

This study identified multiple whole-genome duplication (WGD) events among Brassicaceae species. Remarkably, these events, as well as previously identified WGD events, are synchronized in age, coincident with epoch transitions, adding to the evidence suggesting the environmental instability associated with these transitions favors polyploidy and rapid species diversification.
Abstract
The Brassicaceae (Cruciferae) family, owing to its remarkable species, genetic, and physiological diversity as well as its significant economic potential, has become a model for polyploidy and evolutionary studies. Utilizing extensive transcriptome pyrosequencing of diverse taxa, we established a resolved phylogeny of a subset of crucifer species. We elucidated the frequency, age, and phylogenetic position of polyploidy and lineage separation events that have marked the evolutionary history of the Brassicaceae. Besides the well-known ancient α (47 million years ago [Mya]) and β (124 Mya) paleopolyploidy events, several species were shown to have undergone a further more recent (∼7 to 12 Mya) round of genome multiplication. We identified eight whole-genome duplications corresponding to at least five independent neo/mesopolyploidy events. Although the Brassicaceae family evolved from other eudicots at the beginning of the Cenozoic era of the Earth (60 Mya), major diversification occurred only during the Neogene period (0 to 23 Mya). Remarkably, the widespread species divergence, major polyploidy, and lineage separation events during Brassicaceae evolution are clustered in time around epoch transitions characterized by prolonged unstable climatic conditions. The synchronized diversification of Brassicaceae species suggests that polyploid events may have conferred higher adaptability and increased tolerance toward the drastically changing global environment, thus facilitating species radiation.
INTRODUCTION
Brassicaceae is one of the most diverse plant families, comprising 49 tribes, 321 genera, and over 3660 species (Al-Shehbaz, 2012), including economically important edible and industrial oilseed and vegetable crops as well as highly diverse wild germplasm (Warwick, 2011). The members of this family are distributed worldwide (Lysak and Koch, 2011). Historically, research in the family has focused primarily on the model Arabidopsis thaliana and a few economically important species, such as members of the genera Brassica, Raphanus, and Sinapis. The underutilized wild relatives of crop species within the Brassicaceae offer enormous potential as sources of genetic diversity for agronomic, physiological, and economic traits (Warwick et al., 2009). Yet, many aspects of their genome evolution and the evolutionary trajectories that formed the crucifer species are not well understood.
A sound phylogenetic classification is critical to understand the evolutionary relationships among distantly related crucifer species. Three major phylogenetic lineages (lineages I to III) were proposed in the core Brassicaceae (Beilstein et al., 2006) based on chloroplast NADH dehydrogenase F (ndhF) sequence data. Several subsequent phylogenetic studies performed based on either analysis of single gene sequence data sets (Bailey et al., 2006; Franzke et al., 2009) or supermatrix analysis incorporating five to 10 loci (Bailey et al., 2006; Koch et al., 2007; Lysak et al., 2009) have supported the three lineages, at least on the broad scale. However, resolving deeper nodes and branch points by comparing a larger data set of orthologous sequences is essential to understand intergeneric and intertribal relationships and to improve resolution of the phylogenetic relationships of the major lineages (i.e., the phylogenetic backbone of the family).
Polyploidy (whole-genome duplication [WGD]) has long been recognized as a prominent force driving species evolution and diversification (Doyle et al., 2008; Soltis et al., 2009). Duplication of genomes creates massive levels of genetic redundancy, which is thought to promote evolutionary innovation either through subfunctionalization (partitioning of function; Cusack and Wolfe, 2007) or neofunctionalization (functional diversification; Blanc and Wolfe, 2004b) of duplicated genes. Postpolyploidy gene retention and subsequent genome evolution are also influenced by an interplay of both relative and absolute gene dosage constraints (Birchler and Veitia, 2007; Freeling, 2009; Bekaert et al., 2011; Hudson et al., 2011). Based on the age of WGDs, polyploid species are classified as neo-, meso-, or paleopolyploids (Mandáková et al., 2010b). Neopolyploids are the most recently formed polyploids (for example, Brassica napus), which are characterized by increased genome size, higher chromosome number, redundant gene content, and extant diploid ancestors (Ramsey and Schemske, 2002; Mandáková et al., 2010b). With the passage of time, neopolyploids evolve into mesopolyploids and subsequently into paleopolyploids by undergoing diploidization through gene loss (fractionation) and extensive rearrangement of genetic material (Song et al., 1995; Lynch and Conery, 2000; Wolfe, 2001). Neopolyploids being recent polyploids contain clearly distinguishable subgenomes (Kagale et al., 2014). In comparison, the parental subgenomes in mesopolyploids are only discernible by comparative genetic and genomic approaches (Parkin et al., 2005; Mandáková et al., 2010b; Parkin et al., 2014), which is not possible in paleopolyploids where long-term genome restructuring leads to the assimilation of parental subgenomes (Mandáková et al., 2010b).
Most plant lineages have undergone one or more rounds of recent and/or ancient polyploidy events throughout their evolutionary history (De Bodt et al., 2005; Cui et al., 2006; Soltis et al., 2009). For example, all angiosperms share the remnants of at least two rounds of ancient polyploidy (Jiao et al., 2011; Amborella Genome Project, 2013), and nearly 15% of angiosperm speciation events are estimated to be caused by polyploidization (Wood et al., 2009). Recurrent polyploidization has played a significant role in the evolution of the Brassicaceae (Lysak and Koch, 2011), and nearly half of the crucifer taxa are hypothesized to be of recent polyploid origin (Franzke et al., 2011). The genome of A. thaliana revealed compelling evidence for remnants of at least three paleopolyploidy events, known as α, β, and γ WGDs (Bowers et al., 2003), that are shared by all crucifer taxa (Haudry et al., 2013). Additionally, early comparative genetic mapping (Parkin et al., 2005) and cytogenetic studies (Lysak et al., 2005; Ziolkowski et al., 2006), as well as the recent whole-genome sequencing of Brassica rapa (Wang et al., 2011) have identified an additional later mesopolyploidy (whole-genome triplication) event in diploid Brassicas. Using Bayesian approaches and fossil information as age constraints, the age of the triplication event has now been estimated to be 22.5 million years (Beilstein et al., 2010). Similarly, recent genome sequencing of Leavenworthia alabamica (Haudry et al., 2013), Camelina sativa (Kagale et al., 2014), and Brassica oleracea (Liu et al., 2014; Parkin et al., 2014) have uncovered more recent neo/mesopolyploidy events that formed the basis for the evolution of their hexaploid genomes. Comparative cytogenetic and molecular phylogenetic analyses have unveiled additional mesopolyploidy events in a few Australian and New Zealand crucifer genera belonging to the Microlepidieae and Heliophileae tribes that are endemic to Namibia and South Africa (Joly et al., 2009; Mandáková et al., 2010a, 2010b, 2012), implying a key role for recurring mesopolyploidy events in the diversification of the Brassicaceae. These data and the substantial numerical expansion of the chromosome complement from the base ancestral karyotype across the Brassicaceae suggest that the neo/mesopolyploidy events revealed so far could represent a fraction of the total.
Here, using next-generation sequencing, we assembled gene repertoires from a range of diverse crucifer species and established molecular phylogenetic relationships among these and other fully sequenced Brassicaceae species. We provide insights into Brassicaceae evolution by uncovering neo/mesopolyploid origins of approximately half of these crucifer taxa and establishing coincidental timing of crucifer diversification with geologically significant events that have occurred during the Cenozoic era of the Earth’s history.
RESULTS
Crucifer Taxa Sampling
Nine completely sequenced Brassicaceae species were available for analyses, and data were collected from an additional 14 species, which were selected based on their geographic distribution, evolutionary adaption to niche environments, and their production of secondary metabolites. In total, these species belong to 18 distinct genera, 13 major tribes, and three lineages of the Brassicaceae (Figure 1; Supplemental Tables 1 and 2). They are adapted to varying lengths of growing season (annual, biennial, and perennial; Supplemental Table 1) and are characterized by a high degree of variation in chromosome number and genome size (Table 1; Supplemental Table 2). Most of these species are both edible (used as leafy vegetables, condiments, and oil) and possess medicinal properties (used to treat various common ailments; Supplemental Table 1). Additionally, some of these species have specialized utilities in the production of dyes, preservatives, essential oils, fungicides, insecticides, disinfectants, and soil conditioners (Supplemental Table 1). Collectively, these species represent a broad spectrum of taxonomic, genetic, and physiological variation observed in the Brassicaceae family.
Figure 1.

Molecular Phylogeny of Brassicaceae Species.
A maximum likelihood tree produced from a supermatrix constructed based on 213 orthologous sequences. Clade support values near nodes represent bootstrap proportions in percentages. All unmarked nodes have absolute support. Branch lengths represent estimated nucleotide substitutions per site. Information about tribal and lineage assignments is given on the right.
[See online article for color version of this figure.]
Table 1. Brassicaceae Species Included in This Study and Summary Statistics of Transcriptome Sequencing.
| Crucifer Speciesb,c | Chromosome Number (2n)d | Relative 1C Content (pg)e | Relative Genome Size (Mb)e | Sequencing and de Novo Assemblya |
||
|---|---|---|---|---|---|---|
| Raw Data (Mb) | Clean Data (Mb) | Unigenes | ||||
| Brassicaceae lineage I | ||||||
| Armoracia rusticana | 32 | 0.75 | 738.47 | 104.2 | 99.4 | 32,531 |
| Barbarea verna | 16 | 0.63 | 616.78 | 183.0 | 174.0 | 46,149 |
| Capsella bursa-pastoris | 32 | 0.23 | 198.91 | 220.4 | 196.5 | 29,299 |
| Erysimum cheiri | 14 | 0.24 | 210.22 | 260.0 | 137.5 | 20,776 |
| Lepidium densiflorum | 32 | 0.22 | 192.77 | 284.4 | 162.1 | 24,433 |
| Lepidium meyenii | 64 | 0.85 | 832.07 | 102.9 | 99.0 | 28,872 |
| Lepidium sativum | 24 | 0.75 | 739.09 | 299.6 | 91.2 | 14,372 |
| Brassicaceae lineage II | ||||||
| Cochlearia officinalis | 28 | 0.83 | 814.51 | 156.1 | 146.8 | 26,839 |
| Draba lactea | 32, 48 | 1.60 | 1,573.95 | 268.8 | 236.5 | 33,575 |
| Isatis tinctoria | 14, 28 | 0.84 | 830.57 | 129.9 | 123.4 | 30,803 |
| Pringlea antiscorbutica | 24 | 0.62 | 611.00 | 151.3 | 130.5 | 27,788 |
| Sisymbrium officinale | 14 | 0.83 | 815.81 | 198.7 | 183.6 | 38,819 |
| Stanleya pinnata | 14 | 0.87 | 853.09 | 264.6 | 205.6 | 32,009 |
| Brassicaceae lineage III | ||||||
| Hesperis matronalis | 24 | 1.87 | 1,844.03 | 307.9 | 114.2 | 16,251 |
Complete statistics on transcriptome sequencing and de novo assembly are provided in Supplemental Table 3.
Additional information on each species, including origin, life cycle, and their edible and medicinal uses is provided in Supplemental Table 1.
A list of completely sequenced Brassicaceae species included in this study is provided in Supplemental Table 2.
Based on Warwick and Al-Shehbaz (2006). For each species, most frequently observed chromosome number(s) is presented.
In this study, 1C content was estimated using 4′,6-diamidino-2-phenylindole nuclei staining, with B. napus as the reference sample. The genome sizes were interpolated from a standard curve created using DAPI staining data from seven additional Brassicaceae species with well-established genome sizes (see Methods).
Transcriptome Pyrosequencing of Crucifer Taxa
Whole-genome sequences are available for at least nine of the 23 Brassicaceae species selected in this study (Supplemental Table 2). To assemble gene repertoires for the remaining 14 crucifer species, cDNA libraries were constructed from leaf tissue and sequenced with the Roche 454 GS FLX platform generating on average about half a million reads per species (Table 1; Supplemental Table 3). After filtering low-quality, adapter contaminated, rRNA, and short reads, high-quality reads from each species were assembled de novo as described in Methods. The final assemblies yielded a total of 402,516 unigenes for the 14 crucifer species (Table 1). On average, each species was represented by ∼29,000 unigenes, with a mean length of 600 bp (Table 1; Supplemental Table 3).
Molecular Phylogeny
We compared the assembled unigenes from the crucifer species and gene coding sequences from completely sequenced Brassicaceae species to construct a supermatrix consisting of 213 orthologous genes in a concatenated alignment of 84,727 bp, which was used to define evolutionary relationships among these crucifer species. Maximum likelihood phylogenomic analysis of the supermatrix produced a resolved phylogeny (Figure 1) showing four major groups of taxa. The phylogenetic tree was rooted with Cleome species (included as an outgroup) belonging to Cleomaceae, a sister family of the Brassicaceae. While Aethionema arabicum, a representative of the tribe Aethionemeae, the first diverging group in the family, formed the most basal group, the remaining crucifer species were divided into three well resolved clades: (1) Hesperis matronalis formed an independent clade with 100% bootstrap support; (2) B. rapa, Schrenkiella parvula, Eutrema salsugineum, and six other crucifer species, including Cochlearia officinalis, Draba lactea, Isatis tinctoria, Pringlea antiscorbutica, Sisymbrium officinale, and Stanleya pinnata, formed the second clade; and (3) Arabidopsis species, Camelina sativa, Capsella rubella, and L. alabamica along with seven other crucifers, including Armoracia rusticana, Barbarea verna, Capsella bursa-pastoris, Erysimum cheiri, Lepidium densiflorum, Lepidium meyenii, and Lepidium sativum, constituted the third clade (Figure 1). The pattern of phylogenetic relationships among sampled taxa affirmed previous depictions of the generic and tribal delimitation of the Brassicaceae (Beilstein et al., 2006, 2008; Al-Shehbaz, 2012; Koch et al., 2012). Five tribes representing taxa sampled in this study, including Camelineae, Cardamineae, Eutremeae, Lepidieae, and Thelypodieae, contained multiple representatives; all five of these tribes were recognized as monophyletic entities with absolute support (Figure 1), thus confirming the tribal classification concept employed by Al-Shehbaz (2012), as well as supporting the accuracy of the transcriptome-based supermatrix approach (Rokas et al., 2003; Hittinger et al., 2010) employed in this phylogenetic analysis. There are several tribes in the Brassicaceae (e.g., Arabideae and Cochlearieae) that were not assigned to any of three lineages but are included in the newly introduced expanded lineage II (Franzke et al., 2011). Phylogenetic placement of D. lactea (tribe Arabideae) and C. officinalis (tribe Cochlearieae), adjacent to other lineage II species (Figure 1), clearly establishes their lineage II ancestry.
Identification of Polyploidy Events
The distribution of synonymous substitutions (Ks) along the coding regions of duplicated gene pairs was analyzed to uncover and assess the frequency of polyploidy events in the Brassicaceae. It is generally thought that individual peaks in Ks distributions represent groups of gene pairs with similar synonymous distances and each peak follows a Gaussian (normal) distribution, with each Gaussian component corresponding to a large-scale duplication event (Blanc and Wolfe, 2004a; Schlueter et al., 2004). To account for multiple duplication events within each lineage, mixture model analysis of Ks distributions was performed. Histograms of the distribution of ln (Ks) values fitted with Gaussian mixture models for each species are presented in Figure 2 and Supplemental Figure 1. There is striking evidence of multiple gene duplication events in each of the crucifer species, as reflected by the presence of two to three major peaks in the ln (Ks) distributions (Figure 2; Supplemental Table 4) and multiple Gaussian components revealed by mixture model analysis (Supplemental Data Set 1).
Figure 2.

Age Distribution of Duplicated Genes in Brassicaceae Species.
Gaussian mixture models fitted to frequency distributions of Ks (synonymous substitution) values obtained by comparing pairs of paralogous genes from Brassicaceae species are shown. Histograms of frequency distributions of Ks values for all 23 species are presented in Supplemental Figure 1. The x and y axes are ln(Ks) and the expected frequency in intervals of 0.1 of ln(Ks), respectively.
(A) Crucifer species that have undergone a recent neo/mesopolyploidy event.
(B) Crucifer species without a recent neo/mesopolyploidy event.
(C) Completely sequenced Brassicaceae species.
Paleopolyploidy Events
A scatterplot of the means of the ln (Ks) values against the standard deviations for each Gaussian component of the fitted model for individual crucifer species revealed four distinct clusters (Figure 3A). All crucifer species included in this study have representative values in clusters 2 to 4, suggesting that the polyploidy events associated with each of these clusters have a common origin and these ancestral evolutionary events occurred before the diversification of Brassicaceae clades. The mean Ks values associated with clusters 2 and 3 (0.77 ± 0.016, and 2.05 ± 0.042, respectively; Figure 3A) are consistent with Ks estimates for the α and β paleopolyploidy events, respectively (Figure 3B). Ks distributions of Cleome spinosa and Cleome gynandra also revealed multiple peaks (Supplemental Figure 2). Mixture model analyses recovered three major peaks in both species, with corresponding mean Ks values of 0.39, 1.94, and 3.76. The peak with the lower Ks value of 0.39 represents a previously reported independent mesopolyploidy event (Barker et al., 2009). The peaks at Ks = 1.94 and 3.76 most likely represent the β and γ WGDs, respectively. The Ks distribution for Carica papaya revealed the presence of a peak at Ks = 2.01 (Supplemental Figure 2), which is similar to the Ks value of the β WGD. However, since Carica is evolving at less than half the rate of Brassicaceae and Cleomaceae lineages (Barker et al., 2009), the rate corrected Ks value of this would likely represent the older γ event.
Figure 3.

Major Polyploidy Events Experienced by Brassicaceae Species.
(A) Scatterplot of all Gaussian mixture model parameters from Ks distributions of Brassicales species. The mean of the ln (Ks) value for each Gaussian component of the fitted model were plotted against their sd for each species (colored circles and squares). The diameter of each circle or square is proportional to data fraction (Supplemental Data Set 1). Four independent clusters (large gray circles) represent major meso- and paleopolyploidy events. A table representing cluster average, sd, and estimated age of associated polyploidy events is appended below the scatterplot.
(B) Age distributions of A. thaliana paralogs representing α, β, and γ paleopolyploidy events. Mixture models fitted to Gaussian components in the histograms of frequency distributions of Ks values obtained by comparing pairs of α, β, and γ duplicates (Bowers et al., 2003) are shown.
Cluster 4 (Figure 3A) with a mean Ks value of 8.32 ± 0.48 has a relatively larger standard deviation, which likely reflects slightly skewed Ks estimates caused by saturation of substitutions in duplicate pairs represented in this more ancient WGD. Since this is the only other cluster that is observed in the region beyond the β WGD (cluster 3; Figure 3A) and the mean Ks value of this cluster is consistent with the oldest paleopolyploidy event observed in Arabidopsis (Figure 3B), we believe this represents the γ WGD. Known caveats with inferring WGD events from Ks distributions are the erosion of the ancient WGD signal due to the elimination of a large fraction of duplicated genes and saturation of substitutions, leading to artificial peaks in the Ks distribution beyond a defined threshold of 2 to 2.5 (Vanneste et al., 2013). However, the clear cluster of values at Ks = ∼8.5 (Figure 3A) suggests that by increasing taxonomic and genomic breadth, it is possible to infer paleopolyploidy events from Ks distributions exceeding previously suggested Ks thresholds (Vanneste et al., 2013).
Neo/Mesopolyploidy Events
In addition to the ancient paleopolyploidy events (α, β, and γ WGD), a further peak at Ks = 0.38 (Figure 2; Supplemental Table 4) was found in the B. rapa distribution, representing the now well documented mesopolyploidy event (Parkin et al., 2005; Ziolkowski et al., 2006; Wang et al., 2011). Similarly, mixture model analyses of the Ks distributions of L. alabamica and C. sativa also revealed the presence of a major peak at Ks = 0.33 and 0.09, respectively (Figure 2C), which represent the independent hexaploidy events experienced by these species (Haudry et al., 2013; Kagale et al., 2014). In addition to these events, the major finding of this study was the revelation of further independent neo/mesopolyploidy events in many of the Brassicaceae species (Figure 2A and Cluster 1 in Figure 3A; Supplemental Table 4). The Ks distributions of several crucifer species, including five lineage I species (A. rusticana, C. bursa-pastoris, L. meyenii, L. densiflorum, and L. sativum) and three lineage II species (D. lactea, P. antiscorbutica, and S. pinnata), displayed an additional major peak at Ks = 0.12 to 0.19 (Figure 2A and Cluster 1 in Figure 3A), indicating these species have experienced a further recent neo/mesopolyploidy event. Considering the phylogenetic placement of these crucifer species (Figure 1), these events, at a very minimum, represent five (three in lineage I and two in lineage II) independent neo/mesopolyploidy events: (1) Armoracia event; (2) Capsella event, specific to C. bursa-pastoris as C. rubella did not experience a polyploidy event; (3) Lepidium event, shared by all three Lepidium species; (4) Draba event; and (5) Stanleya-Pringlea event.
Rate Heterogeneity among the Lineages of the Brassicaceae and Cleomaceae
To determine if the age distributions of duplicated genes within the diverse Brassicaceae species are comparable, Ks distributions of orthologs between each of the Brassicaceae species and C. spinosa (outgroup) were compared. Except for C. officinalis, Ks distributions of all other Brassicaceae species were found to be nearly identical (Supplemental Figure 3 and Supplemental Table 5). Furthermore, it was previously shown that the Ks branch lengths of Cleome and Arabidopsis from Gossypium (outgroup) are also nearly identical (Barker et al., 2009). Collectively, these results suggest that both Brassicaceae and Cleomaceae species are evolving with a similar molecular evolutionary rate and, thus, a single synonymous substitution rate of 8.22 × 10−9 substitutions/synonymous site/year for Brassicaceae species, extrapolated using an established age of 22.5 million years ago (Mya) for the whole-genome triplication event in diploid brassicas (Beilstein et al., 2010), is applicable to both Brassicaceae and Cleomaceae-specific polyploidization events.
Ages of Polyploidy Events within the Brassicaceae
Substitutions at synonymous sites, being selectively neutral, should evolve at a rate similar to the mutation rate (Lynch and Conery, 2000) and thus can be used to estimate the age of homolog divergence. Routinely, the age of the mode of secondary peaks in Ks distributions estimated by assuming clock-like rates of synonymous substitution have been used as a proxy for the age of the underlying polyploidy events. However, it should be noted that a secondary peak in a Ks distribution, in particular that representing an allopolyploidy event, likely indicates the timing of the speciation event creating the parental diploids and not their actual merger to form the allopolyploid species. For example, Ks distribution of the allopolyploid B. napus displays the most recent secondary peak at Ks = 0.11 (Figure 4; Supplemental Figure 4), which represents the actual speciation of the diploid progenitors B. rapa and B. oleracea ∼6 Mya rather than the allopolyploidy event that formed B. napus ∼10,000 years ago (Schmidt and Bancroft, 2011). Allopolyploidization may potentially occur immediately or a long time postspeciation. Thus, based on the secondary peaks in Ks distribution, it is impossible to predict the absolute age of allopolyploidy events; however, it can be used as an indirect measure of its maximum age.
Figure 4.

Age Distributions of Duplicated Genes in B. napus.
B. napus genome was reconstituted by combining the genome sequences of B. rapa and B. oleracea. The histogram of frequency distributions of Ks values obtained by comparing pairs of syntenic orthologs from B. rapa and B. oleracea is shown. The red line in the histogram represents mixture model fitted with normal distribution components. The dotted lines represent the individual Gaussian components. The frequency distribution of Ks values obtained by comparing pairs of homoeologs within the actual B. napus genome sequence (obtained from B. Chalhoub; Supplemental Figure 4) produced a peak (Ks = 0.11) identical to that representing speciation of B. rapa and B. oleracea.
Accordingly, based on the geometric mean Ks of peaks and the calibrated synonymous substitution rate for the Brassicaceae, the maximum age of α and β WGDs were estimated at ∼47.1 ± 1.00 and 124.6 ± 2.57 Mya, respectively (Figure 3A). The γ WGD is probably shared by all core eudicots (Bowers et al., 2003; Jaillon et al., 2007; Lyons et al., 2008; Tang et al., 2008; Barker et al., 2009). Considering significant variation in molecular evolutionary rate and lack of precise estimate of nucleotide substitution rate among eudicots, the precise age of the γ WGD could not be established. The recent polyploidy events in the crucifer species were estimated to have occurred within the last 7 to 12 million years (Figure 3A; Supplemental Table 4) and thus are more recent than the mesopolyploidy event experienced by B. rapa.
Major Lineage Separation Events
To assess the relative age of separation of the three lineages within the Brassicaceae, we estimated the level of synonymous substitutions for pairs of orthologous sequences identified between representative species from each lineage of the Brassicaceae (A. thaliana for lineage I, B. rapa for lineage II, and H. matronalis for lineage III; Figure 1). Similar comparisons were performed between A. thaliana, included as a representative of the Brassicaceae, and C. spinosa to determine the relative age of the Brassicaceae-Cleomaceae split point. The Ks distributions of putative orthologous pairs revealed a major peak in each comparison (Figure 5). Based on the mode of Ks values of individual peaks, the ages of lineage I/III–lineage II, lineage I–lineage III, and Brassicaceae–Cleomaceae split points were estimated to be 26.6, 20.8, and 52.6 Mya, respectively (Figure 5).
Figure 5.

Age of Lineage Speciation Events.
(A) Histograms of frequency distributions of Ks values obtained by comparing pairs of orthologous genes among Brassicales species. Mixture models fitted to Gaussian components in the histograms of frequency distributions of Ks values obtained by comparing pairs of orthologous genes are shown.
(B) List of major lineage separation events inferred from each distribution and their estimated ages.
DISCUSSION
Polyploidy is recognized as an important mechanism of plant diversification as it can lead to the creation of new species and have profound effects on subsequent lineage evolution (Wood et al., 2009). Brassicaceae, being one of the most species-rich families, has experienced recurrent polyploidization events during its evolution. The three well-known ancient α, β, and γ paleopolyploidization events that have marked the evolutionary history of the Brassicaceae were initially thought to have occurred around 14.5 to 86, 170 to 235, and 300 Mya, respectively (Bowers et al., 2003). Several subsequent attempts at determining the precise age of these ancient polyploidy events and their relative position in the context of Brassicaceae phylogeny have provided ambiguous estimates (Lynch and Conery, 2000; Blanc et al., 2003; Ermolaeva et al., 2003; De Bodt et al., 2005; Schranz and Mitchell-Olds, 2006; Ming et al., 2008; Tang et al., 2008; Barker et al., 2009). A limitation of many of these previous analyses has been the lack of extensive taxonomic breadth, as many of these studies focused only on A. thaliana. In some studies, multiple species were included, but these analyses were limited by the lack of large sequence data sets and differences in the source of data sets across species.
This study tried to overcome these limitations by including multiple Brassicaceae species and increasing the gene coverage for each species via next-generation sequencing of their transcriptomes. Increased taxonomic breadth and sequence resources facilitated successful identification of the ancient α and β WGDs and estimation of their age of occurrence and their appropriate positioning in the broader context of the Brassicales phylogeny (Figure 6). Our estimates of the age of α and β WGDs, 47 and 124 million years, respectively, are slightly older than some previous reports, but are within 95% confidence interval overlap of estimates from another independent study (Patrick Edger, personal communication). The presence of α, β, and γ WGDs in all sampled crucifer species indicates that these events occurred prior to the diversification of the Brassicaceae family. The lack of an α WGD in Cleome species confirms its Brassicaceae specific prevalence and the fact that the β WGD is shared by Brassicaceae and Cleomaceae species but not by C. papaya indicates its occurrence in the most common recent ancestor of Brassicaceae-Cleomaceae after the divergence from Caricaceae.
Figure 6.

Speciation, Polyploidy, and Lineage Separation Events in the Brassicaceae Presented in Phylogenetic Context.
Brassicaceae chronogram adapted from Beilstein et al. (2010) was slightly modified by adjusting the branch lengths to reflect the ages of major polyploidy and lineage separation events. The positions of the well-known ancient α and β paleopolyploidy events in relation to various lineage divergence events are indicated. The positions of recent speciation and subsequent neo/mesopolyploidy events experienced by various Brassicaceae and a Cleomaceae species are indicated with dark colored ovals. The ages of previously inferred mesopolyploidy events in Australian Brassicaceae species (indicated with red colored asterisks) were recalculated based on the synonymous substitution values of the three nuclear genes reported by Mandáková et al. (2010a) and the calibrated synonymous substitution rate of 8.22 × 10−9. Numbers in parenthesis indicate estimated age of the speciation of parental diploids of each neo/mesopolyploid species. The actual neo/mesopolyploidization in these species may have occurred anytime between the age of speciation mentioned in brackets and the current day. E. Cretaceous, early Cretaceous; L. Cretaceous, late Cretaceous; Pal, Paleocene; Oligo, Oligocene; Pl, Pliocene; Q, Quarternary.
The Brassicaceae family evolved from other eudicot members at the beginning of the Cenozoic era (∼50 Mya; Figure 6); however, substantial diversification of the family occurred only during the Neogene period (23 Mya to the present). Notably, the three lineages of Brassicaceae diverged during the Oligocene-Miocene transition (Figure 6), which is characterized by a major transient glaciation event, Mi-1, and deep-sea cooling (Miller et al., 1991; Zachos et al., 2001b). These data suggest the split between lineage I and II (27 Mya) occurred before the split between lineage I and III (20 Mya). However, the phylogenetic analysis (Figure 1) suggested that lineage I is sister to lineage II. This discrepancy could be due to the inclusion of only a single species from lineage III. Future sampling of additional species from lineage III as well as the paraphyletic expanded lineage II (Franzke et al., 2011) is essential to fully resolve the split points between the three lineages of the Brassicaceae. The genomes and transcriptomes of many Brassicaceae species are currently being sequenced through various community sequencing programs, which when available will aid in increasing the genomic depth and improving family-wide phylogenetic resolution.
The prevalence of neo/mesopolyploidy in 11 out of the 23 (50%) diverse crucifer genera (belonging to both lineages I and II; Figures 1 and 6) analyzed in this study suggests that polyploidization is a widespread phenomenon among the Brassicaceae and may have profoundly influenced its species evolution and diversification. In the absence of full genome sequences, characterization of recent polyploids as neo- or mesopolyploids can be inferred based on elevated chromosome number and ploidy status. Those species whose chromosome number reflects a simple duplication of the base chromosome number can be classified as a neopolyploid. In contrast, mesopolyploidy is implicated when species show substantially different chromosome counts from a simple multiple ploidy event, suggestive of extensive reduction in the chromosome number through rearrangement of ancestral chromosome blocks during diploidization. Of the 11 crucifer species that have experienced recent polyploidy events, at least three species, including A. rusticana, C. bursa-pastoris, and L. densiflorum, with the chromosome number and ploidy level of 2n=4x=32 (Table 1; Warwick and Al-Shehbaz, 2006) are neotetraploids. Similarly, D. lactea (2n=2x,4x,6x=16,32,48; Grundt et al., 2005) and L. meyenii (2n=8x=64; Toledo et al., 1998) can be classified as high neopolyploids. Based on cytological, morphological, and biogeographical analyses, C. bursa-pastoris was reported to be an autotetraploid species (Hurka et al., 2012). Recent genome sequencing of C. sativa has also revealed its neopolyploid nature (Kagale et al., 2014). On the other hand, comparative genomic, cytogenetic, and molecular phylogenetic analyses have confirmed the mesopolyploid nature of crucifer species belonging to the Brassiceae (B. rapa, 2n=20), Microlepidieae (Ballantinnia species and Stenopetalum species), and Heliophileae tribes (Mandáková et al., 2010b, 2012; Wang et al., 2011). Similarly, the lower chromosome count in S. pinnata (2n=14) and P. antiscorbutica (2n=24) suggests their mesopolyploid nature. Although interpretation of the nature of polyploidization based on chromosome count and ploidy status is pragmatic, future genome sequencing of these crucifer species combined with comparative genetic mapping and cytogenetic studies will shed further light on the evolutionary origin and mode of the underlying polyploidization events.
Interestingly, the timing of speciation events forming parental diploids of the uncovered neo/mesopolyploidy events are clustered (Cluster 1 in Figure 3A) at around 10.1 ± 0.40 Mya and thus coincide with the late Miocene era or the Miocene-Pliocene boundary (Figure 6). The Miocene epoch (23.03 to 5.3 Mya) was dominated by warmer global climates, which gradually intensified during the mid to late Miocene, causing the expansion of seasonal aridity (Zachos et al., 2001a; Fortelius et al., 2006; Eronen et al., 2009; Liu et al., 2009). Furthermore, the desiccation of the Mediterranean Sea took place around the Miocene-Pliocene boundary, leading into the geologically catastrophic Messinian salinity crisis (Hsu et al., 1977) when further major climatic changes were experienced. The emergence of the core Oleracea lineage (that includes B. rapa and B. oleracea; 4.2 to 9.3 Mya), and diversification of Crambe and Vella (∼7 Mya) also coincided with the Messinian salinity crisis (Arias et al., 2014). The dramatically increased aridity and salinity during this period had considerable impact on land vegetation, triggering forest cover decline and radiation of the seasonally adapted taxa (Janis, 1993; Cerling et al., 1997; Fortelius et al., 2006). The climatic deterioration during late Miocene may have favored speciation and subsequent polyploidization among Brassicaceae species.
Influence of climatic variations during epoch transitions on speciation and polyploidization in the Brassicaceae is also evident at the Paleocene-Eocene boundary. Notably, the Brassicaceae specific α paleopolyploidization (∼47 Mya) and the split between the Brassicaceae and Cleomaceae (∼52 Mya) coincide with the late Paleocene-Eocene Thermal Maximum (PETM; ∼56 Mya; Figure 6), during which the global temperatures increased by 5 to 8°C (Zachos et al., 2001a). The abrupt global warming during PETM combined with the water stress caused by seasonal precipitation and higher evapotranspiration had a dramatic effect on plant taxa, as angiosperm diversity increased rapidly during and post PETM (Jaramillo et al., 2010; McInerney and Wing, 2011). Coincidence of the widespread speciation, polyploidy, and lineage divergence events within the Brassicaceae with epoch boundaries implies that the rapid climatic changes, including fluctuating temperatures, desiccation, aridity, and salinity, during these major geological events influenced the evolution and diversification of crucifer species.
Historically, there have been two strikingly contrasting perspectives on the importance of WGDs, either regarding polyploidy as an evolutionary dead end (Stebbins, 1950; Wagner, 1970; Soltis and Burleigh, 2009) or as a guarantee of evolutionary success (Levin, 1983; Fawcett et al., 2009; te Beest et al., 2011; Vanneste et al., 2014). Polyploidy has both costs and benefits, and under stable environmental conditions, polyploidy is considered to be disadvantageous due to the reduced fitness of polyploid individuals caused by their reproductive isolation and lower fertility (Comai, 2005). The lower speciation/diversification rates and higher extinction rates observed in neopolyploids compared with diploids (Mayrose et al., 2011) could be due to sampling errors and methodological shortcomings (Soltis et al., 2014) or may also be a consequence of the outweighing costs of polyploidy associated genomic and phenotypic instability, as well as the reproductive disadvantages under normal conditions. However, the adaptive advantages of polyploidy caused by enhanced genetic repertoire (resulting from increased heterozygosity, the buffering effect of gene redundancy on mutations, neofunctionalization, differential expression, or epigenetic reprogramming of duplicated genes) and reproductive plasticity (facilitation of reproduction through self-fertilization or asexual means) would be expected to confer a competitive advantage to polyploid species under extreme and unstable environmental conditions (Comai, 2005; Fawcett and Van de Peer, 2010). Indeed, polyploid species, owing to their better adaptability to adverse environmental conditions and extreme habitats, were proposed to have survived the catastrophic events leading to the Cretaceous-Tertiary extinction event (Fawcett et al., 2009). However, the findings of Fawcett et al. (2009) have proved to be controversial due to restricted taxonomic sampling and some inherent limitations of the methods employed for identifying and ageing polyploidy events (Soltis and Burleigh, 2009). In a recent report, the limitations of Fawcett et al. (2009) were addressed by analyzing 41 taxonomically diverse plant species and employing more sophisticated dating analyses (Vanneste et al., 2014). In accordance with the earlier findings, this study demonstrated a strong nonrandom clustering of WGDs around the Cretaceous-Paleogene boundary (Vanneste et al., 2014). Our findings aligning speciation, as well as neo-, meso-, and paleopolyploidization events among Brassicacaeae species with multiple epoch boundaries (Figure 6) confirm the clustering of polyploidy events in association with geologically significant events and affirm the importance of environmental conditions in the promotion of polyploidization and consequent adaptive evolution. The enormous taxonomic and physiological diversity observed among crucifer species and their adaptability to a wide range of extreme habitats is a consequence of the documented environmental fluctuations during the latter part of the Cenozoic era. The confluence of species divergence with dramatic changes in the Earth’s climate provides evidence for the influence of polyploidy in shaping plant families in the face of extreme stresses.
METHODS
Plant Material
With the exception of Armoracia rusticana (wild genotype collected from Saskatchewan) where a root explant was collected, seed were collected from each species analyzed in this study. Except for the following, all seed were provided by Plant Gene Resources of Canada (http://pgrc3.agr.gc.ca/): Arabidopsis thaliana (Col-4, www.arabidopsis.org), Barbarea verna, Erysimum cheiri, and Hesperis matronalis (Richters Seeds), Brassica napus (DH12075, AAFC), Brassica oleracea (TO1000, Tom Osborn, University of Wisconsin), Brassica rapa (PS270, AAFC), Camelina sativa (Lindo, Rod Snowdon, University of Giessen, Germany), Pringlea antiscorbutica (Australian National Botanical Garden, Canberra, Australia), and Stanleya pinnata (Plants of the Southwest, Santa Fe, NM). Seed from each species were sown in a soilless potting mix and germinated at 20°C. All plants were grown in a Conviron growth chamber providing ∼200 μE of light with a 12-h photoperiod at 20°C for 3 weeks and leaf material was harvested for transcript analysis.
Flow Cytometric Estimation of Genome Size
Nuclei were extracted from young leaf tissue from each of the plant species following the protocol described by Galbraith et al. (1983) with minor alterations. Approximately 2 cm2 of tissue was chopped in 0.5 mL of lysis buffer (0.1 M Tris-HCl, pH 7, 2 mM MgCl2, 0.1 M NaCl, and 0.05% Triton) at room temperature. The volume of buffer was adjusted to 1 mL and filtered through a 30-μm pore prior to analysis. DNA content of the nuclei from each species was estimated using 4′,6-diamidino-2-phenylindole (CyStain DNA 2 step; Partec) staining and fluorescence measurements made using the CyFlow Ploidy analyzer (Partec). The fluorescence obtained from B. napus (DH12075) was used as an external reference to standardize all other measurements; this reference was resampled after every third unknown measurement to reduce errors due to random instrument drift. A standard curve was generated by linear regression using the standardized fluorescence data and genome size estimates for the genomes of A. thaliana (130 Mb), Brassica carinata (1285 Mb), Brassica juncea (1070 Mb), B. napus (1128 Mb), Brassica nigra (638 Mb), B. oleracea (697 Mb), and B. rapa (529 Mb). The genome size for each of the remaining species was interpolated using their fluorescence measurement and the standard curve.
RNA Extraction and mRNA Enrichment
Total cellular RNA was extracted using the ToTALLY RNA kit (Ambion) according to the manufacturer’s instructions. The integrity and quantity of total RNA were assessed using the RNA 6000 Nano labchip on a BioAnalyzer (Agilent). Poly(A)+ RNA was purified from DNase I-treated total RNA samples by two sequential rounds of oligo(dT) enrichment using the Poly(A) Purist mRNA isolation kit (Ambion). The concentration of mRNA was checked using Ribogreen (Molecular Probes) and the quality was verified using RNA 6000 Pico labchip on a BioAnalyzer (Agilent).
cDNA Library Construction and 454 Sequencing
cDNA library construction and sequencing was performed according to standard Roche 454 FLX and Titanium protocols. Briefly, mRNA was fragmented with a zinc chloride based RNA fragmentation solution. Double-stranded (ds)-cDNA was synthesized from purified fragmented mRNA using Roche’s cDNA synthesis system and random hexamer primers. Subsequent steps, including end-polishing of ds-cDNA, adapter ligation, removal of smaller fragments, and quality and quantity assessment of the final library, were performed as per standard instructions provided in cDNA rapid library preparation guide (Roche). For sequencing, adapter-ligated DNA fragments were denatured to generate a single stranded library, which was then amplified by emulsion PCR. Individual titrated single strand DNA libraries were sequenced in one half-plate run on the 454 GS-FLX platform using Titanium chemistry. For each species, on average about half a million reads were generated, for a total of ∼3.1 Gb of sequence data (Table 1; Supplemental Table 3).
De Novo Sequence Assembly
A rigorous read preprocessing pipeline was adapted, utilizing in turn (1) the in-built quality, adapter, and length trimming tool in the CLCbio genomics workbench (http://www.clcbio.com); (2) CD-HIT-454 read clustering program (Li and Godzik, 2006); and (3) the SILVA comprehensive rRNA database (Quast et al., 2013). Briefly, standard flowgram files (SFF) generated by 454 sequencing were imported into the CLCbio genomics workbench 4.7 and reads were trimmed, by removing adapter sequences, ambiguous nucleotides, and low-quality sequence, using default settings. Filtered reads were clustered at 100% identity using CD-HIT-454 (Li and Godzik, 2006), and identical PCR duplicates were removed. Any reads shorter than 100 bp were also removed. Subsequently, rRNA reads were identified based on significant nucleotide similarity (BLASTN with E-value cutoff of 1E-40, identity >85% and match length >50 bp) (Altschul et al., 1990) to the SILVA rRNA database (Quast et al., 2013) and discarded.
The clean, unique, and nonribosomal reads were assembled using the 454 GS de novo Assembler software (Version 2.6). An incremental transcriptome (-cDNA flag) assembly approach was implemented through a command line interface with parameters, including isogroup threshold (-ig) of 10,000, isotig threshold (-it) of 10,000, isotig contig count threshold (-icc) of 200, minimum overlap length (-ml) of 125, and minimum identity (-mi) of 98%. Additionally, the use read tips (-urt) option that helps in obtaining contigs from rare or low coverage transcripts was activated. For each assembly, the 454 GS de novo Assembler generated isogroup (analogous to genes) and corresponding isotig (analogous to individual transcripts) sequences. Different isotigs within an isogroup are considered to represent alternative splice variants of the same gene. In almost all transcript assemblies, there was substantial redundancy among isotigs belonging to a subset of isogroups, some of which were nearly identical. The unusually inflated number of isotigs per isogroup is likely an artifact of the greedy assembly algorithm. To avoid biases in downstream analyses due to redundancy, only one isotig (longest) per isogroup was selected for subsequent phylogenetic and Ks analyses. The assemblies yielded a total of 402,516 unigenes for the 14 crucifer species (Supplemental Table 3).
The 454 GS de novo assembler trims the poly(A) tails prior to assembling, so the true orientation of reads in the assembly cannot be determined. The orientation of isotigs from each species was determined by aligning (BLASTX with E-value cutoff of 1E-20) them against the A. thaliana proteome database, and improperly oriented isotigs were reverse complemented.
Annotation of Open Reading Frames in Isotig Sequences
The open reading frame for each isotig sequence was deduced by translating it with GeneWise2.2.0 (Birney et al., 2004) using the corresponding best-match protein from the A. thaliana proteome database as a guide. From the highest scoring GeneWise DNA–protein alignment, ambiguous and frameshift sites were removed, and the protein coding isotig sequence and corresponding translated amino acid sequence were retrieved. For each species, the ORFeome and proteome data sets were then created by grouping corresponding isotig and protein sequences, respectively.
Cleome cDNA Sequencing Data Sets
Sequencing data for Cleome spinosa and Cleome gynandra were obtained from the NCBI short read archive (www.ncbi.nlm.nih.gov/sra; accessions SRR015531.sra and SRR015532.sra). Standard flowgram files were generated from archived SRA format using the SFF-dump tool within the NCBI SRA toolkit. Preassembly cleanup and de novo assembly of reads was performed as described above.
Construction of Orthologous Data Matrix and Phylogenetic Analysis
Individual isotig sequences from each crucifer species and coding DNA sequences of the fully sequenced Brassicaceae species were mapped (BLASTN [Altschul et al., 1990] with E-value cutoff of 1E-06) onto full-length coding DNA sequence of A. thaliana, which was selected as the reference genome. The reciprocal best BLAST hit method was used to determine putative orthologs between A. thaliana and each of the crucifer species. Based on this analysis, a phylogenomic data matrix consisting of 213 unique orthologous gene sets was constructed. Sequences from individual orthologous gene sets were locally aligned using ClustalW (Larkin et al., 2007). Gaps and missing data from each alignment were removed using an automated alignment trimming tool trimAL (Capella-Gutiérrez et al., 2009) with a gap threshold (-gt) value set to 1. Trimmed sequences were realigned and the alignments of 213 gene sets were concatenated using the Phyutility program (Smith and Dunn, 2008) to produce the final data matrix comprising a total alignment length of 84,727 bp.
Phylogenetic analysis was performed using the optimality criteria of maximum likelihood implemented in RAxML (Stamatakis, 2006). The maximum likelihood tree was calculated assuming GTR+GAMMA model of sequence evolution. Robustness of phylogenetic inference was assessed by running 1000 bootstrap replicates using GTR+CAT approximation. Final tree was visualized using the interactive Tree of Life (Letunic and Bork, 2011) Web server. A text file of alignment information is presented in Supplemental Data Set 2.
Identification of Paralogs and Orthologs
Paralogous genes within each crucifer species were identified by performing an all against all protein sequence similarity (BLASTP with an E-value cutoff of 1E-20) search. Putative orthologs between A. thaliana and individual crucifer species were identified by using a reciprocal best BLAST hit method, as described above. Only those paralogous or orthologous sequence pairs that aligned over a length of at least 150 amino acids and showed a sequence identity >60% were used for Ks estimation.
Ks Analysis
Estimation of the level of synonymous substitution between paralogous and orthologous pairs was performed according to the procedure described by Blanc and Wolfe (2004a). For each pair of paralogs or orthologs, protein sequences were aligned using ClustalW (Larkin et al., 2007). The resulting protein alignments were used to produce the corresponding nucleotide alignments using PAL2NAL (Suyama et al., 2006). Ks values for each sequence pair were calculated based on codon alignments using the maximum likelihood method implemented in codeml of the PAML package (Yang, 2007) under the F3x4 model (Goldman and Yang, 1994). Synonymous substitution values for C. sativa were adapted from Kagale et al. (2014).
Ks Analysis of Leavenworthia alabamica
Coding sequences of representative gene models of another recently sequenced lineage I Brassicaceae species L. alabamica (Haudry et al., 2013) were obtained from the UCSC Genome Browser (http://mustang.biol.mcgill.ca:8885). Paralogous genes were identified by performing an all against all nucleotide sequence similarity (BLASTN with an E-value cutoff of 1E-20) search. Ks analysis was performed as described above.
Mixture Model Analysis of Ks Distributions
The Ks data sets for each species were trimmed by removing values of 0.001 or less because they are affected by rounding errors and result in spurious frequency peaks. Histograms were generated using log transformed Ks values with a bin width of 0.1. To identify significant peaks in the ln (Ks) distribution, Gaussian mixture models were fitted to the ln (Ks) values using the R package Mclust, and the number of Gaussian components (G), the mean of each component and corresponding variance, sd, and fractions of data were calculated. The Bayesian Information Criterion was used to determine the best fitting model to the data; however, in many cases under-fitting was observed. The number of Gaussian components was increased to improve the fit where necessary, except in one case (A. thaliana) where the value of G was reduced from 9 to 7. In all the data sets analyzed, there was a peak observed in the distribution of Ks values near 70, with near-identical mean and variance parameters for each species. This represents an uninformative artifact that did not contribute to the model. All Ks data were included to develop the best fitting model and this allowed the detection of weak Gaussian components with means up to ∼20. Ks values beyond 20 are difficult to meaningfully interpret and we restricted our analyses to this boundary.
The fit of the determined models were confirmed by χ2 tests. The upper limit of ln (Ks) = 3 in the χ2 calculation was used to cut off the Ks values beyond ∼20. The number of degrees of freedom for the model was estimated as 3(G-1) for the single species data sets and 3G for the interspecific comparisons accounting for the number of parameters that significantly affect the model for Ks <20.
The developed models for each species fitted well (smallest P value = 0.02; observed from 17 analyses) with the exception of fully sequenced B. rapa, A. thaliana, A. lyrata, L. alabamica, C. sativa, C. rubella, A. arabicum, S. parvula, and E. salsugineum. A summary plot of the means of the ln (Ks) values against the sd of the ln (Ks) values for each Gaussian component detected in the fitted model for each species is presented (Figure 3A) with the upper limit set to Ks = 20. In specific instances, skewed peaks were observed in the distribution of Ks values, this deviation resulted from the combined effects of multiple overlapping Gaussian components. These complications were resolved by combining overlapping Gaussian components and representing them with the mean and sd of their Gaussian mixtures. Thus, for a Gaussian mixture distribution 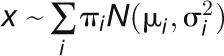 with proportions
with proportions 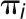 that sum to 1, with means
that sum to 1, with means 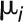 , and standard deviations
, and standard deviations 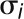 , we determine the combined mean as
, we determine the combined mean as 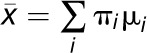 and the combined sd as
and the combined sd as 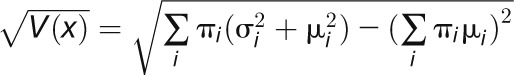 enabling these peaks to be plotted as single points in Figure 3. The se of the mean (SD(m)) of ln (Ks) for each cluster was determined as the sd of the values within each cluster divided by
enabling these peaks to be plotted as single points in Figure 3. The se of the mean (SD(m)) of ln (Ks) for each cluster was determined as the sd of the values within each cluster divided by 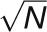 where N is the cluster size. These were converted into their corresponding se of Ks by
where N is the cluster size. These were converted into their corresponding se of Ks by 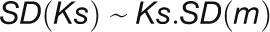 where
where  , and the
, and the 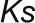 values are presented as
values are presented as 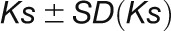 . However, it is likely that these standard deviations are underestimated due to the uncertainty in the limits of the clusters, the number of Gaussian component selected, and effect of merging overlapping Gaussian components.
. However, it is likely that these standard deviations are underestimated due to the uncertainty in the limits of the clusters, the number of Gaussian component selected, and effect of merging overlapping Gaussian components.
Accession Numbers
Sequence data from this article can be found in the GenBank/EMBL data libraries under the following accession numbers: A. rusticana (SRX494797), B. verna (SRX494798), C. bursa-pastoris (SRX494799), C. officinalis (SRX494800), D. lactea (SRX494801), E. cheiri (SRX494802), I. tinctoria (SRX494805), H. matronalis (SRX494803), L. densiflorum (SRX494806), L. meyenii (SRX494807), L. sativum (SRX494809), P. antiscorbutica (SRX494810), S. officinale (SRX494813), and S. pinnata (SRX494815).
Supplemental Data
The following materials are available in the online version of this article.
Supplemental Figure 1. Histograms of Frequency Distributions of Ks Values Obtained by Comparing Pairs of Paralogous Genes of 23 Brassicaceae Species.
Supplemental Figure 2. Age Distributions of Duplicated Genes in Cleomaceae and Caricaceae Species.
Supplemental Figure 3. Combined Ks Distribution Plot of Brassicacaeae versus Cleomaceae Species.
Supplemental Figure 4. Age Distribution of Homoeologous Genes in Brassica napus.
Supplemental Table 1. Information on the Origin, Life Cycle, and Edible and Medicinal Uses of the Brassicaceae Species Included in This Study.
Supplemental Table 2. Completely Sequenced Brassicaceae Genomes Included in This Study.
Supplemental Table 3. Roche 454 Pyrosequencing, Read Filtering, and Assembly Statistics for Crucifer Transcriptomes.
Supplemental Table 4. Estimated Ages of the Inferred Major WGD Events in Brassicaceae Species.
Supplemental Table 5. Pairwise Comparison of Orthologous Ks Distributions between Cleome spinosa and Diverse Brassicaceae Species.
Supplemental Data Set 1. Complete Mixture Model Estimates of Ks Distributions.
Supplemental Data Set 2. Final Concatenated Alignment (Phylip Format) of 213 Orthologous Sequences Used for Generating the Phylogenetic Tree Presented in Figure 1.
Supplementary Material
Acknowledgments
The research was funded through awards from the National Research Council Canada (NRC) Natural Products Genomics Network program and the Agriculture and Agri-Food Canada Canadian Crop Genomics Initiative. We thank members of the NRC DNA Technologies and Bioinformatics Laboratories for technical assistance.
AUTHOR CONTRIBUTIONS
R.X., T.H., and J.C. generated material and sequence data. D.K. collected germplasm. J.N. carried out the statistical analyses. W.E.C. and M.G.L. performed bioinformatic analyses. P.P.E. independently confirmed the data analyses. S.K., S.J.R., A.G.S., and I.A.P.P. designed the study, analyzed the data, and wrote the article. All authors discussed the results and commented on the article.
Glossary
- WGD
whole-genome duplication
- Mya
million years ago
- PETM
Paleocene-Eocene Thermal Maximum
Footnotes
Some figures in this article are displayed in color online but in black and white in the print edition.
Online version contains Web-only data.
Articles can be viewed online without a subscription.
References
- Al-Shehbaz I.A. (2012). A generic and tribal synopsis of the Brassicaceae (Cruciferae). Taxon 61: 931–954. [Google Scholar]
- Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. (1990). Basic local alignment search tool. J. Mol. Biol. 215: 403–410. [DOI] [PubMed] [Google Scholar]
- Amborella Genome Project (2013). The Amborella genome and the evolution of flowering plants. Science 342: 1241089. [DOI] [PubMed] [Google Scholar]
- Arias T., Beilstein M.A., Tang M., McKain M.R., Pires J.C. (2014). Diversification times among Brassica (Brassicaceae) crops suggest hybrid formation after 20 million years of divergence. Am. J. Bot. 101: 86–91. [DOI] [PubMed] [Google Scholar]
- Bailey C.D., Koch M.A., Mayer M., Mummenhoff K., O’Kane S.L., Jr, Warwick S.I., Windham M.D., Al-Shehbaz I.A. (2006). Toward a global phylogeny of the Brassicaceae. Mol. Biol. Evol. 23: 2142–2160. [DOI] [PubMed] [Google Scholar]
- Barker M.S., Vogel H., Schranz M.E. (2009). Paleopolyploidy in the Brassicales: analyses of the Cleome transcriptome elucidate the history of genome duplications in Arabidopsis and other Brassicales. Genome Biol. Evol. 1: 391–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beilstein M.A., Al-Shehbaz I.A., Kellogg E.A. (2006). Brassicaceae phylogeny and trichome evolution. Am. J. Bot. 93: 607–619. [DOI] [PubMed] [Google Scholar]
- Beilstein M.A., Al-Shehbaz I.A., Mathews S., Kellogg E.A. (2008). Brassicaceae phylogeny inferred from phytochrome A and ndhF sequence data: tribes and trichomes revisited. Am. J. Bot. 95: 1307–1327. [DOI] [PubMed] [Google Scholar]
- Beilstein M.A., Nagalingum N.S., Clements M.D., Manchester S.R., Mathews S. (2010). Dated molecular phylogenies indicate a Miocene origin for Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 107: 18724–18728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekaert M., Edger P.P., Pires J.C., Conant G.C. (2011). Two-phase resolution of polyploidy in the Arabidopsis metabolic network gives rise to relative and absolute dosage constraints. Plant Cell 23: 1719–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birchler J.A., Veitia R.A. (2007). The gene balance hypothesis: from classical genetics to modern genomics. Plant Cell 19: 395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birney E., Clamp M., Durbin R. (2004). GeneWise and Genomewise. Genome Res. 14: 988–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc G., Wolfe K.H. (2004a). Widespread paleopolyploidy in model plant species inferred from age distributions of duplicate genes. Plant Cell 16: 1667–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc G., Wolfe K.H. (2004b). Functional divergence of duplicated genes formed by polyploidy during Arabidopsis evolution. Plant Cell 16: 1679–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc G., Hokamp K., Wolfe K.H. (2003). A recent polyploidy superimposed on older large-scale duplications in the Arabidopsis genome. Genome Res. 13: 137–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers J.E., Chapman B.A., Rong J., Paterson A.H. (2003). Unravelling angiosperm genome evolution by phylogenetic analysis of chromosomal duplication events. Nature 422: 433–438. [DOI] [PubMed] [Google Scholar]
- Capella-Gutiérrez S., Silla-Martínez J.M., Gabaldón T. (2009). trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25: 1972–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerling T.E., Harris J.M., MacFadden B.J., Leakey M.G., Quade J., Eisenmann V., Ehleringer J.R. (1997). Global vegetation change through the Miocene/Pliocene boundary. Nature 389: 153–158. [Google Scholar]
- Comai L. (2005). The advantages and disadvantages of being polyploid. Nat. Rev. Genet. 6: 836–846. [DOI] [PubMed] [Google Scholar]
- Cui L., et al. (2006). Widespread genome duplications throughout the history of flowering plants. Genome Res. 16: 738–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cusack B.P., Wolfe K.H. (2007). When gene marriages don’t work out: divorce by subfunctionalization. Trends Genet. 23: 270–272. [DOI] [PubMed] [Google Scholar]
- De Bodt S., Maere S., Van de Peer Y. (2005). Genome duplication and the origin of angiosperms. Trends Ecol. Evol. (Amst.) 20: 591–597. [DOI] [PubMed] [Google Scholar]
- Doyle J.J., Flagel L.E., Paterson A.H., Rapp R.A., Soltis D.E., Soltis P.S., Wendel J.F. (2008). Evolutionary genetics of genome merger and doubling in plants. Annu. Rev. Genet. 42: 443–461. [DOI] [PubMed] [Google Scholar]
- Ermolaeva M.D., Wu M., Eisen J.A., Salzberg S.L. (2003). The age of the Arabidopsis thaliana genome duplication. Plant Mol. Biol. 51: 859–866. [DOI] [PubMed] [Google Scholar]
- Eronen J.T., Ataabadi M.M., Micheels A., Karme A., Bernor R.L., Fortelius M. (2009). Distribution history and climatic controls of the Late Miocene Pikermian chronofauna. Proc. Natl. Acad. Sci. USA 106: 11867–11871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawcett J.A., Van de Peer Y. (2010). Angiosperm polyploids and their road to evolutionary success. Trends Ecol. Evol. 2: e3. [Google Scholar]
- Fawcett J.A., Maere S., Van de Peer Y. (2009). Plants with double genomes might have had a better chance to survive the Cretaceous-Tertiary extinction event. Proc. Natl. Acad. Sci. USA 106: 5737–5742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortelius M., Eronen J., Liu L., Pushkina D., Tesakov A., Vislobokova I., Zhang Z. (2006). Late miocene and pliocene large land mammals and climatic changes in Eurasia. Palaeogeogr. Palaeoclimatol. Palaeoecol. 238: 219–227. [Google Scholar]
- Franzke A., German D., Al-Shehbaz I.A., Mummenhoff K. (2009). Arabidopsis family ties: molecular phylogeny and age estimates in Brassicaceae. Taxon 58: 425–437. [Google Scholar]
- Franzke A., Lysak M.A., Al-Shehbaz I.A., Koch M.A., Mummenhoff K. (2011). Cabbage family affairs: the evolutionary history of Brassicaceae. Trends Plant Sci. 16: 108–116. [DOI] [PubMed] [Google Scholar]
- Freeling M. (2009). Bias in plant gene content following different sorts of duplication: tandem, whole-genome, segmental, or by transposition. Annu. Rev. Plant Biol. 60: 433–453. [DOI] [PubMed] [Google Scholar]
- Galbraith D.W., Harkins K.R., Maddox J.M., Ayres N.M., Sharma D.P., Firoozabady E. (1983). Rapid flow cytometric analysis of the cell cycle in intact plant tissues. Science 220: 1049–1051. [DOI] [PubMed] [Google Scholar]
- Goldman N., Yang Z. (1994). A codon-based model of nucleotide substitution for protein-coding DNA sequences. Mol. Biol. Evol. 11: 725–736. [DOI] [PubMed] [Google Scholar]
- Grundt H.H., Obermayer R., Borgen L.I.V. (2005). Ploidal levels in the arctic-alpine polyploid Draba lactea (Brassicaceae) and its low-ploid relatives. Bot. J. Linn. Soc. 147: 333–347. [Google Scholar]
- Haudry A., et al. (2013). An atlas of over 90,000 conserved noncoding sequences provides insight into crucifer regulatory regions. Nat. Genet. 45: 891–898. [DOI] [PubMed] [Google Scholar]
- Hittinger C.T., Johnston M., Tossberg J.T., Rokas A. (2010). Leveraging skewed transcript abundance by RNA-Seq to increase the genomic depth of the tree of life. Proc. Natl. Acad. Sci. USA 107: 1476–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu K.J., Montadert L., Bernoulli D., Cita M.B., Erickson A., Garrison R.E., Kidd R.B., Melieres F., Muller C., Wright R. (1977). History of the Mediterranean salinity crisis. Nature 267: 399–403. [Google Scholar]
- Hudson C.M., Puckett E.E., Bekaert M., Pires J.C., Conant G.C. (2011). Selection for higher gene copy number after different types of plant gene duplications. Genome Biol. Evol. 3: 1369–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurka H., Friesen N., German D.A., Franzke A., Neuffer B. (2012). ‘Missing link’ species Capsella orientalis and Capsella thracica elucidate evolution of model plant genus Capsella (Brassicaceae). Mol. Ecol. 21: 1223–1238. [DOI] [PubMed] [Google Scholar]
- Jaillon O., et al. French-Italian Public Consortium for Grapevine Genome Characterization (2007). The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 449: 463–467. [DOI] [PubMed] [Google Scholar]
- Janis C.M. (1993). Tertiary mammal evolution in the context of changing climates, vegetation, and tectonic events. Annu. Rev. Ecol. Evol. Syst. 24: 467–500. [Google Scholar]
- Jaramillo C., et al. (2010). Effects of rapid global warming at the Paleocene-Eocene boundary on neotropical vegetation. Science 330: 957–961. [DOI] [PubMed] [Google Scholar]
- Jiao Y., et al. (2011). Ancestral polyploidy in seed plants and angiosperms. Nature 473: 97–100. [DOI] [PubMed] [Google Scholar]
- Joly S., Heenan P.B., Lockhart P.J. (2009). A Pleistocene inter-tribal allopolyploidization event precedes the species radiation of Pachycladon (Brassicaceae) in New Zealand. Mol. Phylogenet. Evol. 51: 365–372. [DOI] [PubMed] [Google Scholar]
- Kagale S., et al. (2014). The emerging biofuel crop Camelina sativa retains a highly undifferentiated hexaploid genome structure. Nat. Commun. 5: 3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch M.A., Dobes C., Kiefer C., Schmickl R., Klimes L., Lysak M.A. (2007). Supernetwork identifies multiple events of plastid trnF(GAA) pseudogene evolution in the Brassicaceae. Mol. Biol. Evol. 24: 63–73. [DOI] [PubMed] [Google Scholar]
- Koch M.A., Kiefer M., German D.A., Al-Shehbaz I.A., Franzke A., Mummenhoff K., Schmickl R. (2012). BrassiBase: Tools and biological resources to study characters and traits in the Brassicaceae-version 1.1. Taxon 61: 1001–1009. [Google Scholar]
- Larkin M.A., et al. (2007). Clustal W and Clustal X version 2.0. Bioinformatics 23: 2947–2948. [DOI] [PubMed] [Google Scholar]
- Letunic I., Bork P. (2011). Interactive Tree Of Life v2: online annotation and display of phylogenetic trees made easy. Nucleic Acids Res. 39: W475–W478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin D.A. (1983). Polyploidy and novelty in flowering plants. Am. Nat. 122: 1–25. [Google Scholar]
- Li W., Godzik A. (2006). Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22: 1658–1659. [DOI] [PubMed] [Google Scholar]
- Liu L., Eronen J.T., Fortelius M. (2009). Significant mid-latitude aridity in the middle Miocene of East Asia. Palaeogeogr. Palaeoclimatol. Palaeoecol. 279: 201–206. [Google Scholar]
- Liu S., et al. (2014). The Brassica oleracea genome reveals the asymmetrical evolution of polyploid genomes. Nat. Commun. 5: 3930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M., Conery J.S. (2000). The evolutionary fate and consequences of duplicate genes. Science 290: 1151–1155. [DOI] [PubMed] [Google Scholar]
- Lyons E., Pedersen B., Kane J., Alam M., Ming R., Tang H., Wang X., Bowers J., Paterson A., Lisch D., Freeling M. (2008). Finding and comparing syntenic regions among Arabidopsis and the outgroups papaya, poplar, and grape: CoGe with rosids. Plant Physiol. 148: 1772–1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lysak, M.A., and Koch, M.A. (2011). Phylogeny, genome, and karyotype evolution of crucifers. In Genetics and Genomics of the Brassicaceae. R. Schmidt and I. Bancroft, eds (Gatersleben, Germany: Springer), pp. 1–32. [Google Scholar]
- Lysak M.A., Koch M.A., Pecinka A., Schubert I. (2005). Chromosome triplication found across the tribe Brassiceae. Genome Res. 15: 516–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lysak M.A., Koch M.A., Beaulieu J.M., Meister A., Leitch I.J. (2009). The dynamic ups and downs of genome size evolution in Brassicaceae. Mol. Biol. Evol. 26: 85–98. [DOI] [PubMed] [Google Scholar]
- Mandáková T., Mummenhoff K., Al-Shehbaz I.A., Mucina L., Mühlhausen A., Lysak M.A. (2012). Whole-genome triplication and species radiation in the southern African tribe Heliophileae (Brassicaceae). Taxon 61: 989–1000. [Google Scholar]
- Mandáková T., Heenan P.B., Lysak M.A. (2010a). Island species radiation and karyotypic stasis in Pachycladon allopolyploids. BMC Evol. Biol. 10: 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandáková T., Joly S., Krzywinski M., Mummenhoff K., Lysak M.A. (2010b). Fast diploidization in close mesopolyploid relatives of Arabidopsis. Plant Cell 22: 2277–2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayrose I., Zhan S.H., Rothfels C.J., Magnuson-Ford K., Barker M.S., Rieseberg L.H., Otto S.P. (2011). Recently formed polyploid plants diversify at lower rates. Science 333: 1257. [DOI] [PubMed] [Google Scholar]
- McInerney F.A., Wing S.L. (2011). The Paleocene-Eocene Thermal Maximum: A perturbation of carbon cycle, climate, and biosphere with implications for the future. Annu. Rev. Earth Planet. Sci. 39: 489–516. [Google Scholar]
- Miller K.G., Wright J.D., Fairbanks R.G. (1991). Unlocking the Ice House: Oligocene-Miocene oxygen isotopes, eustasy, and margin erosion. J. Geophys. Res. 96: 6829–6848. [Google Scholar]
- Ming R., et al. (2008). The draft genome of the transgenic tropical fruit tree papaya (Carica papaya Linnaeus). Nature 452: 991–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkin I.A., Gulden S.M., Sharpe A.G., Lukens L., Trick M., Osborn T.C., Lydiate D.J. (2005). Segmental structure of the Brassica napus genome based on comparative analysis with Arabidopsis thaliana. Genetics 171: 765–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkin I.A.P., et al. (2014). Transcriptome and methylome profiling reveals relics of genome dominance in the mesopolyploid Brassica oleracea. Genome Biol. 15: R77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quast C., Pruesse E., Yilmaz P., Gerken J., Schweer T., Yarza P., Peplies J., Glöckner F.O. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41: D590–D596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey J., Schemske D.W. (2002). Neopolyploidy in flowering plants. Annu. Rev. Ecol. Syst. 33: 589–639. [Google Scholar]
- Rokas A., Williams B.L., King N., Carroll S.B. (2003). Genome-scale approaches to resolving incongruence in molecular phylogenies. Nature 425: 798–804. [DOI] [PubMed] [Google Scholar]
- Schlueter J.A., Dixon P., Granger C., Grant D., Clark L., Doyle J.J., Shoemaker R.C. (2004). Mining EST databases to resolve evolutionary events in major crop species. Genome 47: 868–876. [DOI] [PubMed] [Google Scholar]
- Schmidt, R., and Bancroft, I. (2011). Perspectives on genetics and genomics of the Brassicaceae. In Genetics and Genomics of the Brassicaceae, R. Schmidt and I. Bancroft, eds (New York: Springer), pp. 617–632. [Google Scholar]
- Schranz M.E., Mitchell-Olds T. (2006). Independent ancient polyploidy events in the sister families Brassicaceae and Cleomaceae. Plant Cell 18: 1152–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith S.A., Dunn C.W. (2008). Phyutility: a phyloinformatics tool for trees, alignments and molecular data. Bioinformatics 24: 715–716. [DOI] [PubMed] [Google Scholar]
- Soltis D.E., Burleigh J.G. (2009). Surviving the K-T mass extinction: new perspectives of polyploidization in angiosperms. Proc. Natl. Acad. Sci. USA 106: 5455–5456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soltis D.E., Albert V.A., Leebens-Mack J., Bell C.D., Paterson A.H., Zheng C., Sankoff D., Depamphilis C.W., Wall P.K., Soltis P.S. (2009). Polyploidy and angiosperm diversification. Am. J. Bot. 96: 336–348. [DOI] [PubMed] [Google Scholar]
- Soltis D.E., Segovia-Salcedo M.C., Jordon-Thaden I., Majure L., Miles N.M., Mavrodiev E.V., Mei W., Cortez M.B., Soltis P.S., Gitzendanner M.A. (2014). Are polyploids really evolutionary dead-ends (again)? A critical reappraisal of Mayrose et al. (2011). New Phytol. 202: 1105–1117. [DOI] [PubMed] [Google Scholar]
- Song K., Lu P., Tang K., Osborn T.C. (1995). Rapid genome change in synthetic polyploids of Brassica and its implications for polyploid evolution. Proc. Natl. Acad. Sci. USA 92: 7719–7723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. (2006). RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22: 2688–2690. [DOI] [PubMed] [Google Scholar]
- Stebbins, G.L. (1950). Variation and Evolution in Plants. (New York: Columbia University Press). [Google Scholar]
- Suyama M., Torrents D., Bork P. (2006). PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 34: W609–W612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H., Wang X., Bowers J.E., Ming R., Alam M., Paterson A.H. (2008). Unraveling ancient hexaploidy through multiply-aligned angiosperm gene maps. Genome Res. 18: 1944–1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- te Beest M., Le Roux J.J., Richardson D.M., Brysting A.K., Suda J., Kubešová M., Pyšek P. (2011). The more the better? The role of polyploidy in facilitating plant invasions. Ann. Bot. (Lond.) 109: 19–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo J., Dehal P., Jarrin F., Hu J., Hermann M., Al-Shehbaz I., Quiros C.F. (1998). Genetic variability of Lepidium meyenii and other Andean Lepidium species (Brassicaceae) assessed by molecular markers. Ann. Bot. (Lond.) 82: 523–530. [Google Scholar]
- Vanneste K., Van de Peer Y., Maere S. (2013). Inference of genome duplications from age distributions revisited. Mol. Biol. Evol. 30: 177–190. [DOI] [PubMed] [Google Scholar]
- Vanneste K., Baele G., Maere S., Van de Peer Y. (2014). Analysis of 41 plant genomes supports a wave of successful genome duplications in association with the Cretaceous-Paleogene boundary. Genome Res., in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner W.H. (1970). Biosystematics and evolutionary noise. Taxon 19: 146–151. [Google Scholar]
- Wang X., et al. Brassica rapa Genome Sequencing Project Consortium (2011). The genome of the mesopolyploid crop species Brassica rapa. Nat. Genet. 43: 1035–1039. [DOI] [PubMed] [Google Scholar]
- Warwick, S.I. (2011). Brassicaceae in agriculture. In Genetics and Genomics of the Brassicaceae. R. Schmidt and I. Bancroft, eds (Gatersleben, Germany: Springer), pp. 33–66. [Google Scholar]
- Warwick S.I., Al-Shehbaz I.A. (2006). Brassicaceae: Chromosome number index and database on CD-Rom. Plant Syst. Evol. 259: 237–248. [Google Scholar]
- Warwick, S.I., Francis, A., and Gugel, R.K. (2009). Guide to wild germplasm: Brassica and allied crops (tribe Brassiceae, Brassicaceae), 3rd ed (Ottawa, Canada: Agriculture Agri-Food Research), http://www.brassica.info/info/publications/guide-wild-germplasm.php. [Google Scholar]
- Wolfe K.H. (2001). Yesterday’s polyploids and the mystery of diploidization. Nat. Rev. Genet. 2: 333–341. [DOI] [PubMed] [Google Scholar]
- Wood T.E., Takebayashi N., Barker M.S., Mayrose I., Greenspoon P.B., Rieseberg L.H. (2009). The frequency of polyploid speciation in vascular plants. Proc. Natl. Acad. Sci. USA 106: 13875–13879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. (2007). PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24: 1586–1591. [DOI] [PubMed] [Google Scholar]
- Zachos J., Pagani M., Sloan L., Thomas E., Billups K. (2001a). Trends, rhythms, and aberrations in global climate 65 Ma to present. Science 292: 686–693. [DOI] [PubMed] [Google Scholar]
- Zachos J.C., Shackleton N.J., Revenaugh J.S., Pälike H., Flower B.P. (2001b). Climate response to orbital forcing across the Oligocene-Miocene boundary. Science 292: 274–278. [DOI] [PubMed] [Google Scholar]
- Ziolkowski P.A., Kaczmarek M., Babula D., Sadowski J. (2006). Genome evolution in Arabidopsis/Brassica: conservation and divergence of ancient rearranged segments and their breakpoints. Plant J. 47: 63–74. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.