site (Schaider et al. 2007) and extraction steps specifically targeting Fe and Mn oxides (see Gleyzes et al. 2002 for a review). Table 1 shows the solutions and procedures used for extraction. Additional analyses were conducted on subsequent samples collected in 2009. X-ray diffraction (XRD) was used to analyze mineralogy and look for the presence of crystalline iron oxide phases. Mössbauer spectroscopy was used to identify iron phase and valence state. Scanning electron microscopy (SEM) with element mapping was used to assess morphology and elemental composition. Loss on ignition (LOI; 500°C, 12 h) was used as a measure of OM. While previous studies have noted the potential for LOI to include minor contributions from phases other than OM, such as ignition of carbonates and water loss from clays and metal oxides (Heiri et al. 2001), LOI measurements of a marine sediment standard reference material showed good agreement with certified TOC values (Table S1).
Table 1.
Sequential extraction procedure. The sum of two phases shaded in gray includes all iron oxide phases (FeOX = AmOX + XtalOX).
| Abbreviation | Solution | Procedure | Targeted phase(s) |
|---|---|---|---|
| SOL | 18 MΩ water | 20°C, 2 h | soluble |
| EX | 1 M MgCl2 | 20°C, pH 5.0, 1 h | exchangeable |
| CO3 | 1 M Na acetate | 20°C, pH 5.0 (adjusted with acetic acid), 5 h | carbonates |
| MnOX | 0.1 M hydroxylamine in 0.01 M HNO3 | 20°C, pH 2.1, 30 min | Mn oxides (reducible), carbonates |
| AmOX | 0.25 M hydroxylamine in 0.25 M HNO3 | 50°C, pH 0.75, 30 min | amorphous Fe and Al oxides (reducible) |
| ORG | 0.1 M tetrasodium pyrophosphate | 20°C, 24 h | organic matter (oxidizable) |
| XtalOX | 0.3 M Na dithionite and Na citrate, in 1.0 M Na bicarbonate | 20°C, 3 h | crystalline Fe oxides (reducible) |
| RES | concentrated HNO3 | 20°C, 2 h | residual |
2.3 Flow rate calculations
Flow rates at field sampling locations were calculated based on measured dilution of injected LiBr tracer solution. In all cases, flow rates of the injection solution were <1% of the flow rate of the creek or surface input. Flow calculations were based on Li data because some post-injection Br− concentrations fell below the lowest calibration standard. Flow rates of Lytle Creek and one MWP runoff input were estimated by visual inspection relative to other quantified inputs. The flow rate measured at the furthest downstream location (Point G) was approximately the same as a concurrent real-time measurement (38 L/s) at a USGS gauging station (07185090) 2.2 km downstream (point H), suggesting that our measurements captured most of the inputs associated with mining sources.
2.4 Modeling metal speciation
PHREEQC (Parkhurst and Appelo 1999) was used to model metal speciation of stream water samples. The WATEQ4F database was used as the source of equilibrium constants, including binding constants for metal interactions with iron oxides (Dzombak and Morel 1990) and solubility products for a range of secondary mineral phases, including sulfate, carbonate, and hydroxide minerals, as well as highly crystalline primary mineral phases. For Zn, Cd, Cu, Mn, and Ni, updated stability constants for binding to strong and weak iron oxide binding sites were used (Balistrieri et al. 2003). Initial pH values were based on field measurements but were allowed to vary, while redox potential was calculated by PHREEQC, in equilibrium with atmospheric oxygen. Magnesium was not measured, but was estimated by balancing cation and anion charges. The resulting concentrations (31–98 mg/L) were similar to those reported in Tar Creek in August 2005 (17-73 mg/L) by Cope et al. (2008). The total acid soluble concentration of Fe was used to calculate the amount of Fe(OH)3 formed, and then a density of weak and strong binding sites was specified (Dzombak and Morel 1990). The solution was allowed to equilibrate with the iron oxide phases, and other possible solid phases were specified in the WATEQ4F database.
2.5 Temporal comparisons
Historical concentrations of heavy metals and other water quality parameters in MWP runoff and MD discharges were compiled from reports published in the 1980s, including MWP runoff on the west side of Tar Creek from the Admiralty pile and MD discharge in the vicinity of samples collected in the current study. Statistical significance of differences between 1980s data and our results (2005 and 2006 combined) was assessed using the non-parametric Mann-Whitney U-test in R (Version 2.12.0). For samples with concentrations below the detection limit (DL), a value of 0.5*DL was used for plotting and in statistical comparisons. In some cases, comparisons were not made because of small sample sizes or when DLs for samples with non-detectable concentrations were above measured concentrations in other studies.
3. Results and discussion
3.1 Composition of inputs
Substantial differences in MD discharge and MWP runoff composition (Tables 2, S1) are indicative of differences in the relative abundance of key mineral phases, biogeochemical conditions, and transport processes. Median concentrations of Pb, Zn, and Cd were 3, 7, and 170 times higher, respectively, in MWP runoff than in MD inputs, whereas median Fe concentrations were 600 times higher in MD inputs. The MD inputs had consistently lower and more variable pH (5.0-6.4) than MWP runoff (pH 7.0-7.3), although neither the MD discharges nor MWP runoff were strongly acidic, reflecting the acid neutralizing capacity of carbonate minerals in the host rock. The relatively acidic conditions of the MD inputs may be due to differences in the relative abundance of carbonate-bearing minerals, since higher pyrite:carbonate ratios are associated with more acidic conditions in acid mine drainage and acid rock drainage (Balistrieri et al. 1999), or from incomplete degassing of CO2 from MD inputs (Carroll et al. 1998).
Table 2.
Water quality parameters and concentrations of total acid soluble metals and dissolved anions in mine waste pile runoff and mine drainage discharge samples collected in January and May 2005 and June 2006 and in stream samples collected at the most upstream location (Location A) in June 2006. ND = not detected.
| mine waste pile runoff |
mine drainage discharge |
Upstream Tar C. (location A) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| mean | median | range | N | mean | median | range | N | ||
| pH | 7.3 | 7.4 | 6.97 - 7.56 | 11 | 5.9 | 6.1 | 4.97 - 6.44 | 6 | 7.14 |
| SpC (mS/cm) | 2.5 | 2.6 | 1.91 - 2.79 | 11 | 2.6 | 2.6 | 2.46 - 2.72 | 6 | 1.12 |
| DO (mg/L) | 7.2 | 6.9 | 6.75 - 8.06 | 4 | 3.6 | 4.0 | 2.01 - 4.50 | 4 | 7.27 |
| ANC (mg/L as CaCO3) | 87 | 89 | 66.8 - 111 | 9 | 99 | 99 | 75.9 - 122 | 2 | 201 |
| Cl (mg/L) | 4.3 | 4.3 | 1.61 - 6.70 | 8 | 10 | 11 | 4.13 - 17.3 | 5 | 25.5 |
| SO42- (mg/L) | 1300 | 1200 | 996 - 1560 | 9 | 1400 | 1400 | 1350 - 1500 | 6 | 426 |
| Ca (mg/L) | 520 | 550 | 389 - 584 | 4 | 530 | 520 | 509 - 549 | 3 | 171 |
| Fe (mg/L) | 1.6 | 0.076 | ND - 12.7 | 9 | 39 | 44 | 13.0 - 59.0 | 6 | 1.33 |
| Mn (mg/L) | 0.26 | 0.13 | ND - 1.16 | 11 | 1.5 | 1.4 | 0.919 - 2.43 | 6 | 0.855 |
| Zn (mg/L) | 41 | 42 | 20.1 - 69.7 | 11 | 6.2 | 6.5 | 3.28 - 7.89 | 6 | 3.97 |
| As (μg/L) | 0.36 | 0.37 | 0.309 - 0.410 | 4 | 16 | 4.0 | 3.02 - 40.4 | 3 | 0.841 |
| Cd (μg/L) | 190 | 180 | 15.3 - 443 | 11 | 0.88 | 1.1 | 0.259 - 1.38 | 6 | 4.31 |
| Cu (μg/L) | 1.5 | 1.0 | 0.125 - 5.12 | 11 | 0.32 | 0.30 | 0.159 - 0.500 | 6 | 0.783 |
| Ni (μg/L) | 110 | 100 | 46.9 - 169 | 10 | 190 | 190 | 173 - 212 | 5 | 9.60 |
| Pb (μg/L) | 14 | 9.5 | 4.73 - 29.6 | 11 | 2.6 | 3.2 | 0.179 - 4.99 | 6 | 2.39 |
Sulfate concentrations were similarly elevated in MD discharges and MWP runoff (1000-1600 mg/L), indicating active sulfide oxidation within both the piles and underground mines, but the large differences in trace metal concentrations suggest differences in dominant sulfide mineral phases and biogeochemical conditions. The relative abundance of Fe in the MD suggests that FeS2 oxidation is a more dominant process in the mine workings, whereas oxidation of ZnS and PbS may be more important within the piles. This difference may contribute to the difference in acidity between sources, since Pb and Zn oxidation only releases acidity when Fe is the oxidant, whereas pyrite oxidation releases acidity under all conditions (Plumlee 1999). Although mining operations removed much of the metal ore, the mine waste material remains elevated in labile Zn, Pb, and Cd, especially in fine particle size fractions (Schaider et al. 2007). While mine waste also contains elevated Fe concentrations (up to 3.5% in <37 μm samples, unpublished data) and XRD analyses showed the presence of pyrite as a constituent in <37 μm mine waste (Schaider et al. 2007), there is little visual evidence of Fe oxide formation associated with surface MWP runoff discharges. Oxidation of ferrous iron (Fe(II)) and reprecipitation of Fe3+ within the piles under oxic conditions related to periodic wetting and drying may explain the very low concentrations of Fe in surface MWP runoff. At pH 7.3, Fe(II) oxidation and Fe oxide formation are relatively rapid; Fe(II) has a half life on the order of minutes at pH 7.3, compared with a half life on the order of a day at pH 6 (calculated from Stumm and Morgan 1996). Iron oxides were evident in saturated wetland areas near the bases of MWPs, confirming that Fe can be mobilized from mine waste under saturated conditions. The relatively high affinity of Pb for Fe oxides may partially contribute to the low enrichment of Pb in MWP runoff relative to MD discharges, as well as potential precipitation of Pb-bearing secondary mineral phases, such as PbSO4, that have been observed to form rinds on the surfaces of PbS particles (Diehl et al. 2008).
We used a mass balance approach to estimate metal concentrations in subsurface inputs along a 0.2-km stretch between points B and C where there were no visible surface inputs and flow nearly doubled. We calculated average total acid soluble concentrations of Ca, Cu, Ni, Zn, Cd, As, and Mn in subsurface inputs (Csub, μg/L), assuming conservative behavior, as:
where M and Q are total acid soluble metal fluxes (μg/s) and water flow rates (L/s), respectively, at points B and C (Table S3). Pb and Fe were excluded due to apparent non-conservative behavior, which may include losses to sediments or leaching from mine waste in the streambed. Along this stretch, Csub values were similar to those of MWP runoff, with elevated Zn and Cd (21,000 and 49 μg/L, respectively) and low As (0.57 μg/L). By contrast, Csub for Mn (2,300 μg/L) was relatively high compared to surface MWP runoff (120–1200 μg/L). These estimates suggest that subsurface inputs along Stretch #1 primarily originated from MWPs and that metals were generally transported conservatively through the subsurface, with the addition of Mn mobilized from soils. Manganese concentrations in local mine waste (95-420 μg/g, <37 μm fraction, n=6, data not shown) are similar to those found in background soils (40-900 μg/g, ATSDR 2012).
3.2 Metal concentrations and mass loading in Tar Creek
In June 2006, the chemical composition of Tar Creek water at the most upstream location (Location A) appeared to be a mixture of MWP runoff and groundwater. Concentrations of Zn, Pb, and Cd at Location A were elevated above background levels, likely from upstream MWPs and mine waste in the stream bed, but were 5-45 times lower than average concentrations in MWP runoff (Table 2). By contrast, the concentration of Mn was over threefold higher than the average for MWP runoff, consistent with inputs from groundwater.
The chemical composition of stream water samples along Stretch #1 reflected additional inputs from MWP runoff, while stream water along Stretch #2 was more similar to MD, suggesting that along each stream stretch, metal loading was dominated by a single source. Stretch #1 had higher pH and ANC concentrations than Stretch #2 (Figure 2, Table S4), reflecting the relatively low pH and ANC of MD inputs. Zinc and Cd were primarily present in the dissolved phase along both stretches, and their total acid soluble concentrations increased by a factor of 4 and 10, respectively, along Stretch #1 before being diluted by MD inputs downstream of point D. By contrast, total acid soluble Pb concentrations increased only in close proximity to major surface inputs and decreased between these inputs, indicative of substantial losses to sediments. This trend is consistent with <10% of Pb being present in the dissolved phase and the relatively high affinity of Pb for particle surfaces (Dzombak and Morel 1990) and relatively low solubility products of Pb-containing minerals (Stumm and Morgan 1996). Total acid soluble Fe concentrations were more than an order of magnitude lower along Stretch #1 than along Stretch #2, where there was much more extensive formation of streambed iron oxides. Despite the presence of solid phase ferric iron, >80% of total Fe along Stretch #2 was present as dissolved Fe, suggesting that most Fe(II) originating from the MD had not yet oxidized to Fe(III). This result is reasonable given an estimated half-life for Fe(II) under in-stream conditions (pH 5.8, 5 mg/L O2) of approximately 60 h (calculated from Stumm and Morgan 1996). The one unit decrease in pH from Stretch #1 to #2 is not expected to decrease Pb sorption to Fe oxides, since Pb binding to Fe oxides is fairly constant from pH 5 up to pH at least pH 7 (Dyer et al. 2003).
Figure 2.

Chemical constituents in Tar Creek water samples. (a)-(d) Total acid soluble and dissolved metal concentrations and total acid soluble metal fluxes; (e) pH and acid neutralizing capacity (ANC); and (f) dissolved SO42− and Cl− concentrations. Locations of surface inputs of mine waste pile runoff (R), mine drainage (M), and Lytle Creek (LC) are indicated with arrows showing their junctions with Tar Creek. The shaded area shows Stretch #2, visibly impacted by iron oxides. Note differences in scales of y-axes.
In order to evaluate the kinetics of removal processes and whether metals and anions exhibited conservative mixing and transport over small spatial scales, we assessed mixing at two locations: Location B, 20 m downstream of a MWP input, and Location G, 25 m downstream of a MD discharge. We calculated expected concentrations at Locations B and G using flow rate data and chemical concentrations in the upstream creek water (Locations A and F, respectively) and in the corresponding surface input. We compared these expected concentrations with those measured at the downstream location (Figure S1). Interestingly, at both downstream locations, predicted and measured concentrations were similar, and most predicted concentrations were within 20% of expected values, suggesting that along mixing zones of 20-30 m (approximate time scale of minutes) sorption and precipitation reactions did not substantially limit metal mobility.
The relative importance of surface and subsurface MD discharges and MWP runoff to metal loading along Stretches #1 and 2 varied by metal as a function of flow rate and relative metal concentrations of each input (Figure 3). Stretch #2 contributed five times more flow than did Stretch #1, with subsurface inputs contributing 86% and 35% of added flow along Stretches #1 and #2, respectively. For Cd, Stretch #1 was the major source of Cd loading, driven by the much higher concentrations of Cd in MWP runoff than in MD inputs, whereas Stretch #2 was the major source of Zn loading, given the more similar Zn concentrations in MWP runoff and MD inputs. For both metals, the proportion of subsurface inputs was greater along Stretch #1 (78-86%) than along Stretch #2 (20-55%), reflecting their relative flow rates. For Pb, nearly all loading was attributable to Stretch #2, and the majority of apparent Pb loading along Stretches #1 and #2 could be accounted for by surface inputs. However, subsurface Pb inputs were likely underestimated by this mass balance approach due to precipitation and sorption into sediments. Overall, MWP runoff appears to be the main determinant of metal composition along Stretch #1 and to contribute a majority of Cd and a substantial fraction of Zn, via both surface and subsurface inputs, along the entire highly-impacted stretch of Tar Creek.
Figure 3.

Contributions to water flow and metal fluxes originating from upstream Tar Creek (as measured at location A), surface and subsurface inputs along Stretches #1 and #2. Horizontal bars separate Stretches #1 and #2. Iron flux decreased slightly (0.18 kg day−1) along Stretch #1; for all other stretches, increases in flux and flow rate were observed. Note differences in scales of y-axes.
3.3 Geochemical modeling and solid phase metal speciation
3.3.1 PHREEQC modeling
PHREEQC was used to model metal speciation and sorption to iron oxides in Tar Creek water samples. Overall, the model results suggest that iron oxides exerted a strong influence over Pb concentrations along Stretch #2, while precipitation of Zn-carbonate minerals may limit dissolved Zn concentrations along Stretch #1 (Figure S2).
PHREEQC underpredicted the fraction of particle-associated Pb, especially along Stretch #1. Iron oxide-bound Pb was predicted to account for 1-13% of Pb along Stretch #1 and 70-80% of Pb along Stretch #2, with the remainder of Pb predicted to be present primarily in dissolved carbonate and sulfate complexes. By contrast, measurements of total and dissolved metal concentrations in creek water indicated that nearly all Pb (88-100% for Stretch #1, 98-100% for Stretch #2) was present in the particulate phase. While results of equilibrium-based modeling should be viewed as estimates when modeling a dynamic system, these discrepancies suggest Pb binding to phases other than iron oxides in sediments, such as particulate OM or suspended iron oxide aggregate material, may play a more significant role in speciation than expected.
PHREEQC results indicate that sorption to iron oxides plays a minor role in Zn and Cd speciation. Sorption to iron oxides was predicted to account for <10% of both Zn and Cd along Stretch #1, and 2-4% (Zn) and 9-14% (Cd) along Stretch #2. These results were consistent with empirical measurements indicating that generally <10% of Cd and Zn were present in the particulate phase in creek samples. Other controls on Zn solubility are possible; at point D, 40% of Zn was predicted to precipitate as ZnCO3·H2O, and at points A-C, ZnCO3·H2O was close to saturation (SI -0.02 to -0.18). No Pb or Cd minerals were supersaturated along Stretch #1. Carroll et al. (1998) calculated that Pb carbonate phases are saturated along Tar Creek, although O'Day et al. (1998) observed Pb-O interatomic distances consistent with bonding to iron oxides at high Fe concentrations. Thus, iron and carbonate phases may compete for Pb at the circumneutral pH values documented in Tar Creek. Neither of these papers explicitly considered interaction with OM.
3.3.2 Fe speciation in iron oxide aggregate material
In addition to equilibrium modeling with PHREEQC, we characterized solid-phase mineralogy and metal concentrations and speciation in iron oxide aggregate material collected from streambed surfaces in Tar Creek. Mineralogical analyses confirm the non-crystalline nature of the Fe in these aggregate materials. X-ray diffraction (XRD) analyses (data not shown) confirmed a disordered or amorphous nature with either broadened peaks suggesting two-line ferrihydrite or no identifiable iron phase despite visual evidence of an iron oxide-rich aggregate material. Isomer shifts calculated from Mössbauer spectroscopy analyses show that iron oxide aggregate material is completely ferric. Mössbauer data are best fit as two ferrihydrite-like coordination environments (Figure S3). The development of a doublet in one of the coordination environments in the downstream sample may indicate that the iron minerals in the aggregate material undergo ordering during transport, but a more thorough characterization would be needed to validate this observation.
Analyses of total Fe and Fe speciation reveal impurities in Fe oxide aggregate material and shifts in Fe speciation during downstream transport. Total Fe concentrations ranged from 28% to 49% and decreased from an average of 44% at location E to 33% at location H (Table 3). These concentrations were below theoretical Fe concentrations of pure phase Fe oxide phases (approximately 59% for ferrihydrite assuming the disordered structure (Fe8.2O8.5(OH)7.4·3H2O) proposed by Michel et al. (2010)), suggesting that OM, fine grain mine waste particles, secondary minerals, or other phases were abundant in the aggregate material. Organic matter, as measured by loss on ignition (LOI), constituted around 20% of sample mass and decreased slightly from location E (25%) to location H (18%). Concentrations of Al increased downstream (Table 3), indicating incorporation of sediments and primary mine waste into the aggregate during transport. Textural evidence along with X-ray element mapping obtained from backscatter field-emission electron microscopy images of a single sample collected from location E shows heterogeneity of iron oxide aggregate material and suggests that detrital quartz minerals are serving as nuclei or hosts for Fe oxide precipitation (Figure 4). Furthermore, oxide-based elemental analysis suggests that there may be two populations of iron oxide “phases” with different affinities for heavy metals forming on the surface of host detrital quartz: high contrast material from region (a) containing 36 wt% Fe and 1.2 wt% Zn; and less bright oxides in region (b) containing no measurable Zn, 8.5 wt% P, and 52 wt% Fe. Phosphorus may coprecipitate with or adsorb to iron oxide phases, lowering surface reactivity or preventing Zn adsorption.
Table 3.
Average total metal concentrations (s.d. in parentheses) in iron oxides collected from the Tar Creek streambed at locations E and H and from <37 μm mine waste samples collected from six mine waste piles throughout the Tar Creek site (Schaider et al. 2007).
| Fe % | Mn (mg kg-1) | Zn (mg kg-1) | Pb (mg kg-1) | Cd (mg kg-1) | Al (mg kg-1) | S (mg kg-1) | |
|---|---|---|---|---|---|---|---|
| Location E (N=3) | 44 (4.6) | 490 (42) | 3,200 (710) | 59 (13) | 45a (23) | 2,600 (170) | 9,600 (1,400) |
| Location H (N=2) | 33 (9.3) | 880 (8.3) | 18,000 (13,000) | 330 (320) | 150a (59) | 5,000 (940) | 14,000 (2,300) |
| <37 μm mine waste (N=24) | 2.0 (0.75) | 210b (71) | 59,000 (28,000) | 5,700 (4,800) | 120 (25) | 9,500 (8,100) | 4,200 (2,400) |
Based on total Cd extracted using sequential extractions, N=3 at both locations.
Based on total Mn extracted using sequential extractions, N=6.
Figure 4.
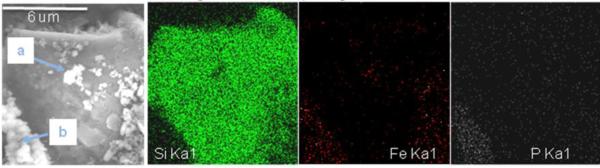
Backscatter field-emission scanning electron microscope image and x-ray maps of iron oxide aggregate material filtered from Tar Creek. A detrital quartz core serves as a host for iron oxides precipitated on its surface. X-ray maps note two geochemically distinct iron oxide phases. Note 6-micron scale bar in BSE image. Regions (a) and (b) are described in the text.
Sequential extractions reveal limited but potentially important changes in Fe speciation during downstream transport. The proportion of Fe extracted from steps targeting Fe oxide phases (FeOX = AmOx + XtalOx) increased slightly from 51% at Location E to 58% at Location H, attributable to an increase in the more crystalline fraction (XtalOx) and simultaneous decrease in the organic-associated fraction (ORG) (Figure 5). While the magnitude of this shift is small and the total concentration of extracted Fe does not change significantly (Figure S4), this trend provides supporting evidence for mineral evolution and ordering during transport. The relatively minor changes in Fe speciation overall, however, suggest relatively conservative behavior, over 1 km of transport, and that a physical mechanism such as dilution of aggregate material by entrainment of sediments and primary mine waste may explain changes in metal concentration and speciation.
Figure 5.

Sequential extraction results for (a) Fe, (b) Mn, (c) Pb, (d), Zn, and (e) Cd at locations E and H, shown as a percentage of total extracted. Data represent the average of three replicate extractions from each location. Total extracted concentrations are presented under the columns.
3.3.3 Geochemical associations of heavy metals in iron oxide aggregate material
Iron oxide aggregate material contained elevated heavy metal concentrations (Table 3), up to 27,200 μg/g Zn, 554 μg/g Pb and 200 μg/g Cd, with substantial portions associated with Fe oxide fractions. Between points E and H, the concentration of Cd increased by more than a factor of three and Zn and Pb concentrations increased more than five-fold, consistent with additional sorption, co-precipitation, or entrainment of fine grain sediments and mine waste during transport. Concentrations of Zn, Pb, and Cd are all highly enriched in <37 μm mine waste collected from piles throughout the Tar Creek site (Table 3), although it is difficult to use these concentrations to calculate the contribution of fine grain mine waste to the iron oxide material given the high degree of heterogeneity among and within piles, variations in metal concentrations as a function of particle size, and in-stream weathering processes that may alter the solid-phase chemical composition of mine waste particles.
Sequential extractions of Fe oxide aggregate material reveal complexity in metal speciation that cannot be modeled using geochemical parameters alone (Figure 5, Table S4). As expected, Fe oxides (extracted in AmOx + XtalOx) sequester a majority of Pb (70% and 73% of total Pb extracted at locations E and H, respectively) and a substantial portion of Zn (49% and 40%) and Cd (31% and 20%). This observation agrees with prior XAS data showing Zn adsorption to Fe oxide phases (O'Day et al. 1998), although our PHREEQC modeling did not predict a significant role for newly-formed Fe oxides in dissolved Zn speciation. A greater percentage of Pb than Zn or Cd is associated with the step targeting more ordered or crystalline iron oxide phases (XtalOx), which may be a function of greater Pb affinity or stronger bonding mechanism (i.e., Pb forming a greater proportion of inner sphere bonds) (Dzombak and Morel 1990). While the percent of Zn, Cd, and Pb extracted by Fe oxide-targeting steps was either the same or lower at Location H compared to Location E, the estimated concentration of these three metals associated with the FeOx phase more than doubled, suggesting continued adsorption during transport despite decreasing total Fe concentrations.
Other sequential extraction steps reveal differing behaviors of Pb, Zn, and Cd. The remaining Pb not extracted by iron oxide targeting steps was primarily associated with the ORG and residual (RES) steps. Despite other evidence for accumulation of primary mine waste in downstream samples, the proportion of Pb in the RES fraction (i.e., in sulfides or silicates) decreased from 19% to 10% while increasing in the ORG fraction, from 9% to 16% of total extracted Pb (Figure S4). Zinc is more labile and partially associated with the carbonate-targeting extraction step (CO3; 28% increasing to 36% at location E and H, respectively), supporting PHREEQC modeling results that predicted saturation of Zn carbonate phases. Like Pb, the proportion of Zn in the RES fraction decreased downstream. Cadmium had the smallest fraction associated with iron oxides and was most labile, with substantial proportion extracted in the exchangeable (EX), CO3, and Mn oxide (MnOx)-targeting steps. These results reflect the relatively low affinity of Cd for iron oxide surfaces, potential competition for adsorption sites with Pb and Zn, or higher Cd affinity for other minerals and adsorption sites. The high proportion of Cd extracted in the CO3 step (24% and 35% at locations E and H respectively) is consistent with EXAFS and SIMS analyses of Tar Creek sediment identifying a cadmium carbonate compound upstream of location A, likely through substitution of Cd into calcite (O'Day et al. 1998). The increase in Cd in the RES fraction is consistent with incorporation of sphalerite-containing mine waste. Previous XRD analyses of fine grained mine waste showed the presence of sphalerite (ZnS) (Schaider et al. 2007), in which Cd is a minor constituent, and O'Day et al. (1998) found Cd associated with sphalerite in Tar Creek sediments.
On the whole, these results suggest that iron oxide aggregate material is responsible for the transport of a significant fraction of particulate metals, and that aggregate material continues to physically and geochemically evolve and scavenge metals with downstream transport. While sequential extractions only provide information about operationally-defined fractions and may lack specificity for key target phases (Kheboian and Bauer 1987), they are useful for assessing speciation and potential environmental mobility on bulk samples, for comparing speciation profiles among metals and locations, and for obtaining a macro-scale understanding of metal transport. Sequential extraction data both contrast and reinforce results from XAS (O'Day et al. 1998) and the PHREEQC modeling conducted in this study, demonstrating their relevance in scaling observations and in validating models. It is clear that treating naturally-occurring iron oxides as analogs of synthetically-formed, well-characterized phases precipitated in vitro is not appropriate for estimating sorption inventories, which, in turn, impacts estimates for both transport and exposure.
3.4 Temporal trends and broader implications
The elevated concentrations of Zn, Pb, and Cd across a range of phases in iron oxide aggregate material implies substantial loadings of all three heavy metals into streambed iron oxides, which has implications for benthic ecosystems and transport into floodplains. Based on flow rates of surface and subsurface MD inputs and the composition of surface MD inputs, total annual Fe loading into the creek was estimated to be 160,000 kg y−1. Assuming complete conversion of all Fe into iron oxides with the same composition as those collected at location H (Table 3), we estimated annual loadings into sediments of 8,800 kg y−1 Zn, 160 kg y−1 Pb, and 15 kg y−1 Cd. While the exact quantities will depend on changes in flow rate and water composition over the course of a year, these results point to the importance of iron oxides on a watershed scale as a vector of metal transport into floodplain soils.
Environmental assessments provide snapshots of conditions at mining-impacted sites, but the relative importance of multiple metal sources and metal speciation may vary substantially over time. At Tar Creek, comparing our results to our compilation of historical data suggests that the relative importance of MWPs and MD as sources of metal loading changed over course of two decades. Our recent measurements of dissolved Pb, Cd, and Zn concentrations in MWP runoff (2005–2006) are comparable to those reported from the same location in the 1980s (Figure 6). In addition to metals, concentrations of bulk dissolved ions (as measured by specific conductivity and alkalinity/ANC) in MWP runoff have not significantly changed over this time period, possibly reflecting a steady state leaching process even as secondary minerals have formed in response to decades of chemical weathering of these surface piles.
Figure 6.

Dissolved metals and other water quality parameters in (a) mine drainage discharge and (b) mine waste pile runoff from the 1980s and 2000s (current study). Comparisons between 1980s and 2000s data were evaluated using the Mann-Whitney U test. Significance level: * (p<0.05), ** (p<0.01), *** (p<0.001), n.s. (not significant). SpC = specific conductance, ANC = acid neutralizing capacity. In the absence of ANC data, alkalinity data were used when available. Statistical comparisons were not conducted for Fe, Pb, and Ni in MWP runoff due to data limitations described in text.
By contrast, dissolved Pb, Cd, and Zn concentrations in MD decreased by a factor of 940, 34, and 29, respectively, over the same time period, accompanied by a modest increase in pH (Figure 6). The decrease in metal concentrations is consistent with a prior study showing that metal concentrations in nearby mineshafts decreased approximately 10-100 fold from 1976/1977 to 2002 (U.S. Geological Survey 2003). Samples of MD collected near Location D in 1983-1985 contained 110-200 mg/L Zn, compared to 5-10 mg/L Zn in 2001-2003 (DeHay et al. 2004) and 5-8 mg/L in 2005-2006 (current study). These decreases in all the major ore-forming metals in the MD discharge may reflect changes in subsurface hydrology over time that result in less contact time between primary ore materials and groundwater, or changes in metal speciation of solid surfaces and redox conditions in the aquifer. At other mining sites, metal concentrations in mine water were highest right after groundwater filled mine workings following the cessation of mining operations (Mayes et al. 2005). However, in the absence of historical flow rate data, it is not possible to fully assess temporal changes in metal loading. It is worth noting that the greater than 10-fold decrease in metal concentrations in mine water far exceeds reasonable estimates for changes in flow rates. This implies that the overall loading of metals into Tar Creek has diminished over time. Archival evidence for these evolving loading patterns may exist in both floodplain soil profiles and in Tar Creek and Neosho River sediments.
4. Conclusions
Our comparisons of MD and MWP runoff highlight the critical interactions of these two metal loading sources into Tar Creek. While >99% of Fe loading can be attributed to surface and subsurface MD inputs, much of the Zn and Cd, and to a lesser degree Pb, originates from MWP runoff. Since iron oxides effectively trap these metals, the MD-derived iron oxides actually serve to decrease the mobility and bioavailability of these metals. These interactions need to be considered when designing remediation strategies. Remediation of MD that does not simultaneously address surface and subsurface inputs of MWP runoff may unintentionally enhance in situ bioavailability and downstream metal transport by decreasing Fe concentrations and thus reducing potential sorption to and settling of iron oxides. By demonstrating statistically significant temporal changes in MD composition over time and geochemical complexity in heavy metal-trapping iron oxides, our work more generally emphasizes the importance of longitudinal evaluations of solid-phase and aqueous metal speciation and bioavailability during risk assessment at contaminated sites.
Supplementary Material
Highlights.
We studied fate of Zn, Pb, and Cd and role of Fe oxides in a mining impacted creek.
Mine waste pile runoff and mine drainage are both major sources of heavy metals.
Natural Fe oxide aggregates contain mine waste, organic matter, and other impurities.
Sequential extractions show multiple phases of Zn, Pb, and Cd in Fe oxide aggregates.
Pile runoff and mine drainage showed different decadal-scale chemical variations.
Acknowledgements
We thank Earl Hatley, Mei Ai Khoo, and Kathleen McCarthy for help with sample collection; Jim Besancon and Matt Neal for XRD analyses; Rosalie Sharp for LOI measurements; Jimmy Hauri for help with lithium measurements; Darby Dyar for Mössbauer analyses; and FEI Instruments for SEM analysis.
The work for this publication was made possible by grants number P01 ES012874 and ES00002 from NIEHS and from a STAR Research Assistance Agreement No. RD-83172501 awarded by the U.S. EPA. It has not been formally reviewed by either the NIEHS or EPA. The views expressed in this document are solely those of the authors and do not necessarily reflect those of either the NIEHS or the EPA. Neither NIEHS nor the EPA endorses any products or commercial services mentioned in this publication.
Abbreviations
- AmOX
non-crystalline iron and aluminum oxide fraction CO3, carbonate fraction
- EX
ion-exchangeable fraction
- FeOX
iron oxides
- MD
mine drainage
- MnOX
manganese oxide fraction
- MWP
mine waste pile
- ORG
organic matter fraction
- RES
residual fraction
- SOL
water soluble fraction
- XtalOX
crystalline iron oxide fraction
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- ATSDR . Toxicological Profile for Manganese. U.S. Department of Human and Health Services, Agency for Toxic Substances and Disease Registry; Atlanta, GA: 2012. [PubMed] [Google Scholar]
- Balistrieri LS, Box SE, Bookstrom AA, Ikramuddin M. Assessing the influence of reacting pyrite and carbonate minerals on the geochemistry of drainage in the Coeur d'Alene mining district. Environ Sci Tech. 1999;33:3347–53. [Google Scholar]
- Balistrieri LS, Box SE, Tonkin JW. Modeling precipitation and sorption of elements during mixing of river water and porewater in the Coeur d'Alene River basin. Environ Sci Tech. 2003;37:4694–701. doi: 10.1021/es0303283. [DOI] [PubMed] [Google Scholar]
- Carroll SA, O'Day PA, Piechowski M. Rock-water interactions controlling zinc, cadmium, and lead concentrations in surface waters and sediments, US Tri-State Mining District. 2. Geochemical interpretation. Environ Sci Tech. 1998;32:956–65. [Google Scholar]
- Cismasu AC, Michel FM, Tcaciuc AP, Tyliszczak T, Brown GE. Composition and structural aspects of naturally occurring ferrihydrite. C R Geosci. 2011;343:210–8. [Google Scholar]
- Cope CC, Becker MF, Andrews WJ, DeHay KL. Scientific Investigations Report. U.S. Geological Survey; Reston, VA: 2008. Streamflow, Water Quality, and Metal Loads from Chat Leachate and Mine Outflow into Tar Creek, Ottawa County, Oklahoma, 2005. pp. 2007–5115. [Google Scholar]
- DeHay KL, Andrews WJ, Sughru MP. Scientific Investigations Report. U.S. Geological Survey; Reston, VA: 2004. Hydrology and Ground-Water Quality in the Mine Workings within the Picher Mining District, Northeastern Oklahoma, 2002–03. pp. 2004–5043. [Google Scholar]
- Diehl SF, Hageman PL, Smith KS. Chapter A: What is weathering in mine waste? Mineralogic evidence for sources of metals in leachates. In: Verplanck PL, editor. Understanding contaminants associated with mineral deposits. U.S. Geological Survey; Reston, VA: 2008. pp. 4–7. Circular 1328. [Google Scholar]
- Dierberg FE, DeBusk TA. An evaluation of two tracers in surface-flow wetlands: Rhodamine-WT and lithium. Wetlands. 2005;25:8–25. [Google Scholar]
- Dyer JA, Trivedi P, Scrivner NC, Sparks DL. Lead sorption onto ferrihydrite. 2. Surface complexation modeling. Environ Sci Tech. 2003;37:915–22. doi: 10.1021/es025794r. [DOI] [PubMed] [Google Scholar]
- Dzombak DA, Morel FMM. Surface Complexation Modeling: Hydrous Ferric Oxide. John Wiley & Sons; New York, NY: 1990. [Google Scholar]
- España JS, Pamo EL, Santofimia E, Aduvire O, Reyes J, Barettino D. Acid mine drainage in the Iberian Pyrite Belt (Odiel river watershed, Huelva, SW Spain): Geochemistry, mineralogy and environmental implications. Appl Geochem. 2005;20:1320–56. [Google Scholar]
- Fields S. The earth's open wounds: Abandoned and orphaned mines. Environ Health Perspect. 2003;111:A154–61. doi: 10.1289/ehp.111-a154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford RG, Bertsch PM, Farley KJ. Changes in transition and heavy metal partitioning during hydrous iron oxide aging. Environ Sci Tech. 1997;31:2028–33. [Google Scholar]
- Gilbert B, Ono RK, Ching KA, Kim CS. The effects of nanoparticle aggregation processes on aggregate structure and metal uptake. J Colloid Interf Sci. 2009;339:285–95. doi: 10.1016/j.jcis.2009.07.058. [DOI] [PubMed] [Google Scholar]
- Gleyzes C, Tellier S, Astruc M. Fractionation studies of trace elements in contaminated soils and sediments: a review of sequential extraction procedures. Trac-Trends Anal Chem. 2002;21:451–67. [Google Scholar]
- Hansel CM, Benner SG, Neiss J, Dohnalkova A, Kukkadapu RK, Fendorf S. Secondary mineralization pathways induced by dissimilatory iron reduction of ferrihydrite under advective flow. Geochim Cosmochim Acta. 2003;67:2977–92. [Google Scholar]
- Heiri O, Lotter AF, Lemcke G. Loss on ignition as a method for estimating organic and carbonate content in sediments: reproducibility and comparability of results. J Paleolimnol. 2001;25:101–110. [Google Scholar]
- Henneberry YK, Kraus TEC, Nico PS, Horwarth WR. Structural stability of coprecipitated natural organic matter and ferric iron under reducing conditions. Org Geochem. 2012;48:81–9. [Google Scholar]
- Hochella MF, Moore JN, Putnis CV, Putnis A, Kasama T, Eberl DD. Direct observation of heavy metal-mineral association from the Clark Fork River Superfund Complex: Implications for metal transport and bioavailability. Geochim Cosmochim Acta. 2005;69:1651–63. [Google Scholar]
- Kheboian C, Bauer CF. Accuracy of selective extraction procedures for metal speciation in model aquatic sediments. Anal Chem. 1987;59:1417–23. [Google Scholar]
- Kimball BA, Runkel RL, Gerner LJ. Quantification of mine-drainage inflows to Little Cottonwood Creek, Utah, using a tracer-injection and synoptic-sampling study. Environ Geol. 2001;40:1390–404. [Google Scholar]
- Lachmar TE, Burk NI, Kolesar PT. Groundwater contribution of metals from an abandoned mine to the North Fork of the American Fork River, Utah. Water Air Soil Pollut. 2006;173:103–20. [Google Scholar]
- Lin AYC, Debroux JF, Cunningham JA, Reinhard M. Comparison of rhodamine WT and bromide in the determination of hydraulic characteristics of constructed wetlands. Ecol Eng. 2003;20:75–88. [Google Scholar]
- Luza KV. Stability Problems Associated with Abandoned Underground Mines in the Picher Field Northeastern Oklahoma. Oklahoma Geological Survey; Norman, OK: 1986. Circular 88. [Google Scholar]
- Masue-Slowey Y, Loeppert RH, Fendorf S. Alteration of ferrihydrite reductive dissolution and transformation by adsorbed As and structural Al: Implications for As retention. Geochim Cosmochim Acta. 2011;75:870–86. [Google Scholar]
- Mayes WM, Jarvis AP, Younger PL. Assessing the importance of diffuse mine water pollution: a case study from Durham County, UK.. 9th International Mine Water Association Congress; Oveido, Spain. International Mine Water Association; 2005. pp. 497–505. [Google Scholar]
- Michel FM, Barrón V, Torrent J, Morales MP, Serna CJ, Boily JF, et al. Ordered ferrimagnetic form of ferrihydrite reveals links among structure, composition, and magnetism. Proceedings of the National Academies of Science. 2010;107:2787–92. doi: 10.1073/pnas.0910170107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Day PA, Carroll SA, Waychunas GA. Rock-water interactions controlling zinc, cadmium, and lead concentrations in surface waters and sediments, US Tri-State Mining District. 1. Molecular identification using X-ray absorption spectroscopy. Environ Sci Tech. 1998;32:943–55. [Google Scholar]
- Ostergren JD, Brown GE, Parks GA, Persson P. Inorganic ligand effects on Pb(II) sorption to goethite ([alpha]-FeOOH): II. Sulfate. J Colloid Interf Sci. 2000;225:483–93. doi: 10.1006/jcis.1999.6702. [DOI] [PubMed] [Google Scholar]
- OWRB . Summary of Data Collected by Governor's Tar Creek Task Force Regarding Groundwater Discharge from Abandoned Lead and Zinc Mines of Ottawa County, Oklahoma, December 1979 to March 1981. Oklahoma Water Resources Board, Water Quality Division; 1981. [Google Scholar]
- OWRB . Water Quality Characteristics of Seepage and Runoff at Two Tailings Piles in the Picher Field, Ottawa County, Oklahoma. Tar Creek Field Investigation Task 1.2. Oklahoma Water Resources Board; 1983. [Google Scholar]
- Parkhurst DL. Open-File Report. U.S. Geological Survey; Oklahoma City, OK: 1987. Chemical Analyses of Water Samples from the Picher Mining Area, Northeast Oklahoma and Southeast Kansas. pp. 87–453. [Google Scholar]
- Parkhurst DL, Appelo CAJ. Water-Resources Investigations Report. U.S. Geological Survey; Denver, CO: 1999. User's Guide to PHREEQC (Version 2)-A Computer Program for Speciation, Batch-Reaction, One-Dimensional Transport, and Inverse Geochemical Calculations. pp. 99–4259. [Google Scholar]
- Perret D, Gaillard JF, Dominik J, Atteia O. The diversity of natural hydrous iron oxides. Environ Sci Tech. 2000;34:3540–6. [Google Scholar]
- Plumlee GS. The environmental geology of mineral deposits. Rev Econ Geol. 1999;6A:71–116. [Google Scholar]
- Schaider LA, Senn DB, Brabander DJ, McCarthy KD, Shine JP. Characterization of zinc, lead and cadmium in mine waste: Implications for transport, exposure and bioavailability. Environ Sci Tech. 2007;41:4164–71. doi: 10.1021/es0626943. [DOI] [PubMed] [Google Scholar]
- Stumm W, Morgan JJ. Aquatic Chemistry: Chemical Equilibria and Rates in Natural Waters. John Wiley & Sons; New York, NY: 1996. [Google Scholar]
- Thompson A, Chadwick OA, Rancourt DG, Chorover J. Iron-oxide crystallinity increases during soil redox oscillations. Geochim Cosmochim Acta. 2006;70:1710–27. [Google Scholar]
- Thompson A, Rancourt DG, Chadwick OA, Chorover J. Iron solid-phase differentiation along a redox gradient in basaltic soils. Geochim Cosmochim Acta. 2011;75:119–33. [Google Scholar]
- Tonkin JW, Balistrieri LS, Murray JW. Modeling metal removal onto natural particles formed during mixing of acid rock drainage with ambient surface water. Environ Sci Tech. 2002;36:484–92. doi: 10.1021/es0109085. [DOI] [PubMed] [Google Scholar]
- U.S. Geological Survey . Assessment and Comparison of 1976-77 and 2002 Water Quality in Mineshafts in the Picher Mining District, Northeastern Oklahoma and Southeastern Kansas. Water-Resources Investigations Report. Denver, CO: 2003. pp. 03–4248. [Google Scholar]
- Younger PL, Banwart SA, Hedin RS. Mine Water - Hydrology, Pollution, Remediation. Kluwer Academic Press; Norwell, MA: 2002. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.