

Abstract
Few other neurotransmitters are of as intense interest to neuropsychiatry and neurology as dopamine, yet existing techniques to monitor dopamine release leave an important spatiotemporal gap in our understanding. Electrochemistry and fluorescence imaging tools have been developed to fill the gap, but these methods have important limitations. We circumvent these limitations by introducing a dopamine-gated chloride channel into rat dorsal striatal medium spiny neurons, targets of strong dopamine innervation, thereby transforming dopamine from a slow transmitter into a fast transmitter and revealing new opportunities for studying moment-to-moment regulation of dopamine release. We demonstrate pharmacological and biophysical properties of the channel that make it suitable for fast, local dopamine measurements, and we demonstrate for the first time spontaneous and evoked responses to vesicular dopamine release in the dorsal striatum. Evoked dopamine currents were separated into a fast, monosynaptic component and a slower-rising and decaying disynaptic component mediated by nicotinic receptor activation. In summary, LGC-53 represents a dopamine biosensor with properties suitable for temporal separation of distinct dopamine signals in targets of dopamine innervation.
Keywords: dopamine release, ligand-gated ion channel, nicotinic, presynaptic, striatum
Introduction
Rapid, moment-to-moment changes in transmitter release underlie behavioral adaptation and short-term forms of memory (Zucker and Regehr, 2002). In the case of dopamine (DA), alteration in DA release represents a common early effect of many addictive drugs, and changes in DA release may underlie early pathology in diseases such as Parkinson's (Garcia-Reitböck et al., 2010; Scott et al., 2010). Unfortunately, there are significant barriers to the study of short-term or rapid changes in DA release. DA activates G-protein-coupled receptors (GPCRs) that are linked indirectly to changes in membrane excitability. This typically makes endogenous GPCR-mediated signals poor sensors of amount and timing of DA release. In contrast, ligand-gated ion channels at “fast” glutamate, GABA, glycine, and peripheral nicotinic synapses (e.g., the neuromuscular junction) are high-fidelity sensors of local, synaptic transmitter release and are directly responsible for such fundamental observations as the quantal basis of release, paired-pulse facilitation/depression, and synchronous versus asynchronous transmitter release.
Limitations hamper other, non-receptor probes of DA release. Electrochemistry and microdialysis lack the spatial and temporal resolution to study rapid changes in transmitter release. They are best suited in situ to measure volumetric, spatiotemporally averaged DA transients because probes are too large to be positioned in the synapse, although amperometry can detect single vesicles when suitably placed (Staal et al., 2004). Fluorescent presynaptic reporters of DA release have been used recently (Sankaranarayanan et al., 2000; Daniel et al., 2009; Gubernator et al., 2009; Garcia-Reitböck et al., 2010; Rodriguez et al., 2013). Single presynaptic terminals can be visualized in dissociated cultures and in some cases tissue slices, but the methods do not have temporal sensitivity comparable with postsynaptic currents (PSCs; Gubernator et al., 2009; Rodriguez et al., 2013). D2-activated potassium channels have sensitivity to detect evoked (Ford et al., 2009) and quantal (Gantz et al., 2013) DA release but apparently only for specialized dendritic release within the substantia nigra/ventral tegmental area.
The above limitations have sustained an important gap in our understanding of basal DA transmission, alterations in release wrought by drugs of abuse, and changes related to pathology. To fill this gap, we introduce the use of a DA-gated chloride channel (LGC-53) from Caenorhabditis elegans (Ringstad et al., 2009) to study DA release from mammalian DA terminals in the dorsal striatum. We first examine pharmacological and biophysical properties of the receptor important in the use of LGC-53 as a biosensor of synaptic DA. By virally introducing LGC-53 into striatal medium spiny neurons, we demonstrate evoked and spontaneous phasic currents attributable to DA release. We distinguish temporally distinct components of DA release evoked by local stimulation: fast monosynaptic DA release and a surprising large and slow disynaptic component mediated by nicotinic receptor activation. We conclude that heterologous expression of LGC-53 in normal targets of DA innervation is a new tool for studying a previously poorly characterized spatiotemporal domain of DA release.
Materials and Methods
Molecular biology.
A wild-type (WT) C. elegans LGC-53 DNA sequence was used for Xenopus oocyte expression studies (Ringstad et al., 2009). The WT LGC-53 sequence can be found at the National Center for Biotechnology Information as accession number NM_171812.5. For mammalian expression, the sequence was optimized for mammalian codon usage, the signal sequence was optimized to that of rat α1 GABAA receptor (GABAAR) subunit, and predicted ER retention sequences were mutated to improve surface expression (Epoch Life Sciences). These changes to the predicted retention sequences were as follows: R398A, R399A, R400A, R439A, R440A, R441A, and K442A.
Solutions.
Bath solutions for the various experiments are referred to by letter below. Solution A (oocyte culture) contained 96 mm NaCl, 1 mm KCl, 1 mm MgCl2, 2 mm CaCl2, and 10 mm HEPES at pH 7.4, supplemented with 5 mm pyruvate, 100 U/ml penicillin, 100 μg/ml streptomycin, and 50 μg/ml gentamycin. Solution B (oocyte recording) contained the following (in mm): 96 NaCl, 1 KCl, 1 MgCl2, 2 CaCl2, and 10 HEPES at pH 7.4. Solution C (culture recording) contained the following (in mm): 138 NaCl, 4 KCl, 1 MgCl2, 2 CaCl2, 10 glucose, and 10 HEPES at pH 7.4. Solution D contained the following (in mm): 87 NaCl, 75 sucrose, 25 NaHCO3,1.25 NaH2PO4, 2.5 KCl, 0.5 CaCl2, 3 MgCl2, and 25 glucose. Solution E contained the following (in mm): 92 choline chloride, 2.5 KCl, 30 NaHCO3,1.25 NaH2PO4, 2 CaCl2, 1 MgCl2, 25 glucose, 20 HEPES, 5 sodium ascorbate, 2 thiourea, and 3 sodium pyruvate. Solution F contained the following (in mm): 125 NaCl, 25 NaHCO3, 1.25 NaH2PO4, 2.5 KCl, 2 CaCl2, 1 MgCl2, and 25 glucose.
Standard whole-cell pipette solutions included the following (in mm): 130 potassium gluconate, 4 NaCl, 5 EGTA, 0.5 CaCl2, and 10 HEPES, pH 7.3. For some experiments, when indicated, cesium gluconate, cesium methanesulfonate, cesium chloride, or potassium chloride substituted for potassium gluconate. For slice studies, 4 mm QX-314 was included in the pipette solution.
Xenopus oocytes.
Oocytes were harvested and prepared by standard methods. Briefly, stage V–VI oocytes were harvested from sexually mature female Xenopus laevis (Xenopus One) under 0.1% tricaine (3-aminobenzoic acid ethyl ester) anesthesia, according to protocols approved by the Washington University Animal Studies Committee. Oocytes were defolliculated by shaking for 20 min at 37°C in collagenase (2 mg/ml) dissolved in calcium-free solution containing the following (in mm): 96 NaCl, 2 KCl, 1 MgCl2, and 5 HEPES at pH 7.4.
Constructs were prepared in pGEMHE plasmids containing Xenopus β-globin 5′ and 3′ UTRs to stabilize mRNA. Capped mRNA encoding WT LGC-53 was transcribed in vitro using the mMESSAGE mMachine Kit (Ambion) from linearized vectors containing receptor coding regions. Subunit transcripts were injected in a volume of up to 20–50 nl and 10–50 ng of total RNA 16–24 h after defolliculation. Oocytes were incubated up to 5 d at 18°C in solution A. Xenopus oocytes were recorded using standard two-electrode voltage-clamp recordings. Oocytes were cultured in bath solution A and recorded in bath solution B using pipettes filled with 3 m KCl.
Two-electrode voltage-clamp experiments were performed with an OC725 amplifier (Warner Instruments) 2–5 d after RNA injection. The extracellular recording solution was solution B. Intracellular recording pipettes were filled with 3 m KCl and had open tip resistances of ∼1 MΩ. DA and modulators were applied from a common tip via a gravity-driven multibarrel delivery system. Except when indicated, cells were voltage clamped at −70 mV, and the peak current or the current at the end of 30 s was measured as indicated. Nicotinic antagonists were preapplied for 5 min before challenge with coapplied DA and antagonist.
Cultures.
HEK 293T and Neuro 2A (N2a) cells were cultured under standard conditions and transiently transfected with pcDNA3.1 vector encoding LGC-53. RFP or GFP was cotransfected as a marker of expression. Primary neuronal culture details have been published previously. Briefly, postnatal rat striatal neurons were seeded onto a monolayer or island of astrocytes prepared as described previously (Moulder et al., 2007) and grown for 10–14 d before use in synaptic studies.
For transfected cell lines and primary neurons, whole-cell recordings were performed in recording solution C. To isolate LGC-53 PSCs, we used 1–5 μm NBQX, 25–50 μm d-APV, and 5–10 μm gabazine. In some experiments, 100 μm picrotoxin was also added as described.
Lentivirus injections and striatal slices.
Lentivirus particles were prepared by the Hope Center Viral Vector Core at Washington University. Constructs used the FCIV vector, a lentiviral expression vector that drives gene expression under control of the ubiquitin C promoter (Li et al., 2010). In some cases, lentivirus was prepared in a vector in which expression was under the control of the synapsin I promotor, which was more effective than FCIV when injected into striatum of more mature animals (older than postnatal week 3). Coexpression of Venus fluorescent protein was controlled by an IRES sequence. Lentiviruses were produced in HEK 293T cells. Viral particles were stereotactically injected into P5 rat pups (FCIV vector) or postnatal week 3 pups (Synapsin I promoter), followed by slice harvest at P20–P22 or P30–P40 for recording. Coronal or horizontal slices were prepared with a Leica 1200S tissue slicer in low-sodium buffer D (see above). Slices were stored for 30 min at 34°C in a low-sodium choline solution E (see above); thereafter, slices were transferred to oxygenated solution F stored at ∼25°C. Slices were perfused at 2–4 ml/min with oxygenated saline F at ∼25°C with antagonists added as indicated. Whole-cell recordings from slices were performed using pipette solutions of the compositions given above.
Stimulation of evoked striatal PSCs was achieved with a whole-cell pipette filled with bath solution and connected to a stimulus isolator, with stimulus amplitudes of 5–300 μA at 100–150 μs. A 500 ms paired-pulse interval was used for the paired-pulse experiments, with a sweep interval of 60 s. For the study of nicotinic acetylcholine receptor (nAChR) and D2 pharmacology in slices, a nearly maximal PSC was elicited before nAChR antagonist or D2 agonist application by varying the stimulus intensity up to 250 μA with a 150 μs pulse width. This helped to ensure the presence of the early PSC. For matched-amplitude comparisons of early and late PSCs, stimulus strength was adjusted to produce a 1000 ± 150 pA PSC1 amplitude in the presence of 1 μm dihydro-β-erythroidine (DHβE) (early PSC) or without antagonist (compound PSC). The standard holding potential was −70 to −80 mV unless otherwise indicated. Voltages, including the current–voltage relationships in the figures, were not corrected for liquid junction potentials.
Imaging.
Images were acquired on an upright Nikon microscope equipped with infrared differential interference contrast optics, a QImaging camera, and MetaMorph software (Molecular Devices). Images were adjusted in NIH ImageJ or Adobe Photoshop (Adobe Systems) for brightness, contrast, and pseudocoloring.
Data analysis.
Electrophysiology data files were analyzed with Clampfit (versions 9.2 and 10.4; Molecular Devices) or imported into Igor (version 6.3; Wavemetrics) for analysis with custom-written routines or the plug-in NeuroMatic. Graphs were constructed with SigmaPlot (version 12.5). PSC amplitudes were measured from the baseline 5 ms before the stimulation. For biphasic responses, the first component (peak within 17 ms from the stimulus onset) was termed the early PSC, with the latter component termed the late PSC. Paired-pulse ratios were calculated after subtracting baseline current 5 ms before each stimulus. The half-decay was calculated from the peak PSC amplitude to the 50% amplitude. Statistical comparisons were performed in GraphPad Prism (version 6; GraphPad Software) using tests as indicated. All data in figures and text are presented as mean ± SE.
Results
Pharmacological and biophysical properties relevant to fast biosensor
To serve as a sensor of quasi-synaptic or synaptic DA release rather than ambient, low-level or volumetric transmitter, LGC-53 should have a relatively low agonist affinity. Consistent with a previous report (Ringstad et al., 2009), we found that LGC-53 peak currents were activated by DA with an EC50 of 6.4 μm (Fig. 1A,B). This value is comparable with receptors for fast transmitters: a slightly higher EC50 than glutamate gating NMDARs and slightly lower than glutamate gating AMPARs or GABA acting at GABAARs (Patneau and Mayer, 1990; Mortensen et al., 2010). The EC50 is much higher than mammalian DA GPCRs, with Kd values ranging from 1 to 30 nm (Seeman and Van Tol, 1994). Although under the non-steady-state conditions of synaptic release in intact cells high DA concentrations may be needed to activate GPCRs (Ford et al., 2009), a high EC50 for LGC-53 suggests that detection of low, submicromolar ambient concentrations will be minimized and that detection of high, synaptic concentrations will be favored.
Figure 1.
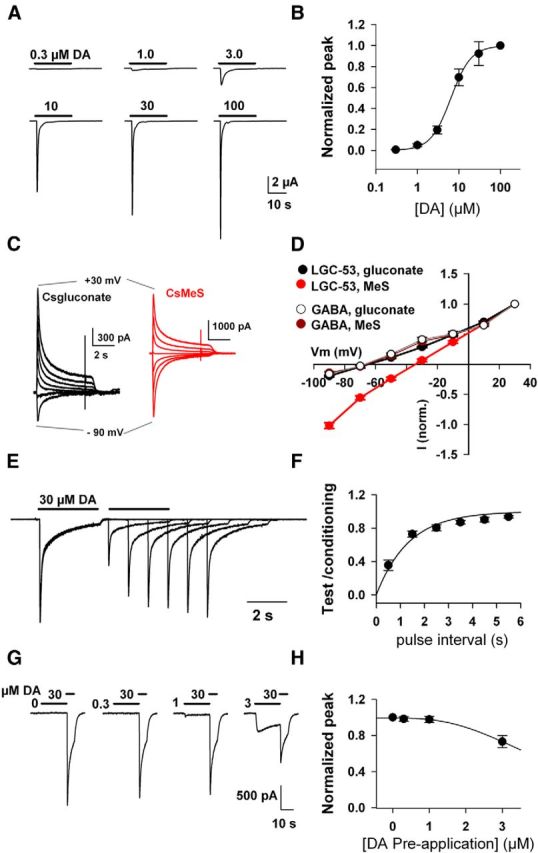
LGC-53 is an atypical anion channel. A, Responses of an oocyte expressing LGC-53 to increasing concentrations of DA. B, Concentration–response curve of peak currents at the indicated DA concentrations, normalized to responses at 100 μm DA (n = 6). The solid line is a fit to the Hill equation, which estimated an EC50 of 6.4 μm and a Hill slope of 1.8. C, N2a cells were transfected with LGC-53 DNA and challenged with DA (30 μm) in cells filled with the indicated whole-cell pipette solutions. D, Current–voltage curves compare anion permeability for LGC-53 expressed in N2a cells (30 μm DA; n = 4 for each pipette solution) and GABAA receptors in hippocampal neurons (10 μm GABA; n = 4 for each solution) in culture. E, F, Recovery from desensitization is rapid. HEK 293T cells transfected with LGC-53 were challenged with 30 μm DA, which evoked biexponential macroscopic desensitization, followed by saline wash for 500 ms. In successive sweeps, the interval was increased as indicated, and the ratio of test response to initial, conditioning response is plotted for each interval (n = 6). G, H, Steady-state desensitization properties of LGC-53 in HEK 293T cells. Significant desensitization with preapplication time of 10 s was not observed at concentrations below 3 μm (n = 5). MeS, methanesulfonate.
Previous work has suggested that LGC-53 is an anion channel. To confirm this, we compared reversal potentials of LGC-53, gated by DA, with GABAARs, gated by GABA and well-characterized anion channels (Bormann et al., 1987). LGC-53 expressed in Xenopus oocytes exhibited a reversal potential near −20 mV and was statistically indistinguishable from the reversal potential of GABA acting at α1β2γ2 GABAAR subunits in the same batches of oocytes (data not shown). However, a difference in anion permeability compared with GABAARs was revealed in whole-cell recordings from transfected mammalian cells, in which alternative intracellular anions substituted for chloride. GABA-gated GABAA channels exhibited weak gluconate and methanesulfonate permeability; these anions led to indistinguishable, negative reversal potentials, consistent with classic studies on GABAARs and glycine receptors (Bormann et al., 1987). However, DA-gated currents from LGC-53 exhibited significantly more positive reversal potentials when methanesulfonate was used as an anion than when gluconate was used (Fig. 1C,D). This result suggests atypical anion permeability for LGC-53 relative to other cys-loop chloride channels, such as the GABAAR (Fig. 1D). The observation has practical implications for the choice of pipette solutions in whole-cell recordings to optimize driving force on DA-activated LGC-53 currents. When LGC-53-transfected cells were filled with CsCl, DA-gated currents exhibited the expected reversal potential near 0 mV (data not shown).
Some transmitter receptors may exhibit limited sensitivity as a result of desensitization properties (Trussell and Fischbach, 1989; Zorumski et al., 1996; Overstreet et al., 2000). In striatal neurons (n = 4) and HEK 293T cells (n = 4) transfected with LGC-53, 30 μm DA elicited characteristic biexponential macroscopic desensitization to application of 30 μm DA (τfast = 128 ± 12 ms, τslow = 1354 ± 189 ms, 48 ± 4% fast; Fig. 1E). After desensitization by 30 μm DA, recovery from desensitization was rapid, with an average time constant of 1.3 s and full recovery by 6 s (Fig. 1E,F). This recovery time constant compares favorably with the slow component of GABAAR and NMDAR recovery from desensitization (Tong et al., 1995) but is slower than AMPAR recovery (Trussell and Fischbach, 1989). Because DA may be tonically present at significant concentrations (Goto et al., 2007), steady-state desensitization could be an impediment to detection of transient, high DA concentrations such as those found at synaptic junctions (Ford et al., 2009). However, when conditioned with concentrations of DA up to ∼3 μm, LGC-53 showed little desensitization (Fig. 1G,H), suggesting that ambient DA levels are unlikely to alter the sensitivity of LGC-53 as a synaptic or quasi-synaptic biosensor.
To serve as a faithful sensor of DA, LGC-53 should exhibit selectivity over other amine neurotransmitters. We found that, when evaluated at 100 μm, norepinephrine (NE) and epinephrine gated ≤10% of the response to DA (Fig. 2A), consistent with high, although not perfect, selectivity for DA. Average peak responsiveness to the alternate agonists (100 μm) was 10 ± 1% (NE) and 6 ± 1% (epinephrine) of the response to DA. Serotonin, histamine, and GABA failed to gate detectable LGC-53 currents at concentrations up to 100 μm (n = 5 oocytes exhibiting responses to 100 μm DA of −5.0 ± 0.5 μA).
Figure 2.

LGC-53 exhibits DA agonist selectivity but unexpected sensitivity to GABAR antagonists. A, Response of an LGC-53-expressing oocyte to 100 μm DA and each of the indicated agonists: NE, epinephrine (Epi), serotonin (5-HT), histamine, and GABA. Summary numbers are in Results. B, Normalized responses showing the effect of various modulators coapplied with 10 μm DA to oocytes expressing LGC-53. n = 4–6 oocytes for each. Screens were performed with an interleaved presentation of DA alone and DA plus test modulator; reversibility verified that small effects did not result from generalized rundown of DA responses. Because the initial screens revealed an interaction between bicuculline and LGC-53 function, other positive and negative modulators of GABAAR function were screened at the indicated concentrations. These included the endogenous neurosteroid allopregnanolone (3α5αP), the sulfated neurosteroid negative modulator pregnenolone sulfate (PS), and the high-potency negative modulator dipicrylamine (DPA) (Chisari et al., 2011). C, The effect of bicuculline, the strongest inhibitor, was overcome by increasing DA concentration (n = 5 oocytes).
We screened LGC-53 against modulators of GluRs and GABAARs. We started with compounds typically used for pharmacological isolation (NBQX to block AMPARs and bicuculline, picrotoxin, and gabazine to block GABAARs; Fig. 2B). LGC-53 exhibited unexpected sensitivity to antagonists of GABAARs. Bicuculline strongly inhibited LGC-53 at concentrations typically used to block GABA transmission (Fig. 2B,C). The antagonism was essentially overcome with increased agonist concentration, suggesting at least a partly competitive mechanism (Fig. 2C). Other antagonists of GABAARs had weaker effects (Fig. 2B), and LGC-53 also exhibited mild sensitivity to neurosteroids that potently and allosterically inhibit (pregnenolone sulfate) or potentiate (allopregnanolone, 3α5αP) GABAAR function (Eisenman et al., 2004; Fig. 2B). These observations suggest that caution is warranted when choosing the pharmacological tools used to isolate LGC-53 responses in native tissue.
Previous results suggested that LGC-53 obeys a pharmacology that is most similar to D2-type mammalian receptors (Ringstad et al., 2009). We confirmed this observation by screening various ligands at 50 μm against currents generated by 10 μm DA. We found that, among D2 antagonists tested, spiperone was the most effective antagonist (Fig. 3A,B). However, the concentration of spiperone required to substantially inhibit LGC-53 responses was significantly higher than the Ki at mammalian receptors, ∼0.06 and 0.08 nm Kd at D2 and D4 receptors, respectively (Seeman and Van Tol, 1994). The antagonism of LGC-53 by spiperone appeared at least partly competitive, because its primary effect was to shift the concentration–response profile of DA to the right (Fig. 3C). Sulpiride (10 μm), used in ensuing pharmacology experiments, weakly suppressed 5 μm DA currents (35 ± 4% depression; n = 4). Nemonapride and haloperidol weakly antagonized 5 μm DA responses (Fig. 3A,B). When tested in LGC-53-transfected striatal neurons, several additional D2 ligands tested had weak or no activity at 50 μm (respiridone, 66 ± 4% inhibition; raclopride, 24 ± 15% potentiation; eticlopride, 19 ± 12% inhibition; n = 4 cells each). The D1 ligand SKF38393 (2,3,4,5-tetrahydro-7,8-dihydroxy-1-phenyl-1H-3-benazepine HCl) also had little effect at 50 μm (Fig. 3A,B).
Figure 3.
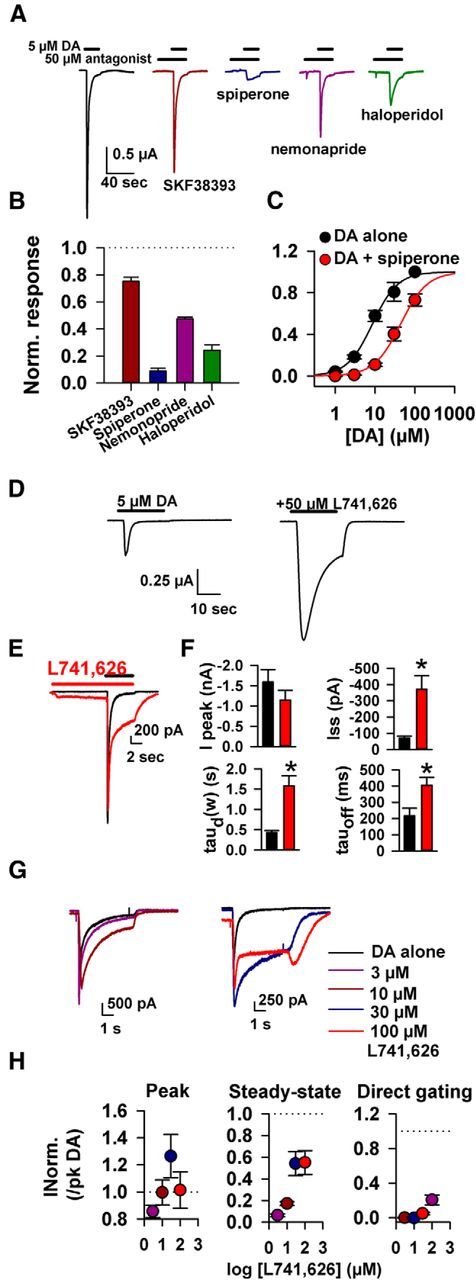
LGC-53 is sensitive to D2 ligands, which can positively or negatively modulate. A, Response of DA alone (5 μm; black trace) and in the presence of 50 μm of the indicated D1 and D2 receptor ligands from an oocyte expressing LGC-53. B, Summary of antagonist pharmacology from four to six oocytes. Antagonists were preapplied and then coapplied with DA, and peak current was normalized to DA alone. C, Spiperone (50 μm) shifted the DA EC50 to the right, suggesting a competitive interaction. Fits to the Hill equation exhibited a shift from an EC50 for DA of 8.6 μm in the absence of spiperone to 43.9 μm in the presence of 50 μm spiperone (n = 4 oocytes). D, The oocyte response to 5 μm DA in the absence and presence of L741,626 (50 μm) revealed potentiation. E, Response of a cultured striatal neuron expressing LGC-53 to 30 μm L741,626 preapplied and then coapplied with 30 μm DA (red trace). A baseline DA response is shown in black. F, Summary of the effect of L741,626 on 30 μm DA-gated LGC-53 currents in striatal neurons. Colors as in E. Measurements were made of peak current (Ipeak), steady-state current (Iss), the weighted desensitization time constant [taud(w)], and the time constant of deactivation (tauoff), measured during agonist washout. *p < 0.05. G, H, Concentration–response characteristics for the effects of L741,626 on responses to 30 μm DA transfected in striatal neurons; n = 6–7). L741,626 at the indicated concentrations was preapplied for 10 s before co-application with DA. The summary in H is color coded to traces in G.
Not all D2 antagonists behaved as LGC-53 antagonists. The D2 antagonist 3-[[4-(4-chlorophenyl)-4-hydroxypiperidin-l-yl]methyl-1H-indole (L741,626) had the interesting property of exhibiting positive allosteric modulation of LGC-53 gating (Fig. 3D–H). At concentrations above 10 μm, L741,626 slowed macroscopic desensitization of LGC-53 currents and enhanced steady-state DA responses (Fig. 3D–H). L741,626 also slowed deactivation of DA-gated currents and directly gated channels at >10 μm (Fig. 3E–H).
Although these observations suggest a potentially rich pharmacology for LGC-53, we were concerned that ligands that modulate LGC-53 may have off-target effects at the concentrations required to modulate channel function (∼50 μm), orders of magnitude higher than concentrations used to modulate D2 receptors. To address this concern, we tested the most active negative and positive modulators, spiperone and L717,626, at 50 μm on evoked PSCs from hippocampal autaptic synapses, which use glutamate as a neurotransmitter. Both compounds depressed evoked AMPAR EPSCs (34 ± 10% for 50 μm spiperone, 55 ± 9% for 50 μm L741,626; n = 3 and 6). In both cases, this depression appeared to arise primarily presynaptically, because it was associated with reversible suppression (25 ± 6%) of voltage-gated sodium current. Similar glutamate EPSC suppression was observed in acutely prepared brain slices containing striatum (50 μm spiperone, 60 ± 8% suppression; n = 5). Thus, at concentrations required to modulate LGC-53, spiperone and L717,626 have significant off-target effects.
DA-gated phasic currents in dissociated culture and brain slices
Aided by biophysical and pharmacological information about the advantages and limitations of LGC-53, we next evaluated the utility of LGC-53 as a biosensor of vesicular DA release. Potential challenges to this utility include expression levels and localization of LGC-53. As a test of principle, we first sparsely transfected primary striatal cultures with LGC-53 and loaded DA as a false transmitter into striatal presynaptic terminals. Using previously published protocols, we incubated cultures with DA during depolarization. Under these conditions, DA enters vesicles through a combination of passive mechanisms and through reloading endocytosing vesicles (Kim et al., 2000; Zhang and Zhou, 2002). After removal of extracellular DA, we established whole-cell recordings from transfected or nontransfected striatal neurons in the presence of antagonists of GluRs and GABAARs. Depolarization of the surrounding network elicited rapid, PSC-like currents in transfected neurons but never in nontransfected neurons or in neurons not previously loaded with DA (Fig. 4). These results suggest that LGC-53 can be heterologously expressed at levels sufficient to detect vesicular DA, albeit from an unknown vesicular concentration.
Figure 4.
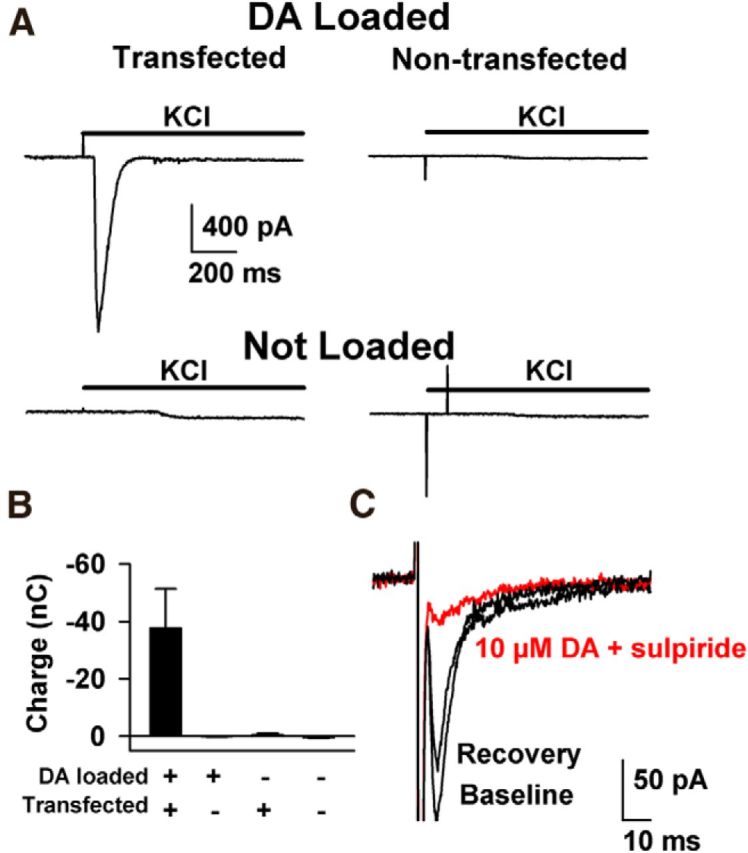
Hijacking striatal synapses to probe LGC-53 as a postsynaptic sensor. A, Cultured striatal neurons were transfected with LGC-53 plasmid DNA and marker GFP. At 10–12 d in vitro, the culture was loaded with 70 mm DA plus 30 mm KCl for 5 min to load striatal synapses with DA (Kim et al., 2000; Zhang and Zhou, 2002). Cells were then transferred to recording medium containing 1 μm NBQX, 5 μm gabazine, and 25 μm d-APV. GFP-expressing cells were clamped at −70 mV. The surrounding network was depolarized with 10 mm total KCl to reveal PSC-like events in transfected, DA-loaded cells but not other control conditions as indicated. B, Summary of the spiperone-sensitive charge obtained from the experimental conditions indicated (n = 7–15 cells per condition). In summary, 12 of 15 transfected cells showed a reversible, spiperone-sensitive response, whereas none of the 9, 8, and 7 cells in the three control conditions, respectively, yielded a response. C, Striatal cells were grown in microcultures as described previously (Moulder et al., 2007) and transduced with virus encoding LGC-53 and Venus. Cultures of one to three neurons were stimulated in the presence of GluR and GABAR blockers to obtain single-axon mediated PSCs, observed in a subset of transfected cells. Fast inward currents preceding the PSC are stimulation currents associated with depolarization. PSCs were sensitive to application of 10 μm DA plus 10 μm sulpiride to help suppress LGC-53 currents and block endogenous D2 presynaptic depression. The sulpiride and DA combination had no effect on autaptic GABA IPSCs (97 ± 5%; n = 7), suggesting no presynaptic effect of the mixture and suggesting that the suppression of LGC-53 PSCs results from DA-mediated suppression. For summary data, see Results.
To determine the time course of LGC-53-mediated currents achieved at bona fide synaptic connections with single action potential stimuli, we extended the false-transmitter loading protocol to striatal neurons cultured on astrocyte islands, which form synaptic junctions that use GABA as a transmitter (Dubinsky, 1989). After DA loading, brief depolarization of solitary neurons evoked inward, PSC-like currents in a subset of LGC-53-positive neurons with confirmed responses to exogenous DA (10 μm, >20 pA, 4 of 13 cells). These events were strongly depressed by 10 μm DA in the presence of 10 μm sulpiride, a mixture designed to desensitize/suppress LGC-53 without engaging D2-mediated presynaptic depression (Fig. 4C). DA-suppressed events were never observed in cells without functionally detectable DA-gated responses (0 of 21 cells; p < 0.05, Fisher's exact test). Thus, we interpret the evoked responses to be LGC-53-mediated PSCs. These DA-sensitive evoked events had best-fit decay time constants of 12.8 ± 4.4 ms (n = 4; range of 4.8–21 ms). Rise times (10–90%) of synaptically presented DA PSCs were 3.6 ± 0.9 ms. These rise and decay times put limits on the expectations for synaptically generated PSCs. The results suggest that, despite the lack of engineered synaptic localization signals, sufficient LGC-53 is trafficked to synapses to mediate synaptic-like PSCs under conditions in which synaptic junctions are hijacked to release DA cargo.
To extend the biosensor approach to a bona fide DA circuit that includes both morphologically synaptic and volumetric junctions (Descarries et al., 1996), we injected lentiviral particles encoding LGC-53 and the fluorescent protein Venus into the dorsal striatum of P5 rat pups. Infected striatal neurons were identified in brain slices 2 weeks after injection by fluorescence and targeted for whole-cell recording (Fig. 5A,B). Presumably as a result of restricted lentiviral expression (Fig. 5A), during normal handling of animals before tissue harvest, we noticed no obvious behavioral abnormalities in LGC-53-injected animals. However, systematic behavioral testing was not performed. We investigated the properties of electrically evoked currents resistant to GluR and GABAR blockers. In nontransduced neurons filled with cesium gluconate pipette solution, evoked PSCs mainly attributable to glutamate were observed at −70 mV. Evoked PSCs attributable to GABA were evoked at +20 mV. PSCs were entirely eliminated by the combination of 1 μm NBQX, 25–50 μm d-APV, and 5 μm gabazine (Fig. 5C; 1.0 ± 0% of baseline; n = 7). In contrast, transduced neurons often exhibited an evoked outward current at +20 mV resistant to GABAAR and GluR blockers (Fig. 5D). Residual current in 10 transduced neurons was 39 ± 14% of the baseline PSC amplitude (p < 0.05 compared with nontransduced controls). Compared with GABAA IPSCs in the same neuron (Fig. 5D), putative LGC-53-mediated currents had a similar latency but rose more slowly (rise time of evoked GABA IPSCs, 5.7 ± 0.8 ms vs 23.5 ± 3 ms for residual currents after stimulation; Fig. 5D, inset; p < 0.05). Many residual PSC-like currents exhibited an inflection on the rising phase (Fig. 5D, inset), reflecting temporally distinct components of DA release explored further below.
Figure 5.
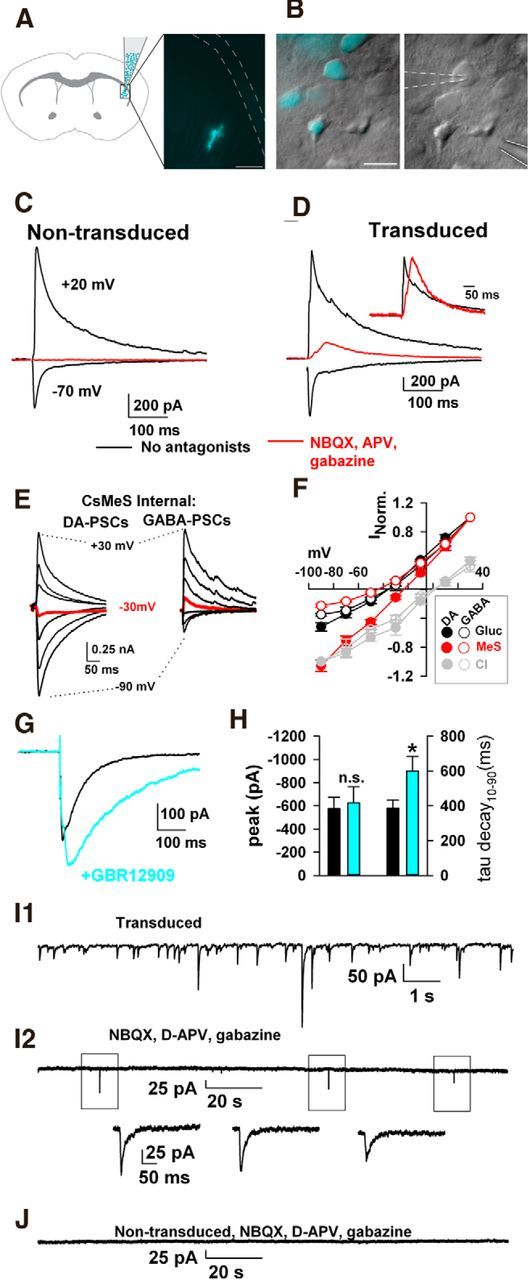
Evoked and spontaneous LGC-53 currents from striatal slices. A, Lentiviral injections into the dorsal striatum produced restricted expression in dorsolateral striatum. Scale bar, 500 μm. B, High-magnification image of transduced and nontransduced neurons in a field from the dorsolateral striatum. Whole-cell recording pipette (dashed outline) and field stimulation pipette (solid outline) are evident in the right. Scale bar, 25 μm. C, Recordings at the indicated membrane potentials from a nontransduced striatal neuron, as identified by Venus fluorescence. The red trace shows the response at +20 mV in the presence of 1 μm NBQX, 50 μm d-APV, and 5 μm gabazine. Stimulus artifacts in this and the subsequent figures have been blanked for clarity. D, In a transduced neuron, antagonist application revealed a residual current. Inset, Scaled traces showing that the residual current has a slower rise to peak than the peak-scaled mixed glutamate/GABA IPSC. E, F, Current–voltage characteristics of the antagonist-insensitive, residual PSCs mirror those in transfected cultures (Fig. 1), including the unique permeability to methanesulfonate (MeS). Curves in D represent PSCs normalized to responses at +30 mV [gluconate (Gluc) and MeS responses] or −90 mV (chloride responses). G, H, Effects of the DA transport inhibitor GBR12909 (1 μm) on residual evoked PSCs. *p < 0.05. I, J, Spontaneous LGC-53-mediated transmission. I1, Total spontaneous PSCs from a transduced neuron before wash in of NBQX, d-APV, and gabazine. I2, In the presence of antagonists, infrequent events remained (boxed events shown at higher resolution in I2). J, In a nontransduced neuron, all events were eliminated by antagonists. For summary data, see Results.
To verify that residual PSCs in transduced neurons had characteristics of LGC-53, we examined reversal potentials of the PSCs using gluconate-based versus methanesulfonate-based pipette solutions. Recordings obtained with a methanesulfonate pipette solution exhibited a reversal characteristically more positive than recordings with a gluconate pipette solution (Fig. 5E,F). In contrast, GABAAR IPSCs in nontransduced neurons exhibited a similar reversal potential regardless of whether gluconate or methanesulfonate was used as the major intracellular anion (Fig. 5E,F). The dopaminergic nature of PSCs was further verified by sensitivity of the PSC decays to the DA transporter inhibitor GBR12909 (1-[2-[bis(4-fluorophenyl)-methoxy]ethyl]-4-[3-phenylpropyl]piperazine; 1 μm; Fig. 5G,H).
We investigated whether LGC-53 imparted sensitivity to detect unitary phasic DA release by examining spontaneous events at −70 mV in chloride-filled cells. Under baseline conditions, spontaneous transmission was evident in both transduced and nontransduced neurons. In both cell types, this was mainly attributable to GABA and glutamate transmission, because most spontaneous events were blocked by a mixture of ionotropic GluR and GABAR blockers (Fig. 5I,J). However, in transduced neurons (n = 9 of 11 cells) but not nontransduced neurons (0 of 7 cells from the same cohort), infrequent events remained that exhibited a time course characteristic of synaptic currents (Fig. 5I2; amplitude range, −10 to −50 pA). Average decay time constant of these spontaneous events was 20 ± 3 ms (n = 9 neurons, 92 events in 40 min of total summated recording time). This decay time constant was significantly faster than decay of evoked responses to even the weakest stimuli presented (best-fit single-exponential time constant for evoked phasic currents, 62 ± 7 ms; amplitude, −119 ± 28 pA; n = 10). However, the decay time constant was similar to that observed in evoked PSCs in culture from false-transmitter loaded cells (Fig. 4C). We conclude that at least a fraction of bona fide spontaneous phasic DA release is captured by the LGC-53 sensor and that spontaneous release exhibits faster transmitter clearance than electrically evoked DA release.
We more closely examined the slow, asynchronous rise times of LGC-53-mediated PSCs. We wondered whether the temporally distinct components could reflect direct and indirect components of DA release, which have not heretofore been temporally distinguished. With strong optogenetic stimulation of the population of cholinergic interneurons, nicotinic-driven DA release has been demonstrated (Cachope et al., 2012; Threlfell et al., 2012; Nelson et al., 2014), occurring presumably through activation of nAChRs on DA fibers (Jones et al., 2001). We examined whether the temporally distinct components might involve direct (monosynaptic) and indirect (disynaptic) nicotinic receptor (nAChR)-driven components of DA release by applying nAChR antagonists. Both mecamylamine (10 μm) and the β2-selective antagonist DHβE (1 μm) reduced the late component of the PSC and left the rapidly rising early component (Fig. 6A,B). The effect of nicotinic antagonists did not result from a direct pharmacological effect of the drugs on LGC-53. In oocytes, LGC-53 currents were not significantly affected by up to 5 min preapplication of 10 μm mecamylamine or 1 μm DHβE (Fig. 6C). The early PSC component in striatal neurons was not a breakthrough glutamate EPSC because its reversal potential was indistinguishable from that of the late component, either when part of the compound PSC or when pharmacologically isolated (Fig. 6D,E). Furthermore, the early component was not a breakthrough GABA current because it persisted in 100 μm picrotoxin added to the omnipresent 10 μm gabazine (Fig. 6F). The early component was almost invariably smaller than the large component and often was visible only with stronger stimulation intensities. We conclude that local electrical stimulation elicits a compound LGC-53-mediated PSC in striatal targets composed of temporally distinct direct (monosynaptic) and a larger indirect (disynaptic), nAChR-driven component of DA release.
Figure 6.
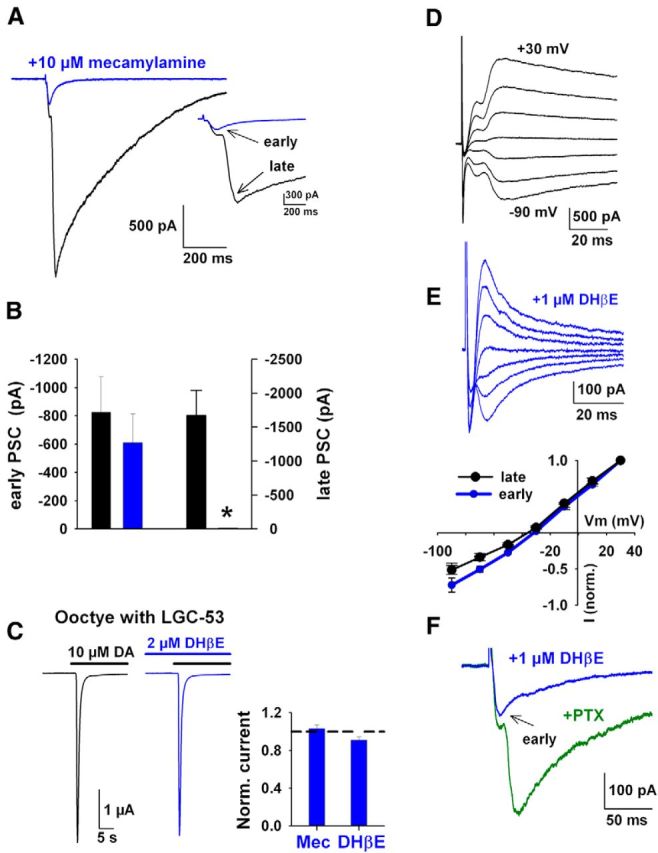
Stimulus-induced activation of nAChRs is responsible for the late phase of DA release. A, A PSC from a transduced neuron evoked before (black) and after application of an nAChR antagonism (blue; 10 μm mecamylamine). Inset shows a higher time resolution to highlight the selective suppression of the late component of the PSC. Arrows indicate the biphasic components as indicated. B, Summary of the change in PSC amplitudes with nAChR antagonism (n = 4, pooled 10 μm mecamylamine and 1 μm DHβE; *p < 0.05). C, LGC-53 is not directly blocked by nAChR antagonists. Response of an oocyte expressing LCG-53 to 10 μm DA (black line) after preapplication for 5 min of 1 μm DHβE (blue line). Inset shows summary of normalized peak currents compared with 10 μm DA alone (dashed line) from the same oocyte (n = 3 for each antagonist). D, Current–voltage characteristics from a transduced neuron with cesium gluconate internal solution before (black) and after (blue) 1 μm DHβE. E, Graph shows summary current–voltage relationship, normalized to peak responses at +30 mV (n = 3). F, The early component PSC (arrow) is retained during application of 1 μm DHβE in the presence of an additional GABAAR antagonist, 100 μm picrotoxin (PTX; 3 of 3 recordings).
The temporal distinction between nicotinic nAChR-evoked and directly evoked release has not been observed previously and highlights the utility of LGC-53. We looked more closely at the temporal characteristics of the two components and relative modulation by paired stimulation (Fig. 7). In the absence of nAChR antagonist, the late PSC peak persisted even when stimulus amplitude was reduced to simulate the overall suppressive effect of nAChR antagonists (Fig. 7A). With weak stimulation, PSCs also decayed more rapidly, consistent with the idea that DA overflow contributes to the decay of large PSCs (Fig. 7A,C). Conversely, PSCs in the presences of nAChR antagonists rose faster and decayed faster than the compound PSC, even when compound and nAChR-antagonized PSCs were matched for amplitude (Fig. 7B,C). This is despite the stronger stimulation required in the presence of nAChR antagonist to generate matched PSC amplitudes. Together, these results suggest a stronger volumetric component to nAChR-driven DA release than to direct release.
Figure 7.
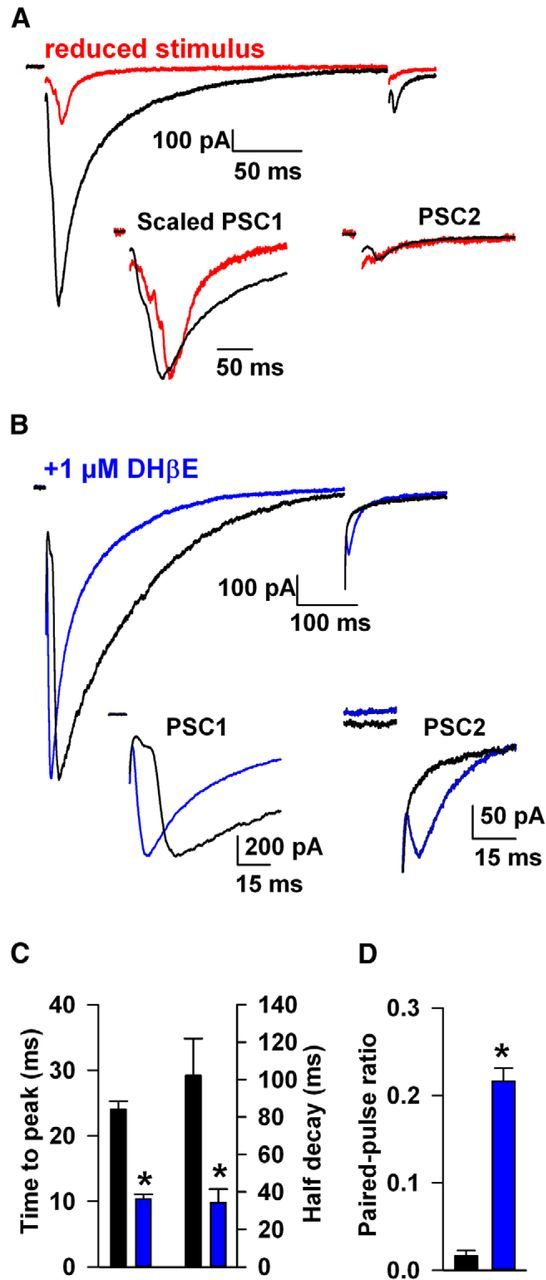
Different temporal characteristics of early and late components of the compound PSC. A, Effect of reduced stimulus amplitude (red trace) on PSC time course and paired-pulse depression. The insets show the two PSCs after scaling the first PSC peak. B, Responses to 500 ms paired stimuli in a neuron recorded in the absence (black) and another in the presence (blue) of 1 μm DHβE, matched in amplitude for the size of the PSC to highlight temporal differences. Insets, Difference in time-to-peak between the antagonist-isolated early component and the late component (absence of antagonist) PSC1 and development of PSC2 after nAChR antagonism (blue). Note the profound depression of PSC2 before nAChR block. C, Summary plot of the difference in the time-to-peak and half-decay for both the antagonist-isolated early PSC1 (−1037 ± 22 pA) and late PSC1 (−1000 ± 22 pA). D, Summary plot of paired-pulse ratio after nAChR antagonism (blue) (n = 8 without antagonist, n = 11 with antagonist; *p < 0.05).
When stimulated with twin pulses at a 500 ms interval, compound LGC-53 responses exhibited profound depression (Fig. 7A,B,D). Although we cannot entirely exclude receptor desensitization as a contributor (see Discussion), the profound depression of the compound PSC did not primarily result from DA overflow and receptor desensitization, because decreasing stimulus intensity to reduce total DA output only mildly relieved the profound paired-pulse depression (Fig. 7A). In nine cells with PSC amplitude averaging 1556 ± 472 pA, we reduced stimulus amplitude to depress the PSC by 74 ± 3%, which only marginally increased the paired-pulse ratio from 0.02 ± 0.02 to 0.10 ± 0.03 (Fig. 7A; p = 0.05, paired t test). In contrast to this small effect, paired-pulse depression was significantly relieved when nicotinic receptors were blocked and the early PSC component was isolated (Fig. 7B,D). This difference persisted even when PSC amplitude was matched across antagonist-treated and untreated cells (Fig. 7B).
Presynaptic D2 receptors are best known as autoreceptors on DA terminals. However, D2 receptors are also localized to axons and axon terminals of cholinergic fibers in the striatum (Alcantara et al., 2003). The relative effect of D2 receptor stimulation on nicotinic-driven DA release versus direct DA release is unknown. We probed the relative contribution by examining the effect of the D2 receptor agonist quinpirole (2 μm) on the two components of evoked DA release. To our surprise, D2 stimulation profoundly depressed the late PSC with a milder effect on the early PSC (Fig. 8A,C). The quinpirole effect was reversed by coaddition of the D2 receptor antagonist sulpiride (10 μm; Fig. 8B,C). As expected from the effect of the D2 agonist on both the direct and indirect components of DA release, the paired-pulse ratio was strongly elevated by quinpirole (Fig. 8A,D). We conclude that D2 activation has a disproportionate effect on the indirect, nAChR-driven component of release, in contrast to canonical views of a primarily autoreceptor function for D2 receptors.
Figure 8.
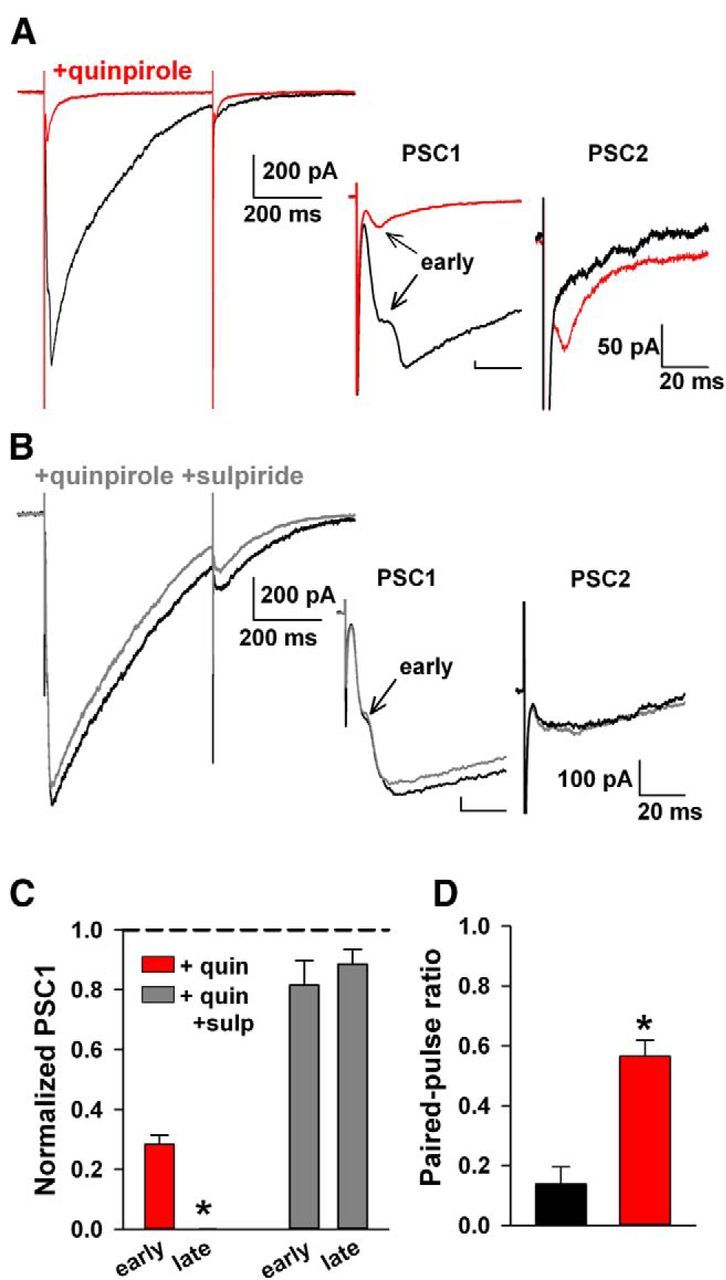
Preferential modulation of disynaptic DA release by exogenous D2 receptor stimulation. A, Paired-pulse response before (black) and after application of a D2 agonist (red; 2 μm quinpirole). Insets show the disappearance of the late PSC1 after quinpirole (red) and a more modest amplitude decrement in the early PSC1 response. Inset shows the development of PSC2 after elevated D2 receptor stimulation (red). PSC2 is displayed after subtracting current 5 ms before stimulation. B, Addition of the D2 antagonist sulpiride (10 μm, gray) blocked the effect of quinpirole. In the inset, the PSC is displayed after subtracting current 5 ms before stimulation. C, Summary of the change in PSC amplitudes with D2 receptor stimulation [n = 4 (red), n = 3 (gray); *p < 0.05]. D, Increase in the paired-pulse ratio after elevated D2 receptor stimulation can be occluded with a D2 antagonist (*p < 0.05). quin, Quinpirole; sulp, sulpiride.
Discussion
Here we introduce a simple method to transform a slow transmitter into a fast transmitter, thereby facilitating study of rapid modulation of DA release, a transmitter important to reward, volitional movement, addiction, attention, and motivation. Unlike nACh, GABA, glycine, or glutamate synapses, nature does not endow mammalian DA target cells with receptors that can be readily exploited to study fundamental features of spike-driven and spontaneous transmitter release. By equipping DA target cells with a fast neurotransmitter receptor, we revealed direct evidence for combined synaptic and volumetric transmission. For the first time, we temporally dissociate disynaptic and monosynaptic components of DA transmission and demonstrate their differential modulation.
Our method has a number of advantages that make it readily applicable. Unlike GABAARs and GluRs, LGC-53 is homomeric and thus requires transduction/transfection of only a single subunit. The low affinity and steady-state desensitization properties render the receptor relatively insensitive to tonic DA levels but sensitive to transient levels of neurotransmitter that exceed ∼1 μm. Desensitization properties are intermediate between AMPA-type GluRs on one hand (desensitization time constant of ∼10 ms) and NMDARs and GABAARs on the other hand (several hundred milliseconds desensitization time constant) and therefore do not hinder sensitivity to synaptic-like pulses of transmitter. LGC-53 currents can be pharmacologically distinguished from glutamate and GABA PSCs. The unique permeability characteristics of LGC-53 suggest that, under carefully chosen experimental conditions, LGC-53 responses can also be distinguished from endogenous glutamate and GABA PSCs on the basis of differential reversal potentials (Figs. 1C,D, 5E,F). Although introduction of a foreign gene has risks of inducing unanticipated functional changes and adaptations, it can be argued that our strategy is less invasive than amperometry, microdialysis, or even optical probes, which can introduce tissue disruption and photodynamic alterations.
However, the LGC-53 expression strategy suffers from some disadvantages. Selectivity for DA is high, but care is needed because of the low sensitivity to NE and epinephrine. Fortunately, NE release in the striatum seems unlikely to be a serious confound (Doucet et al., 1986). Furthermore, overlap in GABAAR and LGC-53 antagonist pharmacology and off-target effects of DA ligands at concentrations needed to modulate LGC-53 are limitations. Surface LGC-53 expression levels can vary. Although some of this variability could result from differences among target cells in the degree of DA innervation, surface expression variability likely also participates.
Are the LGC-53 responses from striatal neurons synaptic? Spontaneous events appear to be closest to true synaptic events, representing single-fiber and possibly even quantal transmission. It seems likely that these events arise from a subset of the ∼30% of true synaptic appositions in striatum (Descarries et al., 1996). Based on the decay time course, most evoked events involve a component of volumetric spillover, with disynaptic nAChR responses exhibiting the strongest volumetric component. This strong volumetric component likely results from the large spatial coverage of the axon collaterals of the cholinergic interneurons (Lehmann and Langer, 1983; Contant et al., 1996). A single local electrical stimulus appears to activate widespread indirect release of DA, probably extending well beyond DA terminals near the site of stimulation. The combined overflow from these terminals is sensed by the target, recorded neuron. In contrast, the same stimulus directly evokes release from a smaller subset of DA terminals (Fig. 6). Even these smaller direct responses likely contain a volumetric component, because a large majority of striatal DA terminals are morphologically nonsynaptic (Descarries et al., 1996). In comparison, only 4% of terminals of the conventional fast transmitter glutamate in the hippocampal CA1 region exhibit a nonsynaptic morphology (Harris and Weinberg, 2012). Thus, although LGC-53 is capable of detecting fast, quasi-synaptic responses (Fig. 4) with estimated “true” synaptic rise times of <5 ms and decay times of ∼20 ms given by false-transmitter experiments, the microanatomy and physiology of DA connections ensure mixed volumetric and quasi-synaptic responses.
Both direct and nAChR-indirect DA release exhibited strong paired-pulse depression, implying a surprisingly high DA vesicular release probability. This is in contrast to views of DA release properties studied recently with optical techniques in dissociated culture (Daniel et al., 2009; but see Cragg, 2003; Adrover et al., 2014). High release probability is often associated with fast synapses that require highly reliable postsynaptic activation (Zucker and Regehr, 2002). The profound, nearly complete depression of compound DA responses (Fig. 7) is consistent with the idea that nAChR activation promotes a very high release probability in terminals, thus usurping and occluding direct release (Threlfell et al., 2012). Because of our use of a ligand-gated ion channel that exhibits desensitization, we cannot exclude a role for desensitization in the paired-pulse depression observed (Figs. 7, 8). However, a very recent study using amperometry, a technique not subject to desensitization, also found evidence for strong paired-pulse depression of DA responses in rodent striatum (Wang et al., 2014). This result validates our conclusion that high presynaptic release probability and vesicle depletion are the main contributors to paired-pulse depression detected by LGC-53.
Our studies also reveal surprising differential modulation of monosynaptic and disynaptic components by D2 receptor activation. DA is known to regulate cholinergic interneuron firing in complex ways. An important effect of D2 receptor activation on cholinergic interneurons is to decrease the high tonic firing rate (Ding et al., 2010). D2 receptors are also present on cholinergic presynaptic elements (Pisani et al., 2000; Alcantara et al., 2003). However, the functionality of these receptors is unclear. Previous results demonstrated that quinpirole reduces nAChR-driven DA release, but the location of the D2 receptors remained unclear (Threlfell et al., 2012). Our results suggest that D2 activation on the ACh fibers themselves dramatically decreases cholinergic output, resulting in nearly complete elimination of the indirect DA response (Fig. 8). The weaker suppression of direct DA release suggests that the quinpirole effect cannot be accounted for solely by D2 autoreceptors on the DA terminals. Thus, direct presynaptic suppression can be added to the mechanisms of negative feedback regulation between the DA and cholinergic systems within the striatum.
Our studies reveal temporal facets of DA release in the dorsal striatum. We temporally distinguish direct DA release from stronger, nAChR-mediated release with longer-lived actions. We demonstrate the differential modulation of monosynaptic and disynaptic components by paired-pulse stimulation and an unexpectedly strong effect of D2 receptor activation on the nAChR-driven component of DA release. We conclude that heterologous LGC-53 expression offers opportunities for studying DA in a spatiotemporal domain that has been limited previously.
Footnotes
This work was supported by the McDonnell Center for Cellular and Molecular Neuroscience, the Taylor Family Institute for Innovative Psychiatric Research, and National Institute on Drug Abuse Grants R21DA032915, and T32DA007261. We thank members of the Mennerick and Zorumski groups and Karen O'Malley for advice and comments. Thanks to Niels Ringstad for the gift of wild-type LGC-53 and Joe Henry Steinbach for suggestion on ER retention and export motifs.
The authors declare no competing financial interests.
References
- Adrover MF, Shin JH, Alvarez VA. Glutamate and dopamine transmission from midbrain dopamine neurons share similar release properties but are differentially affected by cocaine. J Neurosci. 2014;34:3183–3192. doi: 10.1523/JNEUROSCI.4958-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcantara AA, Chen V, Herring BE, Mendenhall JM, Berlanga ML. Localization of dopamine D2 receptors on cholinergic interneurons of the dorsal striatum and nucleus accumbens of the rat. Brain Res. 2003;986:22–29. doi: 10.1016/S0006-8993(03)03165-2. [DOI] [PubMed] [Google Scholar]
- Bormann J, Hamill OP, Sakmann B. Mechanism of anion permeation through channels gated by glycine and gamma-aminobutyric acid in mouse cultured spinal neurones. J Physiol. 1987;385:243–286. doi: 10.1113/jphysiol.1987.sp016493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cachope R, Mateo Y, Mathur BN, Irving J, Wang HL, Morales M, Lovinger DM, Cheer JF. Selective activation of cholinergic interneurons enhances accumbal phasic dopamine release: setting the tone for reward processing. Cell Rep. 2012;2:33–41. doi: 10.1016/j.celrep.2012.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chisari M, Wu K, Zorumski CF, Mennerick S. Hydrophobic anions potently and uncompetitively antagonize GABAA receptor function in the absence of a conventional binding site. Br J Pharmacol. 2011;164:667–680. doi: 10.1111/j.1476-5381.2011.01396.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contant C, Umbriaco D, Garcia S, Watkins KC, Descarries L. Ultrastructural characterization of the acetylcholine innervation in adult rat neostriatum. Neuroscience. 1996;71:937–947. doi: 10.1016/0306-4522(95)00507-2. [DOI] [PubMed] [Google Scholar]
- Cragg SJ. Variable dopamine release probability and short-term plasticity between functional domains of the primate striatum. J Neurosci. 2003;23:4378–4385. doi: 10.1523/JNEUROSCI.23-10-04378.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel JA, Galbraith S, Iacovitti L, Abdipranoto A, Vissel B. Functional heterogeneity at dopamine release sites. J Neurosci. 2009;29:14670–14680. doi: 10.1523/JNEUROSCI.1349-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Descarries L, Watkins KC, Garcia S, Bosler O, Doucet G. Dual character, asynaptic and synaptic, of the dopamine innervation in adult rat neostriatum: a quantitative autoradiographic and immunocytochemical analysis. J Comp Neurol. 1996;375:167–186. doi: 10.1002/(SICI)1096-9861(19961111)375:2<167::AID-CNE1>3.3.CO%3B2-0. [DOI] [PubMed] [Google Scholar]
- Ding JB, Guzman JN, Peterson JD, Goldberg JA, Surmeier DJ. Thalamic gating of corticostriatal signaling by cholinergic interneurons. Neuron. 2010;67:294–307. doi: 10.1016/j.neuron.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doucet G, Descarries L, Garcia S. Quantification of the dopamine innervation in adult rat neostriatum. Neuroscience. 1986;19:427–445. doi: 10.1016/0306-4522(86)90272-1. [DOI] [PubMed] [Google Scholar]
- Dubinsky JM. Development of inhibitory synapses among striatal neurons in vitro. J Neurosci. 1989;9:3955–3965. doi: 10.1523/JNEUROSCI.09-11-03955.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenman LN, He Y, Covey DF, Zorumski CF, Mennerick S. Potentiation and inhibition of GABAA receptor function by neuroactive steroids. In: Smith SS, editor. Neurosteroid effects in the central nervous system. Boca Raton, FL: CRC; 2004. pp. 95–117. [Google Scholar]
- Ford CP, Phillips PE, Williams JT. The time course of dopamine transmission in the ventral tegmental area. J Neurosci. 2009;29:13344–13352. doi: 10.1523/JNEUROSCI.3546-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gantz SC, Bunzow JR, Williams JT. Spontaneous inhibitory synaptic currents mediated by a G protein-coupled receptor. Neuron. 2013;78:807–812. doi: 10.1016/j.neuron.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Reitböck P, Anichtchik O, Bellucci A, Iovino M, Ballini C, Fineberg E, Ghetti B, Della Corte L, Spano P, Tofaris GK, Goedert M, Spillantini MG. SNARE protein redistribution and synaptic failure in a transgenic mouse model of Parkinson's disease. Brain. 2010;133:2032–2044. doi: 10.1093/brain/awq132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto Y, Otani S, Grace AA. The Yin and Yang of dopamine release: a new perspective. Neuropharmacology. 2007;53:583–587. doi: 10.1016/j.neuropharm.2007.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubernator NG, Zhang H, Staal RG, Mosharov EV, Pereira DB, Yue M, Balsanek V, Vadola PA, Mukherjee B, Edwards RH, Sulzer D, Sames D. Fluorescent false neurotransmitters visualize dopamine release from individual presynaptic terminals. Science. 2009;324:1441–1444. doi: 10.1126/science.1172278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris KM, Weinberg RJ. Ultrastructure of synapses in the mammalian brain. Cold Spring Harb Perspect Biol. 2012;4:a005587. doi: 10.1101/cshperspect.a005587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones IW, Bolam JP, Wonnacott S. Presynaptic localisation of the nicotinic acetylcholine receptor beta2 subunit immunoreactivity in rat nigrostriatal dopaminergic neurones. J Comp Neurol. 2001;439:235–247. doi: 10.1002/cne.1345. [DOI] [PubMed] [Google Scholar]
- Kim KT, Koh DS, Hille B. Loading of oxidizable transmitters into secretory vesicles permits carbon-fiber amperometry. J Neurosci. 2000;20:RC101. doi: 10.1523/JNEUROSCI.20-20-j0001.2000. (1–5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann J, Langer SZ. The striatal cholinergic interneuron: synaptic target of dopaminergic terminals? Neuroscience. 1983;10:1105–1120. doi: 10.1016/0306-4522(83)90102-1. [DOI] [PubMed] [Google Scholar]
- Li M, Husic N, Lin Y, Christensen H, Malik I, McIver S, LaPash Daniels CM, Harris DA, Kotzbauer PT, Goldberg MP, Snider BJ. Optimal promoter usage for lentiviral vector-mediated transduction of cultured central nervous system cells. J Neurosci Methods. 2010;189:56–64. doi: 10.1016/j.jneumeth.2010.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortensen M, Ebert B, Wafford K, Smart TG. Distinct activities of GABA agonists at synaptic- and extrasynaptic-type GABAA receptors. J Physiol. 2010;588:1251–1268. doi: 10.1113/jphysiol.2009.182444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulder KL, Jiang X, Taylor AA, Shin W, Gillis KD, Mennerick S. Vesicle pool heterogeneity at hippocampal glutamate and GABA synapses. J Neurosci. 2007;27:9846–9854. doi: 10.1523/JNEUROSCI.2803-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson AB, Hammack N, Yang CF, Shah NM, Seal RP, Kreitzer AC. Striatal cholinergic interneurons Drive GABA release from dopamine terminals. Neuron. 2014;82:63–70. doi: 10.1016/j.neuron.2014.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overstreet LS, Jones MV, Westbrook GL. Slow desensitization regulates the availability of synaptic GABAA receptors. J Neurosci. 2000;20:7914–7921. doi: 10.1523/JNEUROSCI.20-21-07914.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patneau DK, Mayer ML. Structure-activity relationships for amino acid transmitter candidates acting at N-methyl-d-aspartate and quisqualate receptors. J Neurosci. 1990;10:2385–2399. doi: 10.1523/JNEUROSCI.10-07-02385.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisani A, Bonsi P, Centonze D, Calabresi P, Bernardi G. Activation of D2-like dopamine receptors reduces synaptic inputs to striatal cholinergic interneurons. J Neurosci. 2000;20:RC69. doi: 10.1523/JNEUROSCI.20-07-j0003.2000. (1–6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringstad N, Abe N, Horvitz HR. Ligand-gated chloride channels are receptors for biogenic amines in C. elegans. Science. 2009;325:96–100. doi: 10.1126/science.1169243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez PC, Pereira DB, Borgkvist A, Wong MY, Barnard C, Sonders MS, Zhang H, Sames D, Sulzer D. Fluorescent dopamine tracer resolves individual dopaminergic synapses and their activity in the brain. Proc Natl Acad Sci U S A. 2013;110:870–875. doi: 10.1073/pnas.1213569110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaranarayanan S, De Angelis D, Rothman JE, Ryan TA. The use of pHluorins for optical measurements of presynaptic activity. Biophys J. 2000;79:2199–2208. doi: 10.1016/S0006-3495(00)76468-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott DA, Tabarean I, Tang Y, Cartier A, Masliah E, Roy S. A pathologic cascade leading to synaptic dysfunction in α-synuclein-induced neurodegeneration. J Neurosci. 2010;30:8083–8095. doi: 10.1523/JNEUROSCI.1091-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeman P, Van Tol HH. Dopamine receptor pharmacology. Trends Pharmacol Sci. 1994;15:264–270. doi: 10.1016/0165-6147(94)90323-9. [DOI] [PubMed] [Google Scholar]
- Staal RG, Mosharov EV, Sulzer D. Dopamine neurons release transmitter via a flickering fusion pore. Nat Neurosci. 2004;7:341–346. doi: 10.1038/nn1205. [DOI] [PubMed] [Google Scholar]
- Threlfell S, Lalic T, Platt NJ, Jennings KA, Deisseroth K, Cragg SJ. Striatal dopamine release is triggered by synchronized activity in cholinergic interneurons. Neuron. 2012;75:58–64. doi: 10.1016/j.neuron.2012.04.038. [DOI] [PubMed] [Google Scholar]
- Tong G, Shepherd D, Jahr CE. Synaptic desensitization of NMDA receptors by calcineurin. Science. 1995;267:1510–1512. doi: 10.1126/science.7878472. [DOI] [PubMed] [Google Scholar]
- Trussell LO, Fischbach GD. Glutamate receptor desensitization and its role in synaptic transmission. Neuron. 1989;3:209–218. doi: 10.1016/0896-6273(89)90034-2. [DOI] [PubMed] [Google Scholar]
- Wang L, Zhang X, Xu H, Zhou L, Jiao R, Liu W, Zhu F, Kang X, Liu B, Teng S, Wu Q, Li M, Dou H, Zuo P, Wang C, Wang S, Zhou Z. Temporal components of cholinergic terminal to dopaminergic terminal transmission in dorsal striatum slices of mice. J Physiol. 2014 doi: 10.1113/jphysiol.2014.271825. Advance online publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Zhou Z. Ca2+-independent but voltage-dependent secretion in mammalian dorsal root ganglion neurons. Nat Neurosci. 2002;5:425–430. doi: 10.1038/nn845. [DOI] [PubMed] [Google Scholar]
- Zorumski CF, Mennerick S, Que J. Modulation of excitatory synaptic transmission by low concentrations of glutamate in cultured rat hippocampal neurons. J Physiol. 1996;494:465–477. doi: 10.1113/jphysiol.1996.sp021506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]